")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Clinical Utility of Elosulfase Alfa in the Treatment of Morquio A Syndrome
Authors Lee CL, Chuang CK, Chiu HC, Tu RY, Lo YT, Chang YH, Lin SP, Lin HY
Received 27 September 2021
Accepted for publication 7 December 2021
Published 10 January 2022 Volume 2022:16 Pages 143—154
DOI https://doi.org/10.2147/DDDT.S219433
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Chung-Lin Lee,1– 5 Chih-Kuang Chuang,6,7 Huei-Ching Chiu,1 Ru-Yi Tu,6 Yun-Ting Lo,5 Ya-Hui Chang,1,5 Shuan-Pei Lin,1,3,5,6,8 Hsiang-Yu Lin1,3– 6,9
1Department of Pediatrics, MacKay Memorial Hospital, Taipei, Taiwan; 2Institute of Clinical Medicine, National Yang-Ming Chiao-Tung University, Taipei, Taiwan; 3Department of Medicine, MacKay Medical College, New Taipei City, Taiwan; 4MacKay Junior College of Medicine, Nursing and Management, Taipei, Taiwan; 5Department of Rare Disease Center, MacKay Memorial Hospital, Taipei, Taiwan; 6Division of Genetics and Metabolism, Department of Medical Research, MacKay Memorial Hospital, Taipei, Taiwan; 7College of Medicine, Fu-Jen Catholic University, Taipei, Taiwan; 8Department of Infant and Child Care, National Taipei University of Nursing and Health Sciences, Taipei, Taiwan; 9Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, Taiwan
Correspondence: Shuan-Pei Lin; Hsiang-Yu Lin
Department of Pediatrics, MacKay Memorial Hospital, No. 92, Sec. 2, Chung-Shan North Road, Taipei, 10449, Taiwan
Tel +886-2-2543-3535 ext. 3090
; +886-2-2543-3535 ext. 3089
Fax +886-2-2543-3642
Email [email protected]; [email protected]
Abstract: Mucopolysaccharidosis type IVA (MPS IVA or Morquio A) is an autosomal recessive disorder and is one of the lysosomal storage diseases. Patients with MPS IVA have a striking skeletal phenotype but normal intellect. The phenotypic continuum of MPS IVA ranges from severe and rapid progress to mild and slow progress. The diagnosis of MPS IVA is usually suspected based on abnormal bone findings and dysplasia on physical examination and radiographic investigation in the preschool years. In the past, only supportive care was available. Due to the early and severe skeletal abnormalities, the orthopedic specialist was usually the main care provider. However, patients need aggressive monitoring and management of their systemic disease. Therefore, they need an interdisciplinary team for their care, comprising medical geneticists, cardiologists, pulmonary specialists, gastroenterologists, otolaryngologists, audiologists, and ophthalmologists. After the US Food and Drug Administration approved elosulfase alfa in 2014, patients older than 5 years could benefit from this treatment. Clinical trials showed clinically meaningful improvements with once-a-week intravenous dosing (2.0 mg/kg per week), significantly improving the 6min walk test, the 3min stair climb test, and respiratory function when compared with placebo. Elosulfase alfa is well-tolerated, and there is a good response indicated by decreasing urine glycosaminoglycans.
Keywords: elosulfasealfa, enzyme replacement therapy, GALNS, lysosomal storage disorder, Morquio A, MPS IVA, mucopolysaccharidosis
Introduction
Mucopolysaccharidosis IVA (MPS IVA, Morquio A syndrome, Morquio–Brailsford syndrome, OMIM 253000) is a lysosomal storage disease (LSD). It is an inherited autosomal recessive disorder characterized by a deficiency in the enzyme N-acetylgalactosamine-6-sulfatase (GALNS; EC 3.1.6.4) due to mutations of the GALNS gene. The result is an accumulation of the glycosaminoglycans (GAGs), keratan sulfate (KS), and chondroitin-6-sulfate (C6S) in the tissues, bones, and major organs.1‒5
Morquio A syndrome was first described by a pediatric physician, Dr. Luis Morquio, from Uruguay, and an English radiologist, Dr. James Brailsford, from Birmingham, UK, in 1929.6,7 Therefore, the disease is sometimes called the Morquio–Brailsford syndrome. Dr. Morquio described 4 siblings from a Swedish family with corneal clouding, aortic valve disease, and high levels of urinary KS (uKS).6
The prevalence of MPS IVA is 1/323,000 in Denmark, 1/599,000 in the UK, 1/926,000 in Australia, and 1/1,872,000 in Malaysia. The birth prevalence rate of MPS IVA ranges from 1/71,000 in the United Arab Emirates to 1/500,000 in Japan.8,9 Patients with Morquio A syndrome appear healthy at birth and, subsequently, develop multiorgan signs and symptoms. They may be diagnosed as early as 6 months old.
The GALNS gene is located on chromosome 16q24.10 It contains 14 exons with 2339 base pairs and is approximately 50 kb. It encodes a 522-amino acid enzyme. There are numerous heterogeneous mutations of the GALNS gene which can occur throughout the coding sequence. According to ClinVar Database, as of August 2021, 738 known variants of the GALNS gene were found, of which 276 are pathogenic or likely pathogenic, 327 are of unknown clinical significance, and 135 are benign or likely benign.
The signs and symptoms of patients with MPS IVA include short stature for their age with a short neck, abnormal skeletal development and spinal deformities, joint hypermobility and laxity, genu valgum (knock knees), large elbows and wrists, a waddling gait, and a bell-shaped chest. They may have deficient tooth enamel, abnormal heart development, respiratory compromise, corneal clouding, hearing impairment, and mild hepatosplenomegaly11‒16. Patients also have greater anesthetic risks because they have macroglossia, temporomandibular joint stiffness, difficult or failed intubations, abnormal laryngeal anatomy, trachea deformity, and subglottic narrowing.17 Figure 1 showed computed tomography angiogram in a 16-year-old boy with MPS IVA.18 The deficient enzyme activity and resulting abnormal GAG excretion can be detected in the first trimester.19,20
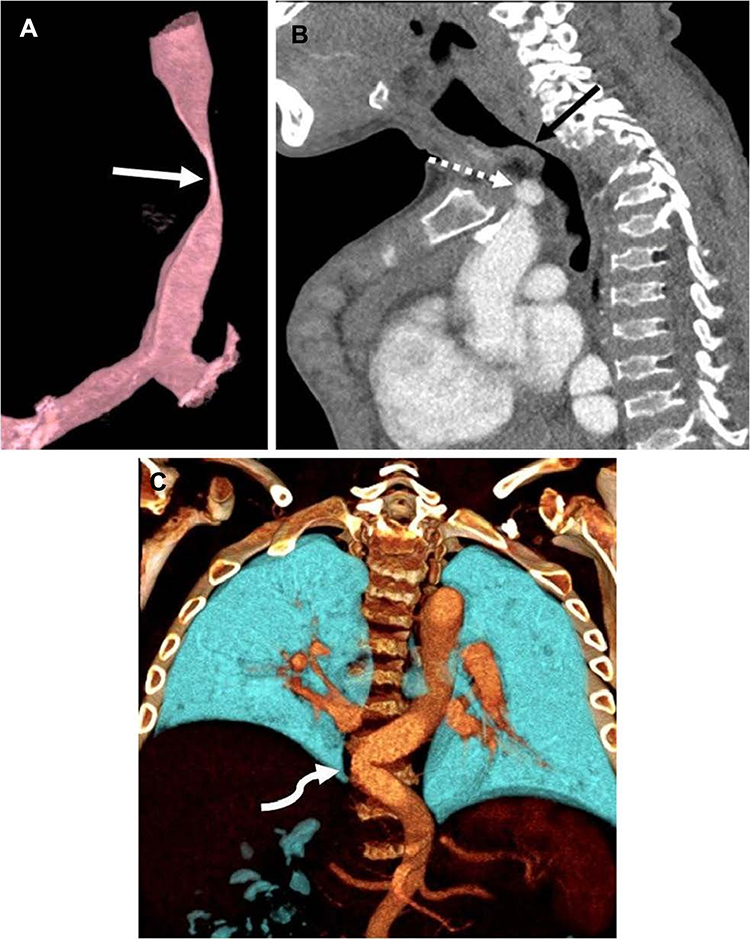 |
Figure 1 Computed tomography angiogram in a 16-year-old boy with mucopolysaccharidosis type IVA. (A) Three-dimensional reconstruction of the trachea in a sagittal oblique projection shows a severe narrowing of the trachea at the thoracic inlet (arrow). (B) Sagittal image shows that the brachiocephalic artery (dashed arrow) contributes to crowding at the thoracic inlet but does not directly indent the trachea (solid arrow). (C) Three-dimensional coronal reconstruction posteriorly in the chest shows tortuosity of the descending thoracic aorta, crossing the midline (arrow). Reprinted by permission from Springer Nature, Averill LW, Kecskemethy HH, Theroux MC, et al. Tracheal narrowing in children and adults with mucopolysaccharidosis type IVA: evaluation with computed tomography angiography. Pediatr Radiol. 2021;51(7):1202–1213. Figure 1 is copyright protected and excluded from the open access licence.18 |
Compared with other types of MPS, Morquio A does not involve the brain or cause significant impairment of cognitive development.21,22 The range of clinical manifestations is diverse in Morquio A. Due to the genetic heterogeneity, a spectrum of disease progression is present (ie, severe/classical, intermediate, and mild/attenuated). Patients with milder “nonclassical/attenuated” forms can have a normal life expectancy.23 According to a study by Lavery et al,24 the main cause of death in nearly two-thirds of patients (63%) with Morquio A is respiratory failure. Other causes are cardiac failure (11%), posttraumatic organ failure (11%), complications of surgery (11%), and myocardial infarction (4%).
Diagnosis of Morquio A
Morquio A syndrome is initially suspected because of the appearance of the symptoms described above. However, differentiating it from other disorders is difficult because of its heterogeneous expressions, which may delay the diagnosis.23,25 At ages 2 to 3 years, the symptoms progress due to a significant GAG accumulation in the tissues, bones, and organs. Because there is a natural decline in uKS levels with age, age-appropriate reference ranges in screening tests are needed to avoid false negatives, especially in older patients.26,27 To confirm the diagnosis of Morquio A syndrome, we must detect low GALNS activity in leukocytes and fibroblasts and perform molecular testing.23,26
Management of Morquio A
A multidisciplinary approach is necessary for managing Morquio A. Having orthopedic intervention in MPS IVA patients is important.28 They need an experienced anesthesia care team because of the possible airway complications secondary to GAGs accumulation in the upper and lower airways. There may be odontoid hypoplasia, impaired respiratory function, and cardiac problems.29,30 Appropriate growth charts should be used to avoid severe growth abnormalities in children.4 Because of the abnormalities of the spine and malalignment of both upper and lower extremities, MPS IVA patients usually have disabilities. They should have a close evaluation by orthopedics specialists and neurosurgeons at least annually. Because of the risk of spinal cord compression, they should have a neurological examination at least every 6 months, including annual radiography of the spine and lower extremities and whole-spine MRI or when clinically indicated.
The involvement of a pulmonary specialist is needed to manage respiratory compromise and sleep apnea.28,31,32 A cardiology team should also closely follow patients to monitor and treat valvular heart disease.33,34 For hearing impairment, an audiology team should treat them aggressively as necessary, and eye examinations and intervention for corneal clouding will help to optimize visual outcomes.3 If patients have conductive hearing loss due to middle ear fluid, they should be treated with ventilation tubes. Patients should have slit lamp biomicroscope of the cornea. If vision is reduced due to corneal clouding, keratoplasty is considered. Although hepatomegaly is not acutely dangerous, other organs may be affected by the size of the liver because MPS IVA patients have short stature. Furthermore, pain and cosmetic difficulties may develop due to dental abnormalities if patients do not have appropriate dental care early.3 To reduce the total number of hospital visits and facilitate care coordination, multidisciplinary clinics can improve patient satisfaction and quality of life.
The care team needs to improve the learning and social environment for MPS IVA patients due to their numerous systemic complications. MPS IVA patients have normal intelligence; thus, optimizing the environment and neurologic protection can prevent damage and facilitate good outcomes for patients.28 The “International guidelines for the management and treatment of MPS IVA syndrome” was released in 2014. The guidelines summarized the extensive experience in managing MPS IVA based on two meetings of international experts. It includes an extensive baseline evaluation and reevaluation at intervals to identify systemic changes early to allow for monitoring and intervention. Table 1 shows the monitoring recommendations for MPS IVA patients.22,28,35
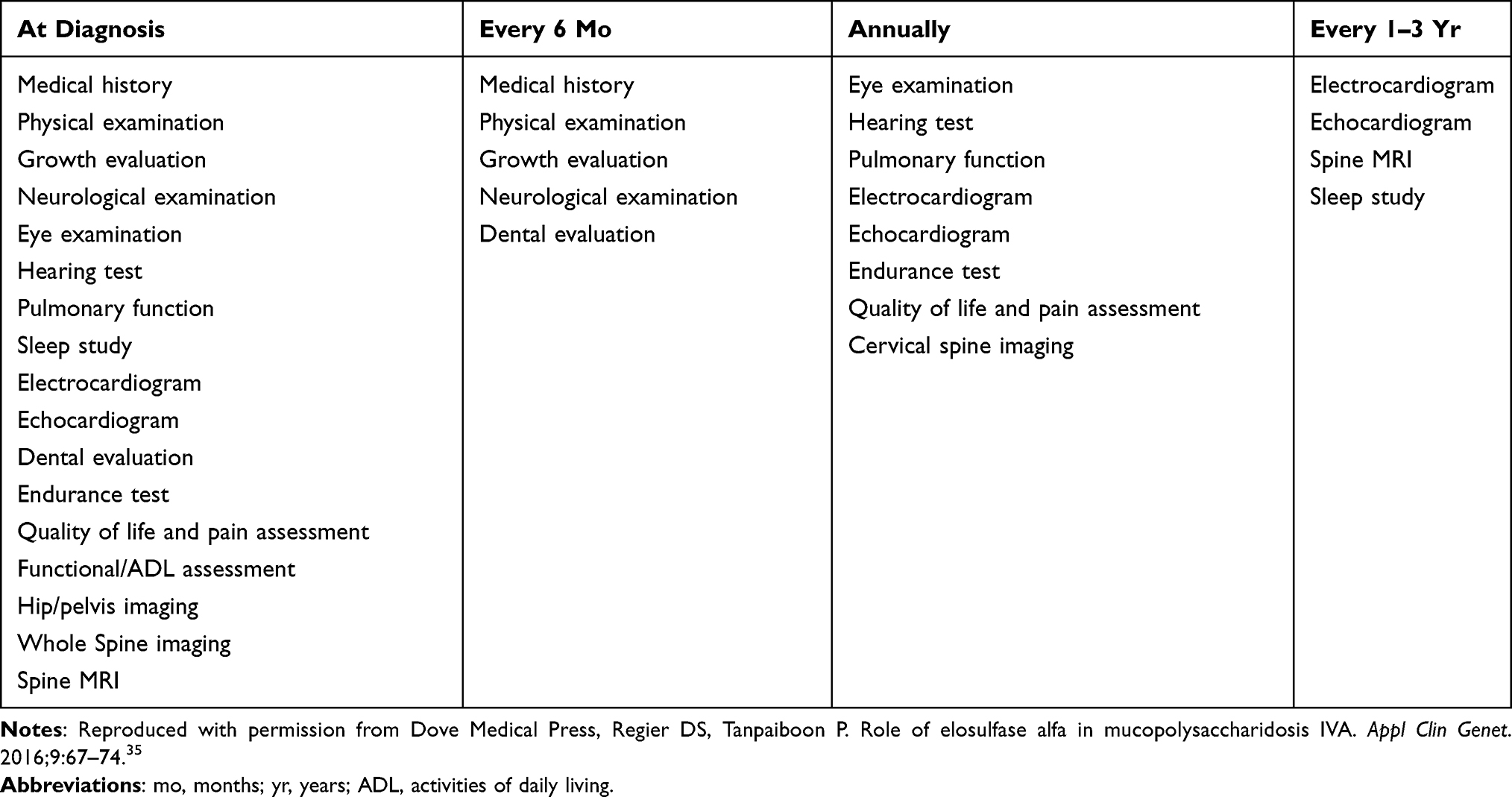 |
Table 1 Monitoring Recommendations for Mucopolysaccharidosis Type IVA Patients |
Until recently, MPS IVA patients only had supportive care for their symptoms like adenotonsillectomy for obstructive sleep apnea, hearing aids for sensorineural hearing loss, and keratoplasty for corneal clouding. Patients also had aggressive supportive and symptomatic surgical and nonsurgical interventions to treat skeletal abnormalities. There are benefits from hematopoietic stem cell transplantation (HSCT) in MPS. According to Yabe et al in 2016,36 there were four MPS IVA patients with HSCT being followed at least 10 years. All of them had normal level of GALNS activity in lymphocytes after HSCT. Only one patient had osteotomy in both legs and others had no orthopedic surgical intervention. Their activity of daily living (ADL) was better than untreated patients. However, due to few MPS IVA patients with HSCT, there are no clear mortalities about HSCT for MPS IVA having been reported.37
For many forms of LSD, enzyme replacement therapy (ERT) has become the standard treatment. In 1996, the first ERT was approved for Gaucher disease. ERT has many limitations, including high cost, low penetration of the central nervous system, and infusion reactions. However, with more clinical experience, infusion reactions could be prevented. The low penetration of enzymes through the blood–brain barrier reduces central nervous system penetration and limits the efficacy of all ERTs. For instance, type 1 Gaucher disease patients have improvements in systemic symptoms after ERT. However, peripheral administration of ERT cannot improve neurological regression in MPS type II and III patients who often die from neurological complications. Because MPS IVA patients do not have intellectual disabilities, ERT can provide beneficial outcomes, including preventing disease progression to a certain extent. Nevertheless, it cannot reverse most pathology that had developed before treatment. There are five types of MPSs for which ERT has currently been approved: MPS I (laronidase; Aldurazyme®, Genzyme, Boston, MA, USA), II (idursulfase; Elaprase®, Shire, Dublin, Republic of Ireland), IVA (elosulfasealfa; Vimizim®, Biomarin, Novato, CA, USA), VI (galsulfase; Naglazyme®, Biomarin), and VII (vestronidase alfa; Mepsevii®, Ultragenyx, Novato, CA, USA).
Elosulfase Alfa Mechanism of Action
ERT can only be administered by intravenous infusion. The mechanism of action of elosulfase alfa is to provide enzymatic activity to reduce the enzyme deficiency caused by MPS IVA. Elosulfase alfa is a purified human enzyme; recombinant DNA technology (recombinant human enzyme or rhGALNS) in the Chinese hamster ovary cell line is used to produce it. It is identical in sequence and enzymatic activity with the human form of GALNS. Elosulfase alfa and human GALNS have the same amino acid sequence and N-linked glycosylation.38,39 Because it contains bis-mannose 6 phosphate, one of the oligosaccharide chains can bind to a cell surface receptor (cation-independent mannose-6-phosphate receptor). Elosulfase alfa metabolizes GAGs, C6S, and KS.40,41 Elosulfase alfa is active in a low pH environment; thus, it has a low activity outside the lysosome.
Pharmacokinetics and Pharmacodynamics
Qi et al reported the pharmacokinetics of elosulfase alfa42 based on a Phase III trial for 24 weeks in 2014.2 They analyzed the pharmacokinetics during and after the initial infusion of elosulfase alfa. Figure 2 shows the pharmacokinetics during and after week 22 of infusion.42,43 This figure reflects the mean plasma concentration profiles of elosulfase alfa during and after infusion. They collected blood samples before and 15, 60, and 120 min after beginning the infusion and 5, 15, 30, 60, 120, and 180 min after completing the infusion. Noncompartmental analysis of pharmacokinetic parameters was done with WinNonlin® (Certara, L.P., Princeton, NJ, USA) software. According to this figure, it shows that after 22 weeks treatment, the dose of 2 mg/kg/QW could keep higher level of elosulfase alfa than the dose of 2 mg/kg/QOW. Table 2 shows that after repeated administration over 22 weeks, the plasma half-life of elosulfase alfa increased from approximately 7 min in the first infusion to approximately 36 min.42,43 The intracellular half-life of elosulfase alfa is estimated to be 5–7 days. This was determined in vitro in human Morquio A fibroblasts.44 The appearance of neutralizing anti-elosulfase alfa antibodies in patients is thought to be associated with the increase in the plasma half-life.42 The total exposure assessed by AUC0-last increased 181% or 192% after 22 weeks of administration. The pharmacodynamics, efficacy, or safety outcomes in patients are not influenced by this apparent increase in exposure.
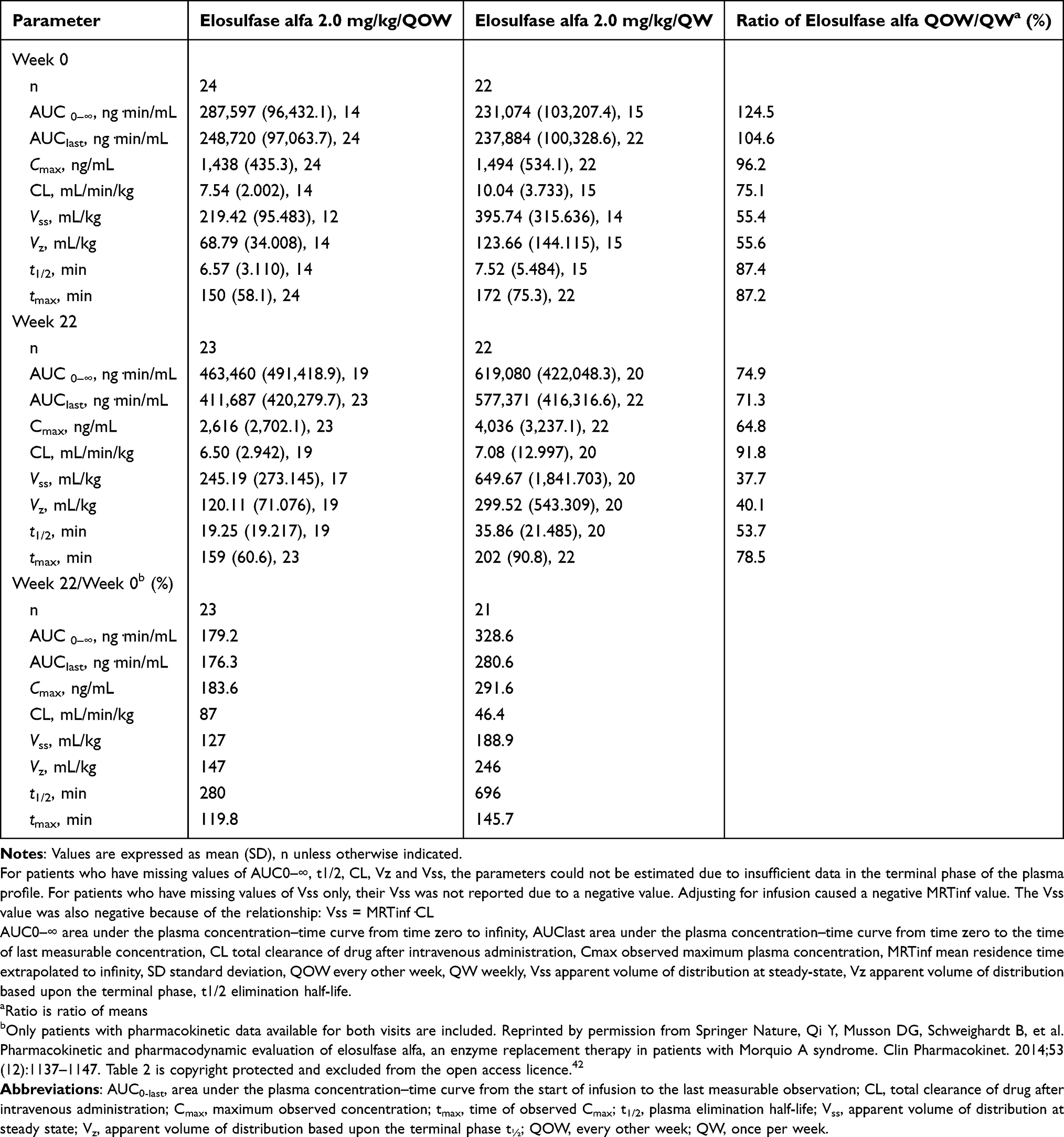 |
Table 2 Pharmacokinetic Parameters for Elosulfase Alfa in Patients with Morquio a Syndrome |
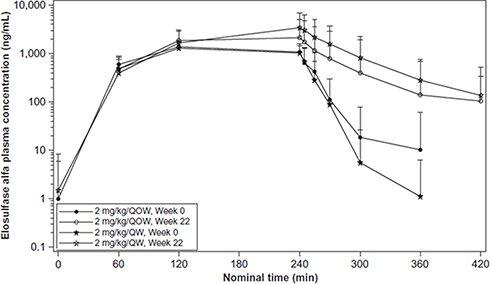 |
Figure 2 Mean plasma concentration profiles of elosulfase alfa during and after infusion. QOW: every other week; QW: once per week. Reprinted by permission from Springer Nature, Qi Y, Musson DG, Schweighardt B, et al. Pharmacokinetic and pharmacodynamic evaluation of elosulfase alfa, an enzyme replacement therapy in patients with Morquio A syndrome. Clin Pharmacokinet. 2014;53(12):1137–1147. Figure 2 is copyright protected and excluded from the open access licence.42 |
Elosulfase alfa normalizes the levels of the deficient endogenous enzyme in lysosomes via a mannose-6-phosphate-mediated mechanism.45 According to preclinical and clinical studies, the major pharmacodynamic effect of elosulfase alfa is to reduce the uKS by increasing the lysosomal degradation of this metabolite45‒47. Although we could use uKS to differentiate MPS IVA from other forms of MPS and to demonstrate pharmacodynamic effects of ERT treatment, we could not predict the skeletal improvement during ERT for MPS IVA by uKS.48 Biodistribution throughout the heart layers, the entire growth plate thickness, and in macrophages and hepatocytes was observed with the intravenous administration of elosulfase alfa to wild-type mice in preclinical studies.45
Preclinical Data
In a study to determine the safety, efficacy, and tolerability of elosulfase alfa, using MPS IVA mice, after 12 weeks of treatment, there was an apparent reduction of GAGs storage in the visceral organs, bone marrow, heart valves, connective tissues, and ligaments.49 There was further evaluation in newborn MPS IVA mice at birth. According to the bone pathology study, although the chondrocytes were vacuolated, the columnar structure remained organized. This result showed that before the cartilage cell layer became mature bone in MPS IVA, the enzyme entered the cartilage and prevented the disorganization of the cartilage structure. It also showed that prompt treatment with ERT could prevent the bone pathology of MPS IVA.50 According to Tomatsu et al in 2010,51 the treatment effect is better when we treat mice for a longer term or from birth. GALNS with hexaglutamate sequence (E6) tagged (E6-GALNS) could clear the storage materials in bone, bone marrow, and heart valves substantially, especially after 24 weekly infusions.
Clinical Trial Data
In MOR-002 trial (Patients with MPS IVA, who enrolled in a prior BioMarin sponsored clinical study of BMN 110; NCT00884949; Study Identification Number MOR-002; Baseline and every 12 weeks for up to 72 weeks), elosulfase alfa was generally well tolerated at all doses. Over 5 years of treatment, no new safety concerns emerged during sustained treatment with elosulfase alfa 2.0 mg/kg per week. Adverse events occurred in patients in all studies; however, most were mild or moderate in severity. Hypersensitivity reactions were frequent; however, they could be managed by additional medications and altering the infusion rate. There was one patient with a type I hypersensitivity reaction and another with recurrent infusion reactions. These patients discontinued the study. Nevertheless, in the subsequent Phase 3 trial, no hypersensitivity adverse events were seen.2
In the MOR-002 trial (72 weeks) and the MOR-100 long-term extension (192 weeks), patients could maintain their performance in the 6min walk test (6MWT) and 3min stair climb test (3MSCT). One patient even had slightly better results in both tests. However, there was no exclusion of surgical procedures during the studies. The surgical procedures could affect the 6MWT and 3MSCT results. Due to the small sample size and absence of a control group, we could not evaluate efficacy. In the Morquio A Clinical Assessment Program (MorCAP) natural history study, which followed the progression of untreated Morquio A syndrome over 2 years, 6MWT distances in untreated patients decreased over this period.52 These results contrast the MOR-002 trial and the MOR-100 long-term extension. However, we should note that the mean age of patients at baseline was higher in the MorCAP observational study than in the MOR-002 study (14.4 ± 11.97 years vs 8.4 ± 2.90 years).
MPS IVA patients risk declining pulmonary function as the condition progresses; however, in the MOR-002 and MOR-100 studies, there were increases in forced vital capacity (FVC), forced expiratory volume in 1s (FEV1), and maximum voluntary ventilation (MVV). However, since there was no comparison with an age-matched group of untreated patients, these results could be partially attributed to growth.53 Therefore, the true impact of treatment on pulmonary function is yet to be determined. Furthermore, there were no negative effects in the quality-of-life measures in children with Morquio A syndrome,54 even though the quality-of-life measures would be expected to decline as the disease progresses. In the MOR-002 and MOR-100 studies, patients maintained a consistent level of function in the domains of self-care, mobility, and caregiver assistance over the 5-year study according to the MPS health assessment questionnaire (MPS-HAQ).
The 2.0 mg/kg per week elosulfase alfa dosing demonstrated the greatest reduction of uKS levels. Additionally, there was superior plasma availability with the 2.0 mg/kg per week dose. Concerning pharmacokinetic parameters, the maximum observed concentration in plasma (Cmax) increased. The areas under the curve from time 0 to last measurable concentration (AUC0−t) were nonlinear and far exceeded the dose increases from 0.1 to 1.0 to 2.0 mg/kg per week. We also observed that the decreases of clearance, the volume of distribution based on the terminal rate constant (Vdz), and the volume of distribution at steady state (Vdss) were also nonlinear. This indicated the possible saturation of clearance mechanisms such as protease degradation and/or cation-independent mannose 6-phosphate receptor (CI-M6PR) receptor-mediated cellular uptake.
The impact of neutralizing antibodies on the pharmacokinetics, seen in MOR-002 and MOR-100, is consistent with the placebo-controlled MOR-004 study.55,56 In the MOR-004 study, we were also aware that there were no relationships between efficacy outcomes and neutralizing antibody positivity or titer according to the alternative cell-based flow cytometry neutralizing antibodies assay.57 This result indicated that neither total antibodies nor neutralizing antibodies are related to the incidence of adverse events, loss of efficacy, or altered pharmacodynamics.
Treatment
Based on infusion volume, 2.0 mg/kg per week intravenous infusion over 3.5–4.5 h is currently the recommended dose. For patients <25 kg and >25 kg, the final volume is 100 mL and 250 mL respectively. The ERT is diluted with 0.9% sodium chloride injection. For patients <25 kg and ≥25 kg, the infusion rate should be 3 mL/h and 6 mL/h for the first 15 min, increasing to 6 mL/h and 12 mL/h for the next 15 min. If this rate is tolerated, the rate can be increased by 6 mL/h for patients <25 kg and 12 mL/h for patients ≥25 kg every 15 min. The maximum rates are 36 mL/h for patients <25 kg and 72 mL/h for patients ≥25 kg. An antihistamine with or without antipyretics should be administered 30–60 minutes before infusion. Due to the risk of sleep apnea and airways difficulties, a nonsedating antihistamine is preferred.
Adverse Events and Reactions
Clinical studies investigating adverse events enrolled 235 patients in all.39,58 Symptoms and signs of anaphylaxis (cough, erythema, throat tightness, urticaria, hypotension, dyspnea, gastrointestinal symptoms) occurred in 16 (6.8%) patients. Anaphylaxis occurred as early as 30 minutes after beginning the infusion to 3 hours after completing it. Subsequent elosulfase alfa infusion with infusion rate adjustments and/or medical intervention were completed in all patients except two. The rate of hypersensitivity reactions was 18.7%, and the reactions were observed as early as 30 minutes after starting the infusion to as late as 6 days afterward. According to the MOR-004 clinical trial (Time Frame: Baseline to Week 24), the most common adverse events were headache (20.3%), pyrexia (18.6%), vomiting (15.3%), nausea (13.6%), diarrhea (11.9%), fatigue (11.9%), upper abdominal pain (8.5%), cough (8.5%), oropharyngeal pain (5.1%), abdominal pain (3.4%), and chills (1.7%).2 Based on the study of children under 5 years of age who received 2.0 mg/kg per week for 52 weeks,59 the rate of adverse events requiring infusion interruption and medical intervention was 0.8%. After treatment, growth velocity and uKS levels were improved.
Prevention and Management of Adverse Reactions
The adverse reaction risk in phase I/II clinical trials could be reduced by nonsedating antihistamines with or without antipyretics.2 If patients have a history of adverse reactions or allergies, we should consider premedication like sedating antihistamines, antipyretics, H2 blockers, leukotriene receptor antagonists, oral cromolyn sodium (personal communication), and/or steroids. Additionally, the infusion rate should be decreased by 25%–50%, especially during the first 1–2 hours.36 Patients should have an airway patency evaluation before beginning ERT if they have airway management or positive pressure during sleep to manage sleep apnea. These patients also need oxygen during infusion therapy.
Because of the increased risk of hypersensitivity reactions, ERT should be avoided if patients have an acute infection, a fever, or respiratory illness.40 Patients should have immediate medical care if they develop an acute hypersensitivity reaction. For mild-to-moderate reactions, the infusion could be slowed or stopped and antihistamines, antipyretics, and/or steroids administered. For severe anaphylaxis reactions, the infusion should be stopped immediately and breathing, and circulation evaluated. One should stabilize the airway, give oxygen supplementation, and administer intramuscular epinephrine and intravenous fluids for hypotension.
Expert Commentary
Pharmacokinetic studies outside of clinical trials and clinical settings are needed. Immunogenicity is not correlated with the efficacy of elosulfase alfa. Although only a small proportion of patients are hypersensitive,60 some clinicians remain concerned about the drug’s immunological risk. They believe the risk of infusion-associated reactions and infusion-site reactions may increase due to the presence of anti-elosulfase alfa antibodies. However, clinical trials have shown no correlation between the immune response and infusion-associated or infusion-site reactions.39 Only a small minority of patients have an elevated immune response.
We cannot currently predict which Morquio A patients will respond to ERT.61 About 10% of patients have an excellent response that exceeds expectations. There is also a small group of patients who do not respond to ERT at all. Therefore, Morquio A patients receiving elosulfase alfa should have at least 12 months of treatment before assessing their response. The current quality-of-life assessment tools do not adequately capture the treatment benefits for patients.62 We must develop improved and more innovative daily lifestyle measures to adequately assess clinical response. Lifestyle tracker apps could provide an all-day record of activity and sleep habits in the future63‒69. Blood KS directly derived from the bone is an important biomarker of improvement in true clinical endpoints including bone pathology or any other skeletal signs and symptoms.70
Orthopedic surgeries are necessary because ERT have provided only modest benefit for skeletal deformities.71 X-ray and MRI of spine showed that ERT had limited effects on bones72 (Figures 3 and 4). Patients with MPS IVA usually have odontoid hypoplasia with subluxation, gibbus deformity, hip dysplasia and osteonecrosis, and lower extremity valgus. Due to these symptoms, patients should necessitate monitoring and often surgical intervention like realignment osteotomy, corrective knee surgery and hip replacement.
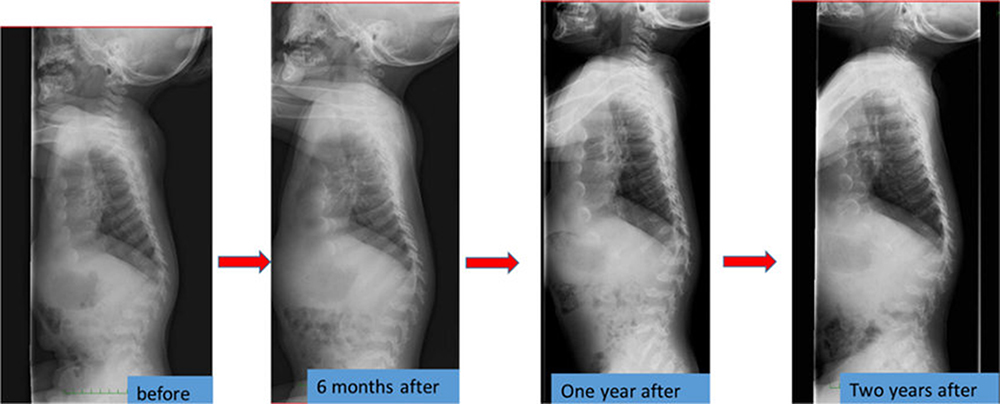 |
Figure 3 The spine X-ray findings before and after the enzyme replacement therapy in a Japanese boy with MPS IVA. Reproduced from Nakamura-Utsunomiya A, Nakamae T, Kagawa R, et al. A case report of a Japanese boy with MorquioA syndrome: effects of enzyme replacement therapy initiated at the age of 24 months. Int J Mol Sci. 2020;21(3):989.72 |
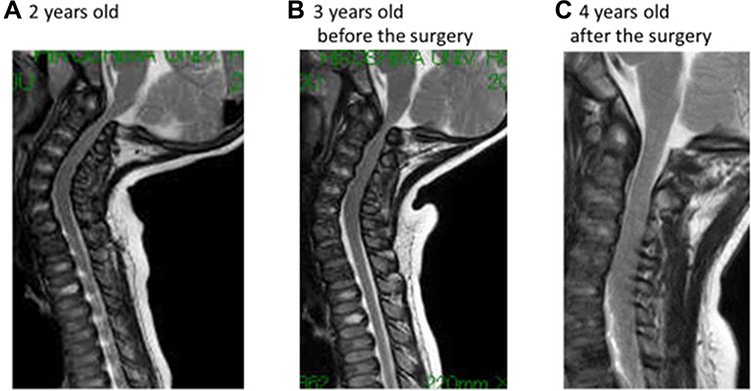 |
Figure 4 The changes in MRI findings. (A) The spinal cord compression at C1 level was already present at the first examination. (B) The lesion was slowly progressive after the ERT started, (C) which was relieved by resecting the posterior arch of the atlas vertebra. Reproduced from Nakamura-Utsunomiya A, Nakamae T, Kagawa R, et al. A case report of a Japanese boy with MorquioA syndrome: effects of enzyme replacement therapy initiated at the age of 24 months. Int J Mol Sci. 2020;21(3):989.72. |
Most patients in the UK and Canada are infused at home. Conversely, patients in the USA are infused in infusion centers which increases treatment costs. Nevertheless, infusion centers offer additional benefits like emotional support from other patients, families, and healthcare workers. Experienced clinicians and healthcare providers, including infusion nurses, are important. They can provide the proper and effective rate of ERT infusion and drug dose and act to prevent or manage adverse reactions.
Elosulfase alfa has improved the walking distance of MPS IVA patients and their respiratory function. However, compared with the growth charts for untreated patients, patients treated with ERT did not show any significant increase in growth in any age group, even if they had early ERT intervention before 5 years of age.73 We still have much to learn to improve our management of Morquio A syndrome. There was no effective drug for Morquio A before elosulfase alfa; however, now, there is hope for the future in this field.
Five-Year View
ERT could only slow skeletal disease progression even when startedin infancy before 2 months of age.74 There are studies aimed at increasing the effectiveness of ERT with targeted therapies.69,75 In preclinical research, gene-targeted therapy has shown some efficacy in treating Morquio A75‒77. It may be more effective to use small, specific, targeted molecules in addition to ERT to treat Morquio A, which will solve the problems of immunogenicity and bone uptake in the future.75,76 In the next 5 years, we should be able to predict which mutations of the GALNS gene are responsive to ERT.
According to a study by Donida et al in a cohort of MPS IVA patients receiving ERT therapy,78 interleukin-6 and decreased glutathione levels were elevated. This suggests a proinflammatory and prooxidant state. However, there were no MPS IVA patients who had not received ERT for comparison. Further investigation of combined antioxidant and anti-inflammatory agent therapy with ERT is needed to optimize patient outcomes.
Conclusions
Morquio A is not just a skeletal disorder; there is a multiorgan involvement. Elosulfase alfa could be effective in improving patient endurance, pulmonary function, and quality of life. It is important to have long-term outcome studies to determine the effect of elosulfase alfa on the bone outcomes of patients. Moreover, we must evaluate the efficacy of ERT treatment in moderating the long-term progression of MPS IVA.
Acknowledgments
Medical writing and editorial was supported by research grants from the Ministry of Science and Technology, Executive Yuan, Taiwan (MOST-110-2314-B-195-010-MY3, MOST-110-2314-B-195-014, MOST-110-2314-B-195-029, MOST-109-2314-B-195-024, MOST-108-2314-B-195-012, and MOST-108-2314-B-195-014) and from MacKay Memorial Hospital (MMH-E-111-13, MMH-E-110-16, MMH-E-109-16, MMH-E-108-16, MMH-MM-10801, and MMH-107-82).
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Matalon R, Arbogast B, Justice P, et al. Morquio’s syndrome: deficiency of a chondroitin sulfate N-acetylhexosamine sulfate sulfatase. Biochem Biophys Res Commun. 1974;61(2):759–765. doi:10.1016/0006-291X(74)91022-5
2. Hendriksz CJ, Burton B, Fleming TR, et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis. 2014;37(6):979–990. doi:10.1007/s10545-014-9715-6
3. Hendriksz CJ, Al-Jawad M, Berger KI, et al. Clinical overview, and treatment options for non-skeletal manifestations of mucopolysaccharidosis type IVA. J Inherit Metab Dis. 2013;36(2):309–322. doi:10.1007/s10545-012-9459-0
4. Montano AM, Tomatsu S, Brusius A, et al. Growth charts for patients affected with Morquio A disease. Am J Med Genet A. 2008;146A(10):1286–1295. doi:10.1002/ajmg.a.32281
5. Lin HY, Chuang CK, Chen MR, et al. Natural history and clinical assessment of Taiwanese patients with mucopolysaccharidosis IVA. Orphanet J Rare Dis. 2014;9:21. doi:10.1186/1750-1172-9-21
6. Morquio L. Sur uneforme de dystrophieosseusefamiliale [Article in French]. Arch Med Des Infants. 1929;32:129–135.
7. Brailsford JF. The classics: chondro-osteo-dystrophy. roentgenographic and clinical features of a child with dislocation of vertebrae. Clin Orthop Relat Res. 1976;114:4–9.
8. Leadley RM, Lang S, Misso K, et al. A systematic review of the prevalence of Morquio A syndrome: challenges for study reporting in rare diseases. Orphanet J Rare Dis. 2014;9:173. doi:10.1186/s13023-014-0173-x
9. Lin HY, Lin SP, Chuang CK, et al. Incidence of the mucopolysaccharidoses in Taiwan, 1984–2004. Am J Med Genet A. 2009;149A:960–964. doi:10.1002/ajmg.a.32781
10. Masuno M, Tomatsu S, Nakashima Y, et al. Mucopolysaccharidosis IVA: assignment of the human N-acetylgalactosamine-6-sulfate sulfatase (GALNS) gene to chromosome 16q24. Genomics. 1993;16(3):777–778. doi:10.1006/geno.1993.1266
11. Neufield EF, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet AL, Valle D, Sly WS, editors. The Metabolic and Molecular Bases of Inherited Disease. Vol. 136.
12. Lin SP, Shih SC, Chuang CK, et al. Characterization of pulmonary function impairments in patients with mucopolysaccharidoses—changes with age and treatment. Pediatr Pulmonol. 2014;49:277–284.
13. Lin HY, Shih SC, Chuang CK, et al. Assessment of hearing loss by pure-tone audiometry in patients with mucopolysaccharidoses. Mol Genet Metab. 2014;111:533–538. doi:10.1016/j.ymgme.2014.02.003
14. Lin HY, Chan WC, Chen LJ, et al. Ophthalmologic manifestations in Taiwanese patients with mucopolysaccharidoses. Mol Genet Genom Med. 2019;7(5):e00617. doi:10.1002/mgg3.617
15. Lin HY, Lee CL, Chiu PC, et al. Relationships among height, weight, body mass index, and age in Taiwanese children with different types of mucopolysaccharidoses. Diagnostics (Basel). 2019;9(4):148. doi:10.3390/diagnostics9040148
16. Lin HY, Chuang CK, Lee CL, et al. Cardiac evaluation using two-dimensional speckle-tracking echocardiography and conventional echocardiography in Taiwanese patients with mucopolysaccharidoses. Diagnostics. 2020;10(2):62. doi:10.3390/diagnostics10020062
17. Khan S, Alméciga-Díaz CJ, Sawamoto K, et al. Mucopolysaccharidosis IVA and glycosaminoglycans. Mol Genet Metab. 2017;120:78–95. doi:10.1016/j.ymgme.2016.11.007
18. Averill LW, Kecskemethy HH, Theroux MC, et al. Tracheal narrowing in children and adults with mucopolysaccharidosis type IVA: evaluation with computed tomography angiography. Pediatr Radiol. 2021;51(7):1202–1213. doi:10.1007/s00247-020-04946-0
19. Kleijer WJ, Geilen GC, Garritsen V, et al. First-trimester diagnosis of Morquio disease type A. Prenat Diagn. 2000;20(3):183–185. doi:10.1002/(SICI)1097-0223(200003)20:3<183::AID-PD774>3.0.CO;2-J
20. Zhao H, van Diggelen OP, Thoomes R, et al. Prenatal diagnosis of Morquio disease type A using a simple fluorometric enzyme assay. Prenat Diagn. 1990;10(2):85–91. doi:10.1002/pd.1970100204
21. Davison JE, Kearney S, Horton J, et al. Intellectual and neurological functioning in Morquio syndrome (MPS IVa). J Inherit Metab Dis. 2013;36(2):323–328. doi:10.1007/s10545-011-9430-5
22. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet A. 2015;167A(1):11–25. doi:10.1002/ajmg.a.36833
23. Hendriksz CJ, Harmatz P, Beck M, et al. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab. 2013;110(1–2):54–64. doi:10.1016/j.ymgme.2013.04.002
24. Lavery C, Hendriksz C. Mortality in patients with morquio syndrome A. JIMD Rep. 2015;15:59–66. doi:10.1007/8904_2014_298
25. Bhattacharya K, Balasubramaniam S, Choy Y, et al. Overcoming the barriers to diagnosis of Morquio A syndrome. Orphanet J Rare Dis. 2014;9:192. doi:10.1186/s13023-014-0192-7
26. Wood TC, Harvey K, Beck M, et al. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36(2):293–307. doi:10.1007/s10545-013-9587-1
27. Lin HY, Lee CL, Lo YT, et al. The relationships between urinary glycosaminoglycan levels and phenotypes of mucopolysaccharidoses. Mol Genet Genom Med. 2018;6:982–992. doi:10.1002/mgg3.471
28. Regier DS, Oetgen M, Tanpaiboon P. Mucopolysaccharidosis type IVA. In: Pagon RA, Adam MP, Ardinger HH, et al. editors. GeneReviews®. Seattle: University of Washington; 2016.
29. Theroux MC, Nerker T, Ditro C, Mackenzie WG. Anesthetic care and perioperative complications of children with Morquio syndrome. PaediatrAnaesth. 2012;22:901–907.
30. Shih SL, Lee YJ, Lin SP, et al. Airway changes in children with mucopolysaccharidoses. Acta Radiol. 2002;43:40–43. doi:10.1034/j.1600-0455.2002.430108.x
31. Lee CL, Lee KS, Chuang CK, et al. Otorhinolaryngological management in Taiwanese patients with mucopolysaccharidoses. Int J Med Sci. 2021;18(15):3373–3379. doi:10.7150/ijms.61827
32. Lin HY, Chen MR, Lin CC, et al. Polysomnographic characteristics in patients with mucopolysaccharidoses. Pediatr Pulmonol. 2010;45:1205–1212.
33. Harmatz P, Mengel KE, Giugliani R, et al. The MorquioA clinical assessment program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109:54–61. doi:10.1016/j.ymgme.2013.01.021
34. Lin SM, Lin HY, Chuang CK, Lin SP, Chen MR. Cardiovascular abnormalities in Taiwanese patients with mucopolysaccharidosis. Mol Genet Metab. 2014;111(4):493–498. doi:10.1016/j.ymgme.2014.02.009
35. Regier DS, Tanpaiboon P. Role of elosulfase alfa in mucopolysaccharidosis IVA. Appl Clin Genet. 2016;9:67–74. doi:10.2147/TACG.S69080
36. Yabe H, Tanaka A, Chinen Y, et al. Hematopoietic stem cell transplantation for Morquio A syndrome. Mol Genet Metab. 2016;117(2):84–94. doi:10.1016/j.ymgme.2015.09.011
37. Taylor M, Khan S, Stapleton M, et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: past, present, and future. Biol Blood Marrow Transplant. 2019;25(7):e226–e246. doi:10.1016/j.bbmt.2019.02.012
38. Biomarin Pharmaceuticals. Vimizim (elosulfase alfa) for the treatment of mucopolysaccharidosis type IVA (Morquio syndrome): briefing document for the endocrinologic and metabolic drugs advisory committee. 2013.
39. Food and Drug Administration. Vimizim (elosulfase alpha) injection, for intravenous use: US prescribing information. 2014.
40. Goldenberg MM. Pharmaceutical approval update. P T. 2014;39(5):337–344.
41. Sanford M, Lo JH. Elosulfase alfa: first global approval. Drugs. 2014;74:713–718. doi:10.1007/s40265-014-0210-z
42. Qi Y, Musson DG, Schweighardt B, et al. Pharmacokinetic and pharmacodynamic evaluation of elosulfase alfa, an enzyme replacement therapy in patients with Morquio A syndrome. Clin Pharmacokinet. 2014;53(12):1137–1147. doi:10.1007/s40262-014-0173-y
43. Hendriksz CJ. Elosulfase alfa (BMN 110) for the treatment of mucopolysaccharidosis IVA (Morquio A Syndrome). Expert Rev Clin Pharmacol. 2016;9(12):1521–1532. doi:10.1080/17512433.2017.1260000
44. Morquiosity; 2021. Available from: http://www.morquiosity.com/.
45. Dvorak-Ewell M, Wendt D, Hague C, et al. Enzyme replacement in a human model of mucopolysaccharidosis IVA in vitro and its biodistribution in the cartilage of wild type mice. PLoS One. 2010;5(8):e12194. doi:10.1371/journal.pone.0012194
46. Hendriksz CJ, Vellodi A, Jones S, et al. Long term outcomes of a Phase 1/2, multicenter, open-label, dose-escalation study to evaluate the safety, tolerability, and efficacy of BMN 110 in patients with Mucopolysaccharidosis IVA (Morquio A syndrome) [abstract]. Mol Genet Metab. 2012;105(2):S35. doi:10.1016/j.ymgme.2011.11.076
47. Lin HY, Chen MR, Lin SM, et al. Cardiac features, and effects of enzyme replacement therapy in Taiwanese patients with Mucopolysaccharidosis IVA. Orphanet J Rare Dis. 2018;13(1):148. doi:10.1186/s13023-018-0883-6
48. Tomatsu S, Sawamoto K, Shimada T, et al. Enzyme replacement therapy for treating mucopolysaccharidosis type IVA (Morquio A syndrome): effect and limitations. Expert Opin Orphan Drugs. 2015;3(11):1279–1290. doi:10.1517/21678707.2015.1086640
49. Tomatsu S, Montano AM, Ohashi A, et al. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum Mol Genet. 2008;17:815–824. doi:10.1093/hmg/ddm353
50. Tomatsu S, Montano AM, Oikawa H, et al. Enzyme replacement therapy in newborn mucopolysaccharidosis IVA mice: early treatment rescues bone lesions? Mol Genet Metab. 2015;114:195–202. doi:10.1016/j.ymgme.2014.05.013
51. Tomatsu S, Montaño AM, Dung VC, et al. Enhancement of drug delivery: enzyme-replacement therapy for murine Morquio A syndrome. Mol Ther. 2010;18(6):1094–1102. doi:10.1038/mt.2010.32
52. Harmatz PR, Mengel KE, Giugliani R, et al. Longitudinal analysis of endurance and respiratory function from a natural history study of Morquio A syndrome. Mol Genet Metab. 2015;114(2):186–194. doi:10.1016/j.ymgme.2014.10.015
53. Hendriksz CJ, Berger KI, Parini R, et al. Impact of long-term elosulfase alfa treatment on respiratory function in patients with Morquio A syndrome. J Inherit Metab Dis. 2016;39(6):839–847. doi:10.1007/s10545-016-9973-6
54. Hendriksz CJ, Lavery C, Coker M, et al. Burden of disease in patients with Morquio A syndrome: results from an international patient-reported outcomes survey. Orphanet J Rare Dis. 2014;9:32. doi:10.1186/1750-1172-9-32
55. Qi Y, Musson D, Martell L, et al. Phase 1/2 pharmacokinetic studies of recombinant human N-acetylgalactosamine-6-sulfatase (rhGALNS) in MPS IVA patients. In:
56. Hendriksz CJ, Santra S, Jones SA, et al. Safety, immunogenicity, and clinical outcomes in patients with Morquio A syndrome participating in 2 sequential open-label studies of elosulfase alfa enzyme replacement therapy (MOR-002/MOR-100), representing 5 years of treatment. Mol Genet Metab. 2018;123(4):479–487. doi:10.1016/j.ymgme.2018.02.011
57. Melton AC, Soon RK, Tompkins T, et al. Antibodies that neutralize cellular uptake of elosulfase alfa are not associated with reduced efficacy or pharmacodynamic effect in individuals with Morquio A syndrome. J Immunol Methods. 2017;440:41–51. doi:10.1016/j.jim.2016.10.006
58. Biomarin Pharmaceuticals. Vimizim (elosulfase alfa) for the treatment of mucopolysaccharidosis type IVA (Morquio syndrome): briefing document for the endocrinologic and metabolic drugs advisory committee 2013; 2013.
59. Jones SA, Bialer M, Parini R, et al. Safety and clinical activity of elosulfase alfa in pediatric patients with Morquio A syndrome (mucopolysaccharidosis IVA) less than 5 y. Pediatr Res. 2015;78(6):717–722. doi:10.1038/pr.2015.169
60. Schweighardt B, Tompkins T, Lau K, et al. Immunogenicity of elosulfase alfa, an enzyme replacement therapy in patients with Morquio A syndrome: results from MOR-004, a phase III trial. Clin Ther. 2015;37(5):1012–1021. doi:10.1016/j.clinthera.2014.11.005
61. National Institute for Health and Care Excellence. Elosulfase alfa for treating mucopolysaccharidosis type IVa. London (UK). 2015.
62. Harmatz PR, Mengel E, Geberhiwot T, et al. Impact of elosulfase alfa in patients with Morquio A syndrome who have limited ambulation: an open-label, Phase 2 study. Am J Med Genet A. 2016;173:375–383. doi:10.1002/ajmg.a.38014
63. Dempsey W, Liao P, Klasnja P, et al. Randomised trials for the Fitbit generation. Signif (Oxf). 2015;12(6):20–23. doi:10.1111/j.1740-9713.2015.00863.x
64. Dhillon SS, Sima CA, Kirkham AR, et al. Physical activity measurement accuracy in individuals with chronic lung disease: a systematic review with meta-analysis of method comparison studies. Arch Phys Med Rehabil. 2015;96(11):2079–2088. doi:10.1016/j.apmr.2015.05.015
65. Diaz KM, Krupka DJ, Chang MJ, et al. Fitbit®: an accurate and reliable device for wireless physical activity tracking. Int J Cardiol. 2015;185:138–140. doi:10.1016/j.ijcard.2015.03.038
66. Feldhege F, Mau-Moeller A, Lindner T, et al. Accuracy of a custom physical activity and knee angle measurement sensor system for patients with neuromuscular disorders and gait abnormalities. Sensors. 2015;15(5):10734–10752. doi:10.3390/s150510734
67. Alharbi M, Bauman A, Neubeck L, et al. Validation of Fitbit-Flex as ameasure of free-living physical activity in a community-basedphase III cardiac rehabilitation population. Eur J Prev Cardiol. 2016;23(14):1476–1485.
68. Hibbing PR, Kim Y, Saint-Maurice PF, et al. Impact of activity outcome and measurement instrument on estimates of youth compliance with physical activity guidelines: a cross-sectional study. BMC Public Health. 2016;16:223. doi:10.1186/s12889-016-2901-8
69. Zhang JH, Macfarlane DJ, Sobko T. Feasibility of a chest-worn accelerometer for physical activity measurement. J Sci Med Sport. 2016;19(12):1015–1019. doi:10.1016/j.jsams.2016.03.004
70. Tomatsu S, Sawamoto K, Alméciga-Díaz CJ, et al. Impact of enzyme replacement therapy and hematopoietic stem cell transplantation in patients with Morquio A syndrome. Drug Des Dev Ther. 2015;9:1937–1953.
71. Tomatsu S, Montaño AM, Oikawa H, et al. Mucopolysaccharidosis type IVA (Morquio A disease): clinical review and current treatment. Curr Pharm Biotechnol. 2011;12(6):931–945. doi:10.2174/138920111795542615
72. Nakamura-Utsunomiya A, Nakamae T, Kagawa R, et al. A case report of a Japanese boy with MorquioA syndrome: effects of enzyme replacement therapy initiated at the age of 24 months. Int J Mol Sci. 2020;21(3):989. doi:10.3390/ijms21030989
73. Doherty C, Stapleton M, Piechnik M, et al. Effect of enzyme replacement therapy on the growth of patients with Morquio A. J Hum Genet. 2019;64(7):625–635. doi:10.1038/s10038-019-0604-6
74. Frigeni M, Rodriguez-Buritica DF, Saavedra H, et al. The youngest pair of siblings with Mucopolysaccharidosis type IVA to receive enzyme replacement therapy to date: a case report. Am J Med Genet A. 2021;185(11):3510–3516. doi:10.1002/ajmg.a.62469
75. Tomatsu S, Mackenzie WG, Theroux MC, et al. Current and emerging treatments and surgical interventions for Morquio A syndrome: a review. Res Rep Endocr Disord. 2012;2012(2):65–77. doi:10.2147/RRED.S37278
76. Toietta G, Severini GM, Traversari C, et al. Various cells retrovirally transduced with N-acetylgalactosoamine-6-sulfate sulfatase correctMorquio skin fibroblasts in vitro. Hum Gene Ther. 2001;12(16):2007–2016. doi:10.1089/104303401753204571
77. Almeciga-Diaz CJ, Montano AM, Tomatsu S, et al. Adeno-associatedvirus gene transfer in Morquio A disease-effect of promoters andsulfatase-modifying factor1. FEBS J. 2010;277(17):3608–3619. doi:10.1111/j.1742-4658.2010.07769.x
78. Donida B, Marchetti DP, Biancini GB, et al. Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy. Biochim Biophys Acta. 2015;1852(5):1012–1019. doi:10.1016/j.bbadis.2015.02.004
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.