")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Clinical utility of alpha-1 proteinase inhibitor in the management of adult patients with severe alpha-1 antitrypsin deficiency: a review of the current literature
Received 4 January 2017
Accepted for publication 16 March 2017
Published 14 July 2017 Volume 2017:11 Pages 2149—2162
DOI https://doi.org/10.2147/DDDT.S105207
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
David G Parr, Beatriz Lara
Department of Respiratory Medicine, Cardio-Respiratory Division, University Hospital Coventry, Coventry, UK
Abstract: Alpha-1 antitrypsin (AAT) functions primarily to inhibit neutrophil elastase, and its deficiency predisposes individuals to the development of chronic obstructive pulmonary disease (COPD). The putative protective serum concentration is generally considered to be above a threshold of 11 µM/L, and therapeutic augmentation of AAT above this value is believed to retard the progression of emphysema. Several AAT preparations, all derived from human donor plasma, have been commercialized since approval by the US Food and Drug Administration (FDA) in 1987. Biochemical efficacy has been demonstrated by augmentation of pulmonary antiprotease activity, but demonstration of clinical efficacy in randomized, placebo-controlled trials has been hampered by the practical difficulties of performing conventional studies in a rare disease with a relatively long natural history. Computed tomography has been applied to measure lung density as a more specific and sensitive surrogate outcome measure of emphysema than physiologic indices, such as forced expiratory volume in 1 second, and studies consistently show a therapeutic reduction in the rate of lung density decline. However, convincing evidence of benefit using traditional clinical measures remains elusive. Intravenous administration of AAT at a dose of 60 mg/kg/week is the commonest regime in use and has well-documented safety and tolerability. International and national guidelines on the management of AAT deficiency recommend intravenous augmentation therapy to supplement optimized usual COPD treatment in patients with severe deficiency and evidence of lung function impairment.
Keywords: alpha-1 antitrypsin deficiency, augmentation or replacement therapy, computed tomography, emphysema, COPD
Introduction to alpha-1 antitrypsin deficiency (AATD) and currently available treatment options
Alpha-1 antitrypsin (AAT) is an acute-phase reactant protein that functions primarily as a protease inhibitor, with neutrophil elastase as the main target. In addition, AAT modifies mucociliary clearance through matriptase inhibition and cellular apoptosis through caspase 3 inhibition and has anti-infective, anti-inflammatory and anti-oxidant effects.1,2 Hepatic production of AAT may reach up to 34 mg/kg per day; 40% of this production remains in the circulation and 60% reaches the tissues.3
AAT is a member of the SERPIN (SERin Proteinase INhibitors) superfamily, which shares a common structure consisting of a core with an active site that is composed of a hinged loop mechanism. This complex structure is very sensitive to conformational changes induced by mutations, and these changes have a variable, but mutation-specific, impact on the biological activity of the protein, its ability to bind with other proteins and its cellular degradation and elimination.4
Low circulating levels of AAT are associated with low AAT concentration in the tissues, and the consequent reduction in the inhibition of inflammatory mediators leads to tissue damage, particularly in the lung. Some abnormal variants spontaneously polymerize leading to intracellular accumulation, endoplasmic reticulum stress and, ultimately, cell death. Polymer accumulation within hepatocytes is believed to be the principal cause of AATD-related liver disease, but AAT polymerization within the lung may also contribute to pulmonary inflammation.5
AAT is coded at the protease inhibitor locus (14q32.1), which is part of a cluster of genes located on chromosome 14.6 The group of protein variants is referred to using the abbreviation Pi (protease inhibitor) followed by a single letter classifier that relates to the electrophoretic migration speed of the relevant protein (M, S, Z, etc).7 The M variant, present in 95% of Caucasian populations, is considered to be the normal variant, and the S and Z variants are the commonest cause of deficiency. An autosomal codominant pattern of inheritance generates the following common potential genotypes: MM, MS, SS, MZ, SZ and ZZ, which relate to the serum protein levels shown in Table 1 (a detailed review of the different AAT variants can be found at OMIM® – Online Mendelian Inheritance in Man® McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine).8,9
 | Table 1 Relationship between the commonest genotypes and AAT serum concentration |
Prevalence of the Z allele is geographically dependent, with a north-to-south and east-to-west gradient in Europe. The highest prevalence of PiZZ corresponds to Sweden and Norway (1:2,000), and in the Balkans, it is around 1:10,000, but the Z allele is extremely rare in non-Caucasian populations. In contrast, the S allele shows the opposite gradient, being more frequent in the Iberian Peninsula and decreasing progressively toward the north and east of Europe. The prevalence of PiSZ in Spain is around 1:300.10 In the “New World”, the prevalence of Z and S alleles is related principally with the pattern of migration from Europe, and in Asia and Africa, the prevalence is extremely low.
It should be recognized, however, that the prevalence of a deficient genotype in a population does not equate with disease prevalence. In particular, the risk of developing lung disease is dependent on serum AAT concentration in combination with environmental factors.11–14 Approximately 60% of PiZZ patients develop lung disease in adulthood, but this percentage varies depending mainly on the extent of tobacco smoking and inhalation of other pollutants.15–17 More than one-third of individuals will remain asymptomatic or experience mild symptoms without any evident impact on life expectancy.18 Less than 5% of PiZZ individuals will develop clinically relevant liver disease.19 Consequently, accurate estimation of the number of patients with AATD-associated liver or lung disease is problematic, and different approaches have been adopted to estimate disease prevalence. Studies of chronic obstructive pulmonary disease (COPD) populations have estimated that 2%–3% of individuals diagnosed with COPD have AATD.9
The relationship between the serum concentration of AAT and the severity of emphysema supports the concept of a pulmonary protective threshold that is generally considered to be 11 μM/L.11 The corollary of this is that correction of the deficiency through therapeutic augmentation should be protective against the development of emphysema. As a consequence of this concept, techniques for the isolation and purification of AAT from plasma donors were developed and, on the basis of biochemical efficacy, the US Food and Drug Administration (FDA) approved the use of intravenous Prolastin® in 1987.20,21 Since then, several products have been approved for the intravenous administration of AAT (Table 2) and >5,000 patients worldwide have received this form of augmentation therapy.22–29 These products are licensed for the treatment of individuals with severe AATD-associated lung disease but have also been used successfully for other indications, including panniculitis, difficult asthma and vasculitis.30 The therapy may also have a role in patients with AATD who have undergone lung transplantation.31
 | Table 2 Purified AAT products currently licensed for intravenous administration |
Purification methods, pharmacology, mode of action, pharmacokinetics and administration of currently available alpha-1 antiproteinase inhibitors
AAT is obtained from human donor plasma (supernatant A, which is equivalent to COHN fraction II + III). The methods for protein isolation and prevention of infectious disease transmission, as well as the properties of the purified AAT, were initially described by Coan et al32 in 1985. One year later, Burnout et al reported obtaining 80%–90% purity in a 65%–75% yield of supernatant using a large-scale chromatographic procedure that included heat treatment to prevent transmission of blood-borne viruses. This method produced a purified AAT that exhibited inhibitory activity against trypsin and elastase that was equivalent to native serum protein.33
Wewers et al22 evaluated the feasibility and biochemical efficacy of purified AAT infusions in a dosage that could maintain serum levels above the putative protective threshold. The concentration of AAT and anti-elastase capacity were assessed in the epithelial lining fluid of the lungs in a population of 21 patients with severe deficiency who were administered AAT at a dose of 60 mg/kg of body weight once a week for a 6-month period. Serum AAT concentration peaked above 300 mg/dL in all patients and was maintained above the putative protective level with a calculated serum half-life of 4.4±0.2 days (mean ± standard deviation). A sustained elevation of AAT was seen in the lower respiratory tract and was associated with a significant increase in the pulmonary antineutrophil elastase capacity. Computer modeling estimated the average serum level over 1 month to be 163±4 mg/dL (mean ± standard deviation), and after 3–4 weeks of therapy, all patients had trough serum levels and serum anti-neutrophil elastase capacity that remained stable throughout the course of the study.
Products commercialized subsequently have comparable efficacy; for instance, Prolastin-C® (based on modifications to the Prolastin manufacturing process) is bioequivalent to Prolastin with a similar mean area under the plasma alpha1-PI concentration vs time curve over 7 days post-infusion (AUC0–7 days) at steady state (155.9 mg·h/mL vs 152.4 mg·h/mL, respectively); a similar mean first observed peak plasma alpha1-PI concentration following drug infusion (Cmax) (1,797 vs 1,848 mg/mL, respectively); adjusted time to reach observed Cmax (tmax) (0.673 vs 0.820 h, respectively) and mean terminal half-life (t1/2 h) (139.3 vs 146.3 h, respectively).34 Trypsone® has shown an in vivo half-life of 5.9±1.2 days, renal clearance of 0.3±0.06 mL/min, Cmax of 37.1±4.8 μmol/L and a distribution volume of 3.5±0.61.27
CSL Behring conducted a multi-center, double-blind, crossover study to demonstrate that the steady-state trough serum AAT level achieved by Zemaira® shows bioequivalence with Prolastin. The study included 44 individuals who received either the study drug or the control drug for a 10-week double-blind phase prior to the control group receiving the study drug. The difference in mean trough serum antigenic AAT concentration between the treatment groups was 1.45 μM (90% CI 2.77, −0.13), and a level of 17.7 μM during the steady-state period was achieved.29
Alternative administration patterns to weekly infusion of 60 mg/kg body weight are in use. Using a Bayesian analysis of a population pharmacokinetic approach to determine whether intervals between doses of >7 days were effective in maintaining steady-state total AAT levels >0.5 g/L, Soy et al concluded that it is feasible to extend the dose interval to 14–21 days, yet still achieve adequate trough concentrations. Their model showed a mean expected exogenous minimum AAT concentration at steady state (Cminss) of 0.73 g/L for 60 mg/kg weekly, 0.34 g/L for 120 mg/kg fortnightly and 0.23 g/L for 180 mg/kg thrice weekly (ie, every 21 days), the last being slightly below the target level. Intervals of 28 days or more were considered unsatisfactory because the putative protective threshold was not maintained.35
Purified AAT is commercialized as a dry powder for reconstitution into the manufacturers’ solvent using an aseptic technique, which must be performed within 3 hours of intravenous administration and infused at the recommended speed of 0.08 mL/kg/min. A significant disadvantage to the use of the intravenous route of delivery is that only 2% of the administered drug is delivered to lung tissue and, given the high costs of using pooled human plasma, alternative approaches are being sought.
The use of alternative sources of AAT, including transgenic human AAT production in other mammals, and the application of recombinant technology for AAT production in yeast may ultimately reduce production costs.3 Unfortunately, the use of AAT isolated from transgenic sheep milk may cause allergic reactions in humans, including possible hypersensitivity pneumonitis, and a trial of this product was consequently terminated.36,37 More recently, Ross et al38 have published data on AAT obtained from PER.C6 cells, but this protein remains to be tested in human studies.
Alternative approaches to the intravenous route of administration have been attempted, although these are not yet available for routine use. Inhaled AAT has the benefit of convenience and the potential benefits of achieving high concentrations of drug in the target tissue using much smaller quantities than when employing the intravenous route of administration. Several studies have demonstrated anti-elastolytic capacity;39 however, the presence of airflow obstruction and heterogeneous ventilation may lead to unpredictable and inconsistent drug deposition with consequent variable clinical efficacy.40
Intravenous augmentation therapy: efficacy studies, case reports and case series
The infusion of donor-derived, purified AAT has been shown to augment pulmonary antiprotease activity as evidenced by the correlation between serum AAT concentration and lung anti-elastase activity as well as decrease in some inflammatory markers and markers of tissue damage.41–43 The outcomes of key clinical studies that have sought to demonstrate biochemical, clinical and radiological efficacy of intravenous augmentation therapy are summarized later and in Table 3.
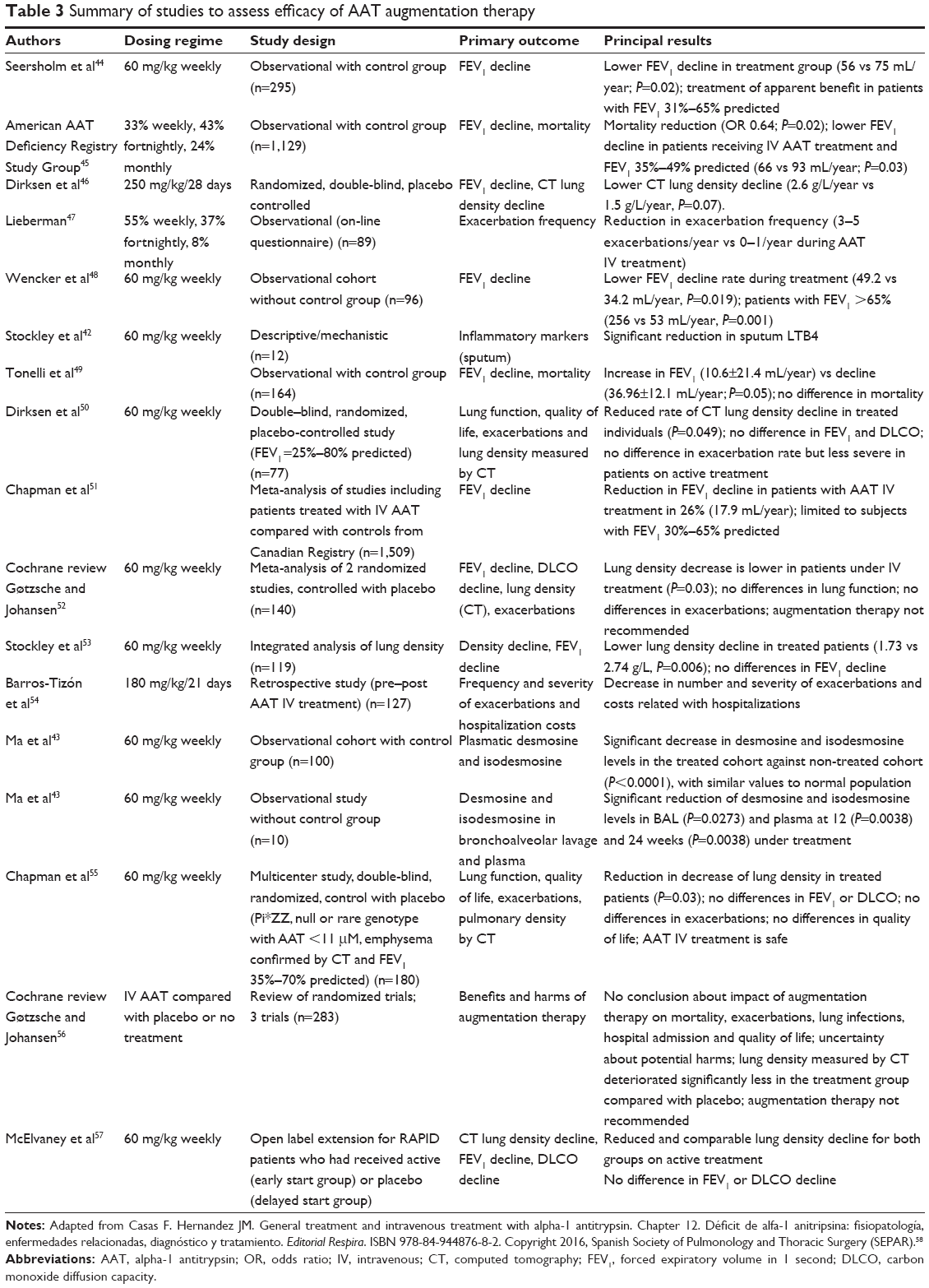 | Table 3 Summary of studies to assess efficacy of AAT augmentation therapy |
Decrease in the rate and severity of exacerbations
Two observational studies have identified a therapeutic reduction in the number of exacerbations: Lieberman47 described a reduction in the rate of exacerbations from 3 to 5 per year prior to treatment to 0–1 per year while receiving treatment. Wencker et al48 identified similar benefits. In a retrospective study, Barros-Tizón et al54 showed a decrease in the number and severity of exacerbations and a correlation with the cost of hospitalizations. The severity of exacerbations was also shown to be reduced by augmentation therapy in the EXACTLE study (please also see “Decrease in CT-measured lung density decline” section).50
Decrease in forced expiratory volume in 1 second (FEV1) decline and mortality
Data obtained from the US NHLBI registry, which included a population of >1,000 patients followed over 3.5 to 7 years, demonstrated a significant reduction in mortality in the patients receiving either continuous or intermittent augmentation therapy compared with those who never received treatment (odds ratio [OR] 0.64; P=0.02).45 In addition, FEV1 decline was shown to be lower in those individuals on therapy with a FEV1 35%–49% predicted (66 vs 93 mL/year; P=0.03). This study was pivotal in demonstrating the potential beneficial impact of augmentation therapy on lung function and mortality, but these findings were recognized to have come from observational data, rather than from a randomized, placebo-controlled clinical trial. Nevertheless, it prompted the development of other national registries purposed with characterizing the natural history of AATD-associated lung disease. Other studies that have evaluated the effect of therapy on FEV1 decline are detailed in Table 3.
In addition to illustrating the likely benefits of intravenous augmentation therapy, data from the NHLBI registry highlighted the practical difficulties of performing a randomized, placebo-controlled trial of intravenous augmentation therapy using FEV1 as the primary outcome measure. At that time, FEV1 was regarded as the “gold standard” surrogate marker for emphysema. However, the study findings indicated that 494 subjects with a baseline FEV1 of 35%–79% predicted would be required per treatment arm over 3 years to detect a difference in FEV1 decline of 15 mL/year.59 As a consequence, the search began for more sensitive and specific surrogate end points for use in randomized, placebo-controlled trials. Imaging biomarkers were recognized as being more sensitive and reproducible than traditional end points and in providing specific quantification of emphysema in a way that non-specific, global measures, such as FEV1, cannot.
Decrease in computed tomography (CT)-measured lung density decline
CT accurately characterizes the morphological changes in the lung arising from emphysematous destruction and can provide objective metrics of disease severity. Visually based severity-scoring systems and densitometric indices derived from the lung voxel histogram (Figure 1) both relate closely to the histopathological features of emphysema. Lung density declines progressively with worsening emphysema due to loss of lung mass from continuing proteolytic tissue destruction and the increasing hyperinflation that is secondary to changes in lung compliance. Because CT images are densitometric maps, CT lung densitometry can be used to monitor disease progression. A number of indices derived from the lung voxel histogram have been utilized for densitometry, but the most sensitive index for monitoring studies has been shown to be the 15th percentile method (PD15).60–62
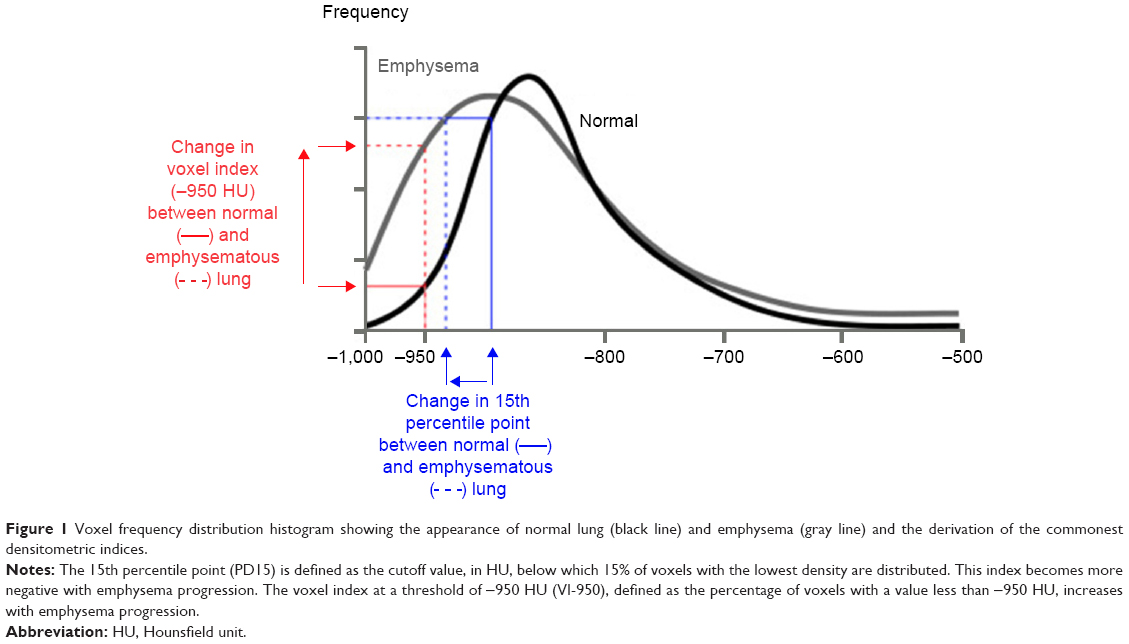 | Figure 1 Voxel frequency distribution histogram showing the appearance of normal lung (black line) and emphysema (gray line) and the derivation of the commonest densitometric indices. |
CT densitometry has been validated extensively for use as a surrogate end point in studies of emphysema-modifying therapy. Densitometric indices correlate with morphometry of resected histopathological specimens, static lung function, exercise physiology and health status indices: they predict mortality better than physiology or health status and are more sensitive measures of disease progression than health status and physiology.63–74 Most importantly, in observational studies of disease progression, CT lung density loss correlates with deterioration in health status and with FEV1 decline.61,62
CT densitometry offers significant advantages over traditional outcome measures; however, repeatability of the measure is limited by the natural variation that arises from differences in lung volume because the lung is a distensible organ that expands and contracts with respiratory movement. It is possible to reduce lung volume variability by exerting control over inspiration using, eg, a “gating” method.75–77 Spirometric control to maintain a constant level of inspiration during image acquisition constituted an early attempt to standardize for lung volume, but this proved difficult to accomplish in practice because of the need for a high level of patient compliance. A preferable method is the use of lung volume measurements obtained from volumetric CT imaging to adjust densitometry values using modeling.78–81 Since the lungs may be viewed as a sponge-like structure with preserved mass, a decrease in lung volume should effect a proportional and predictable increase in lung density. A log-transform of both lung volume and lung density would be expected to produce a linear relationship with a slope of −1. These so-called “volume correction” methods have been recommended for the reduction of variability in longitudinal densitometric studies of emphysema, and although the optimal approach to obtaining repeatable measurements in longitudinal studies has been contentious, recent interventional studies have provided methodological clarification (see later).
Coincident with the developments detailed in the aforementioned studies, a number of pilot studies were performed to explore the use of novel methodologies as alternatives to traditional outcome measures. Between 1991 and 1995, 26 Danish and 30 Dutch registry patients who were ex-smokers with a PiZ phenotype and moderate emphysema (FEV1 between 30% and 80% predicted) participated in a randomized, parallel, double-blind, placebo-controlled study of intravenous augmentation therapy.46 This was performed at two centers, and patients received infusions of either AAT (250 mg/kg body weight) or placebo (human albumin) every 4 weeks for at least 3 years. Full lung function testing was performed at inclusion and at subsequent 3-month intervals. In addition, “patient-administered spirometry” (PASS) was performed every morning and evening throughout the study. Annual CT imaging was performed at both study sites for quantitative analysis to assess lung density, but different, non-contrast imaging acquisition protocols were employed at each site: of particular note, in Denmark, subjects performed tidal breathing at a lung volume close to functional residual capacity (FRC) in the sitting position, whereas in the Netherlands, subjects were ventilated through a pneumotachograph containing a valve that was closed at 75% of the total lung capacity (TLC). Quantitative analysis was performed on the whole lung and, separately, on image slices 5 cm below the carina. Several CT densitometric indices were employed and, in order to reduce the variability in CT lung density arising from differences in inspiratory volume between imaging series, standardization was achieved by the use of log-transformed lung volume. There was no treatment effect evident in the primary outcome measure, FEV1, as would be expected given the underpowered design. However, the secondary outcome parameter, the PD15 of the whole lung, suggested that treatment reduced the annual loss of lung tissue by 1.07 g/L compared with placebo (P=0.07). In the placebo group, CT was more than twice as sensitive at detecting progression of emphysema as lung function, and furthermore, there was a correlation between the change in PD15 and the decline in both FEV1 and KCO. These relationships provided further important validation of CT densitometry as a suitable outcome measure for use in emphysema-modifying trials. The authors concluded that the twice-daily monitoring of patients by PASS added little additional information to the conventional approach of using spirometry because the measurements were heavily interdependent. However, statistical power calculations estimated that a suitably powered study could be conducted over a period of 3 years in just 130 patients using CT densitometry as the primary outcome. This estimate compared very favorably with the power calculations that had been performed for FEV1 as outcome using the NHLBI registry data.59
A second randomized, placebo-controlled study of intravenous AATD augmentation therapy was the EXACTLE study, which was undertaken for the purpose of assessing various surrogate outcome measures for the progression of emphysema.50 The study was conducted at three European sites, and the stated primary outcome was CT densitometry to measure the differential in whole lung density decline between the two treatment groups. Different methods for lung volume correction and regional lung analysis were explored. Other outcomes included exacerbations of COPD, captured using patient diary cards, lung function and health status. Patients with a serum concentration <11 μM were recruited from registries in the UK, Sweden and Denmark and received infusions every week of either AAT (60 mg/kg body weight) or placebo (human albumin) for 2 years, with an optional extension to 30 months. The progression rate of emphysema was determined by the change in the PD15 of the whole lung from CT imaging acquired at baseline and 12 and 24 months. Two methods to standardize density values for total lung volume (TLV) derived from CT imaging were utilized: a “physiological” method adjusted lung density using the ratio of TLV to the individual patient’s TLC and a “statistical” method incorporated log(TLV) as a covariate in the statistical model. Densitometric data were analyzed using both an end point and a slope analysis that, when combined with the two methods of volume adjustment, generated four different analytical approaches.
The modified intention-to-treat population included 36 patients in the active arm and 35 patients on placebo. A beneficial treatment effect of augmentation therapy was suggested by the difference in decline of lung density between the active and placebo groups. This difference was statistically significant in one of the four analytical methods that combined a statistical method for volume correction with an end point analysis (treatment difference 1.472 g/L, P=0.049). As seen in the Dutch–Danish study, the change in volume-adjusted lung density from baseline over the course of the study showed that the difference in lung density between the two treatment groups increased with time, suggesting a therapeutic retardation in the progression of emphysema. Lung function and health status declined over the course of the study, but they were less sensitive measures of emphysema progression than PD15, and there were no significant differences found between the groups in these conventional indices. Nevertheless, in the collective study population, the rates of decline in PD15 and FEV1 were correlated. When treatment efficacy was assessed in a post hoc analysis using targeted regional analysis of the lower lung, which is the predominant location of panlobular emphysema, the retardation of decline in lung density seen in the active arm was statistically significant (P=0.04).82
Exacerbation rates were similar in the active and placebo groups; however, there were proportionately fewer severe exacerbations in the active group. This post hoc analysis finding concurred with there being fewer subjects in the active group with at least one hospitalization due to an exacerbation during the study. The total number of serious adverse events (SAEs) and number of patients with any SAE in the active arm were 28 and 10, respectively, compared with 40 and 18, respectively, in the placebo arm.
The EXACTLE study provided confirmatory evidence of the utility of CT densitometry as a surrogate outcome measure for studies of emphysema modifying therapy in AATD. It also provided some supporting evidence with a patient-reported outcome that suggests that intravenous augmentation may have a beneficial treatment effect on exacerbations. As a consequence of the collective evidence detailed earlier, a meeting of the Blood Products Advisory Committee of the FDA83 concluded that it accepted
serial lung density measurements by HRCT as an appropriate clinically meaningful endpoint to assess the efficacy of augmentation therapy with intravenous alpha 1-proteinase inhibitor products on emphysema disease progression
and permitted its use as a primary end point in Phase IV studies.
Subsequent to the FDA’s acceptance of CT densitometry as a surrogate outcome measure, data from the Dutch–Danish and EXACTLE studies were combined and reanalyzed. The design of these two studies was sufficiently comparable53 as to allow integration of the data with the primary purpose of being able to increase the number of study participants and, consequently, the statistical power. Subjects who had participated in both studies were included only once in this integrated analysis, leaving a total of 119 (n=60, active and n=59, placebo). Mean follow-up was ~2.5 years. The mean change in PD15 from baseline to final CT was −4.082 g/L for active therapy and −6.379 g/L for placebo, with a treatment difference of 2.297 g/L (95% confidence interval, 0.669–3.926: P=0.006). The corresponding rates of annual decline were −1.73 and −2.74 g/L/year, respectively. While there were minor differences between the two studies in CT scanning protocol, the two studies were comparable with regard to patient characteristics, study drug and duration of treatment. Furthermore, the design of both studies met the standards applied for inclusion in an integrated or meta-analysis.
In the wake of EXACTLE, a further study (RAPID) was performed that was powered to demonstrate a treatment effect of intravenous augmentation therapy utilizing CT densitometry as the primary outcome measure.55 Between 2006 and 2010, 180 patients with severe AATD (serum concentration <11 μM) were randomized to receive either intravenous augmentation therapy (60 mg/kg) or placebo over 2 years (RAPID-RCT). Patients in the non-US countries who completed this initial double-blind portion were eligible to receive open-label augmentation treatment at 60 mg/kg for a further 2 years (RAPID-OLE).57 The CT imaging protocol, which was more complex than that employed in the previous two studies, obtained scans at full inspiration (TLC) and relaxed expiration (FRC). In spite of the findings of the EXACTLE study, the chosen primary end point was a combination of CT lung density (PD15) measured at TLC and FRC, but the separate imaging series at TLC and FRC were also utilized. Secondary end points included the number, duration and severity of exacerbations, FEV1, single breath diffusion capacity, incremental shuttle walk test, health status (using the St George’s Respiratory Questionnaire) and mortality. Of the 180 patients, 168 (93%) were ZZ genotype; 93 (52%) received active treatment and 87 (48%) received placebo. Trial withdrawal was slightly higher in the placebo group (18 vs 9, P=0.04). Recruitment was necessary across 28 centers in 13 countries to achieve these numbers and reflected the “real-world” practical difficulties of performing interventional studies in a rare disease, such as AATD.
The absolute differences in lung density decline between the active and placebo groups, when measured at TLC and FRC combined and at FRC alone, were not statistically significant. However, when measured at TLC alone (ie, using the same method advocated by the EXACTLE study), the mean annual rate of decline was lower in the active group (−1.45 g/L/year vs −2.19 g/L/year, P=0.03, with an absolute difference of 0.75 g/L/year) (Figure 2). The only secondary outcome variable that differed significantly between the groups was the serum AAT concentration. A post hoc pharmacometric analysis using modeled data showed that the annual rate of lung density decline was inversely related to the trough serum AAT concentrations. In those patients taking placebo who continued in the open-label extension (ie, RAPID-OLE), the rate of lung density decline was seen to slow and run in parallel with that of patients receiving active treatment throughout the study.57 Furthermore, the correlation between lung density decline and FEV1 decline that was seen in the EXACTLE study was also evident when data over 4 years were examined from the RAPID-RCT and RAPID-OLE studies.
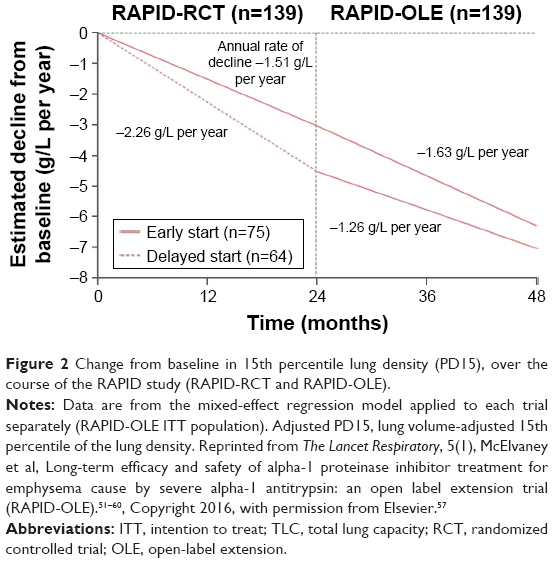 | Figure 2 Change from baseline in 15th percentile lung density (PD15), over the course of the RAPID study (RAPID-RCT and RAPID-OLE). |
The EXACTLE and RAPID studies both demonstrated the critical importance of employing an appropriate CT lung densitometry protocol for the monitoring of emphysema progression and assessment of potential therapeutic disease modification. In particular, the method of volume correction was shown in both studies to influence repeatability of the technique. Furthermore, in the RAPID study, the choice of the level of inspiration at FRC during image acquisition likely deleteriously altered the specificity of lung densitometry as a surrogate measure of emphysema. Emphysema has consistently been defined and quantified on the basis of measurements performed on inflated lung. Therefore, it was entirely logical that the histopathological validation of CT densitometric indices as surrogate measures of emphysema was demonstrated using imaging acquired at full inspiration (ie, approximating to TLC) and compared with morphometry of lung tissue specimens fixed at full inflation. In contrast, CT imaging acquired in an expiratory breath hold is primarily employed for the assessment of air trapping, which is an indirect measure of small airways disease. Consequently, densitometry performed at a volume equivalent to FRC will fail to distinguish the low-density areas arising due to air-trapping from the low-density areas that relate to emphysema.
Inhaled augmentation therapy: rationale and potential applications
A number of lung diseases in which neutrophilic inflammation with an unfavorable elastase–antielastase imbalance plays a predominant role have been considered as potentially benefiting from inhaled AAT augmentation therapy, including AATD-related emphysema and bronchiectasis, cystic fibrosis and post lung transplantation bronchiolitis obliterans syndrome.
The inhaled route of drug delivery obviates the need for intravenous access and is familiar to patients who commonly use aerosol therapy, but the principal rationale for this approach is the need to reduce the amount of protein required to achieve therapeutic concentrations within the lung. There may also be the added benefit of prolonging the half-life of recombinant AAT when using the inhaled route. It is nearly 30 years since this approach was first explored, and initial studies in individuals with AATD suggested that the lung epithelial lining fluid levels of AAT could be raised and the anti-neutrophil elastase capacity maintained. It was estimated that twice-daily administration of 100 mg of human AAT would result in normalization of lung anti-elastase defenses at the alveolar surface.39 It is yet to be determined what the optimal delivery device and dose should be, and it is likely that both will be dependent on the disease being treated and whether the airways or alveoli are the target. However, it is evident that the use of “intelligent” devices, which have the ability to time dose delivery to the inspiratory phase of the respiratory cycle and adjust the pattern of inhalation, can significantly improve efficiency of protein utilization.84,85 Safety and tolerability reports are promising, but suitably designed Phase III trials are needed to determine efficacy of inhaled AAT in AATD and other chronic airway diseases.86
Augmentation therapy in conditions other than emphysema
Extensive data indicate that the principal function of AAT is elastase inhibition, but recent research has demonstrated other biological properties, including anti-inflammatory and immunomodulatory effects. Based on these findings, augmentation therapy has been successfully used in a small number of patients (19 PiZZ and two PiMZ) with vasculitis (2 patients), panniculitis (13 cases), fibromyalgia (3 cases) and refractory asthma (1 case), commonly following the repetitive failure of conventional therapies.30,87 The administration regime used in these conditions has been the same as for the licensed indication, but, in some cases, treatment was discontinued following resolution.
Meta-analyses
Three meta-analyses have evaluated the efficacy of intravenous augmentation therapy. In the first of these, patients treated with intravenous AAT were compared with controls from the Canadian Registry. Randomized and non-randomized clinical studies with either a parallel-group design or a single cohort, pre–post design were eligible if augmentation therapy had been compared with a control regimen and if long-term (>1 year) longitudinal FEV1 follow-up data had been collected. Data from five trials, including a total of 1,509 subjects, revealed a mean reduction in FEV1 decline of 17.9 mL/year in those patients receiving treatment (limited to subjects with FEV1 30%–65% predicted) using a random-effect, meta-analysis.51
In 2010, Gøtzsche and Johansen52 published a review on behalf of the Cochrane Collaboration of the benefits and harms of intravenous augmentation therapy in lung disease: a total population of 140 patients was identified from the Dutch-Danish46 and EXACTLE50 studies, which had been selected independently by the two authors (although some patients included in Gøtzsche and Johansen’s analysis appear to have participated in both studies). Primary outcomes were mortality, FEV1 and harms of the intervention; secondary outcomes included exacerbations, lung infections, hospital admissions, quality of life, carbon monoxide diffusion and CT lung densitometry. The review could not identify any reported information on mortality, although there were no deaths in either study (Professor Dirksen, personal communication). FEV1 deterioration was reported to be slightly greater in the active group, but this was not significant (−20 mL/year more; 95% confidence interval −41 to 1; P=0.06), and for carbon monoxide diffusion, the difference was −0.06 mmol/min/kPa per year (95% confidence interval −0.17 to 0.05; P=0.31). The authors of the review reported that information relating to harms could not be found from the first trial, but in the second trial, SAEs were reported to have occurred in 10 patients in the active group and in 18 patients in the placebo group. They considered that these adverse events were unlikely to have any relation to the drugs used. The review reported a similar quality of life and number of exacerbations annually in the two groups but stated that information on hospital admissions or lung infections was not included in either trial. CT lung density data, combined from both studies, declined less in the active group (difference 1.14 g/L; 95% confidence interval 0.14–2.14; P=0.03). However, the authors concluded that augmentation therapy cannot be recommended because of a lack of convincing evidence of clinical benefit and the cost of treatment. Furthermore, they considered that further studies with surrogate markers cannot be recommended if the aim would be to establish whether augmentation therapy is of clinical benefit and advised that future studies should be powered to detect a possible effect on mortality.
Expert commentary on this publication has proposed a number of important deficiencies in the methodology employed by Gøtzsche and Johansen88 and highlighted that critically important data considered by relevant pulmonology societies to be evidence of efficacy (Table 3) were inappropriately excluded from the meta-analysis.
A second review by the same authors employed comparable methodology to the first review but added data from a third randomized, placebo-controlled trial, the RAPID study,55 to increase the sample population size to 283 (although some patients appear to have participated in more than one of the three studies). It was concluded that, although augmentation therapy does have a statistically significant benefit in reducing the loss of lung tissue measured by CT densitometry, lung function decline was not retarded with therapy. The husband and wife team of authors was unable to reach a conclusion about the impact on mortality, exacerbations, lung infections, hospital admission and quality of life because data on these outcomes were considered to be sparse and, consequently, their recommendation on the use of augmentation therapy was unchanged from their first review.56
Safety and tolerability
The safety and tolerability of AAT augmentation therapy have been well documented. In a 7-year observational cohort study of 747 participants receiving replacement therapy, the overall rate of adverse events was 0.02 per patient-month, and in 83% of patients, no events were reported.89 In participants who received augmentation therapy less frequently than weekly, the rate of adverse events was lower, with 0.03 events per patient-month for weekly infusions compared with 0.024 for fortnightly or three-weekly infusions and 0.005 for monthly infusions. In a further study, only five severe side effects (anaphylactic reaction and congestive cardiac failure) were reported from a total of 58,000 infusions, and the majority of adverse events were mild and self-resolving. These side effects included chills, urticarial rashes, fatigue, nausea and vomiting. No deaths have ever been reported and, since the approval of the use of human AAT by the FDA, there have been no reported cases of hematologic viral transmission.90 A modification of Prolastin, Prolastin-C, was made following the report of a case of anaphylaxis caused by IgE antibodies against Prolastin-HS®: this product has been shown to have equivalence and comparable safety, and there have been no reports of SAEs.91 In the more recent Cochrane review, the potential harms of augmentation therapy were evaluated with the conclusion that most of the reported adverse events were unlikely to be related to treatment, and consequently, no further analysis was conducted.56 Data for the safety and tolerability of Respreeza® are awaited.
Patient-focused perspectives such as quality of life, patient satisfaction/acceptability, adherence and uptake
Emphysema associated with severe AATD causes significant morbidity and mortality, which may possibly be alleviated through protracted augmentation therapy. However, the considerable cost of treatment over many years requires that real-world evidence of efficacy must be obtained, such as through the development and collection of tailored, patient-reported outcomes.92 To date, this issue has not been addressed effectively and there is a pressing need for closer collaboration between industry and clinicians, ideally with effective utilization of the rich data that are available in patient registries.
Administration patterns that differ from weekly infusion are commonly employed because patients often prefer to increase the interval between infusions to 2, 3 or 4 weeks. This approach has been shown in pharmacokinetic studies to be valid,35 but the effect on adherence has not been published.
Conclusion
Intravenous AAT augmentation therapy has been employed for >20 years and appears to be very safe. Trials using lung density as the primary outcome confirm definitively the benefit of this therapy on the progression of emphysema in patients with severe AATD.
The place of replacement therapy in the management of AATD varies between different international and national guidelines (Table 4) but commonly is indicated for use in patients with severe deficiency and evidence of lung function impairment to supplement optimized therapy for COPD.
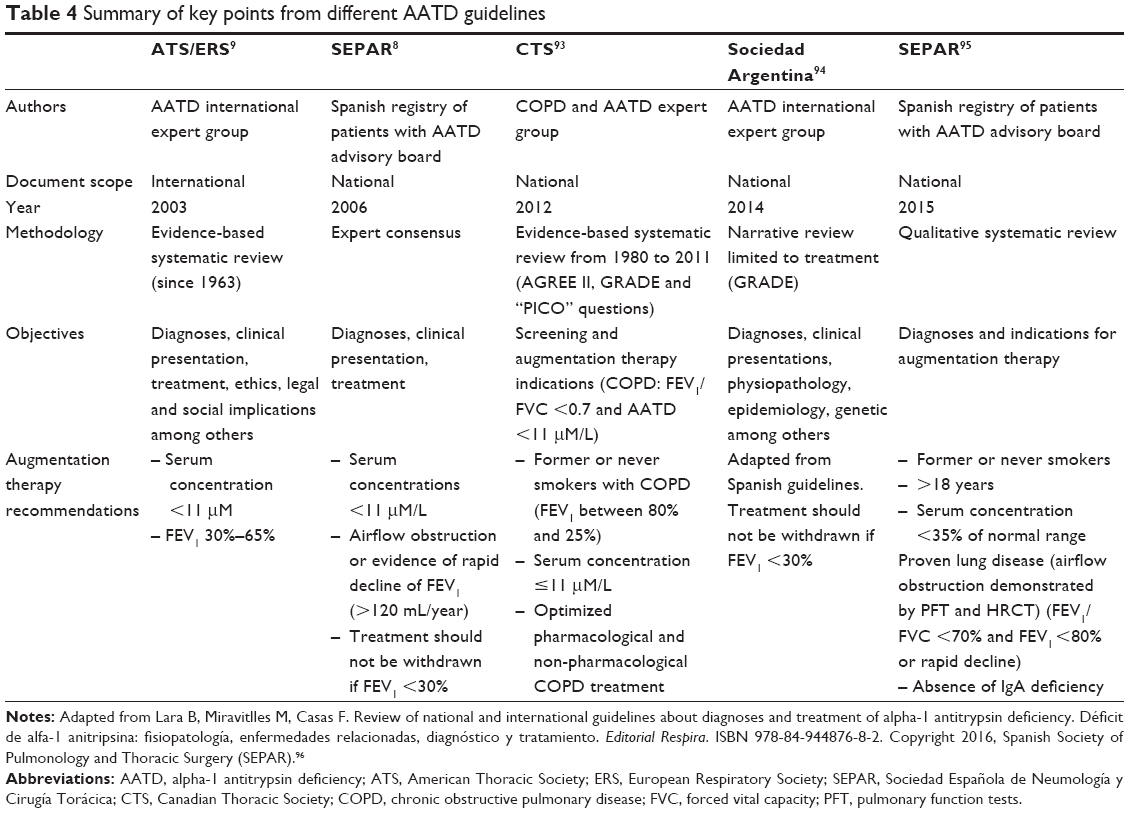 | Table 4 Summary of key points from different AATD guidelines |
Disclosure
DGP has received fees from CSL Behring, Grifols and Kamada for advisory boards and received lecture fees from CSL Behring and Grifols. BL has received speaker’s fees from AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Grifols, Merck, Novartis, Pfizer in the past but not related with the content of this article. The authors report no other conflicts of interest in this work.
References
Janciauskiene S, Nita I, Subramaniyam D, Li Q, Lanacaster JR, Matalon S. Alpha-1 antitrypsin inhibits the activity of the matriptasa catalytic domain in vitro. Am J Respir Cell Mol Biol. 2008;39:631–637. | ||
Petrache I, Fijalkowska I, Medler TR, et al. Alpha-1 antitrypsin inhibits caspase-3 activity preventing lung endothelial cell apoptosis. Am J Pathol. 2006;169:1155–1166. | ||
Carroll T, O’Connor C, Reves E, McElvaney N. Alpha-1 antitrypsin deficiency: a genetic risk factor for COPD. COPD Current Concepts and Practice. Dr Kian-Chung Ong Ed. ISBN: 978-953-51-0163-5. | ||
Gooptu B, Lomas DA. Conformational Pathology of the Serpins: themes, variations and therapeutic strategies. Annu Rev Biochem. 2009;78:147–176. | ||
Perlmutter DH, Brodsky JL, Balisteri WF, Trapnell BC. Molecular pathogenesis of alpha-1-antitrypsin deficiency associated liver disease: a meeting review. Hepatology. 2007;45:1313–1323. | ||
Janciuskiene SM, Bals R, Koczulla R, Volgelmeier C, Köhnlein T, Welte T. The discovery of alpha-1 antitrypsin and its role in health and disease. Respir Med. 2011;105(8):1129–1139. | ||
Shows TB, McAlpine PJ. The 1981 catalogue of assigned human genetic markers and report of the nomenclature committee. Oslo Conference: Sixth International Workshop on human Gene Mpping. Cytogenet Cell Genet. 1982;32:221–245. | ||
Vidal R, Blanco I, Casas F, Jardí R, Miravitlles M. Diagnoses and treatment of alpha-1 antitrypsin deficiency. Arch Bronconeumol. 2006;42(12):645–659. | ||
Alpha-1 antitrypsin deficiency Task force. American Thoracic Society/ European Respiratory Society Statement: Standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168:818–900. | ||
De Serres F, Blanco I, Fernández-Bustillo E. PI*S and PI*Z Alpha-1 antitrypsin deficiency worldwide. A review of existing genetic epidemiological data. Monaldi Arch Chest Dis. 2007;67(4):184–208. | ||
Turino GM, Barker AF, Brantly ML, Cohen AB, Conelly RP, Crystal RG. Clinical features of individuals with PI*SZ phenotype of alpha-1 antitrypsin deficiency. Alpha-1 antitrypsin deficiency Registry Study Group. Am J Respir Crit Care Med. 1996;154:1718–1725. | ||
Lee P, Gildea TR, Stoller JK. Emphysema in non smokers: alpha-1-antitrypsin deficiency and other causes. Cleve Cli J Med. 2002;69:928–929. | ||
Cox D, Levison H. Emphysema of early onset associated with a complete deficiency of alpha-1 antitrypsin (null homozygotes). Am Rev Respir Dis. 1988;137:371–375. | ||
DeMeo DL, Silverman EK. Alpha-1 antitrypsin defieicny. Genetic aspects of alpha-1 antitrypsin deficiency: phenotypes and genetic modifiers of emphysema risk. Thorax. 2004;59:259–264. | ||
Seersholm N, Kok-Jensen A, Dirksen A. Decline in FEV1 among patients with severe hereditary alpha-1 antitrypsin deficiency type PiZ. Am J Respir Crit Care Med. 1995;152:1922–1925. | ||
Piituilainen E, Tornling G, Eriksson S. Enviromental correlates of impaired lung function in non-smokers with severe alpha-1 antitrypsin deficiency (PiZZ). Thorax. 1998;53:939–943. | ||
DeMeo DL, Sandhaus RA, Barker AF, et al. Determinants of airflow obstruction in severe alpha-1 antitrypsin deficiency. Thorax. 2007;62:806–813. | ||
Seersholm N, Kok-Jensen A, Dirksen A. Survival of patients with severe alpha-1 antitrypsin deficiency with special reference to non-index cases. Thorax. 1994;94:695–698. | ||
Tanash HA, Nilsson PM, Nilsson JA, Piituilainen E. Clinical course and prognosis of never smokers with severe AATD (PiZ). Thorax. 2008;63:1091–1095. | ||
Gadek JE, Klin HG, Hholland PV, Crystal RG. Replacement therapy of alpha-1 antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjects. J Clin Invest. 1981;68:1158–1165. | ||
From NIH: Intravenous replacement therapy for patients with severe alpha-1 antitrypsin deficiency. JAMA. 1982;248:1693. | ||
Wewers MD, Casolaro MA, Sellers SE, Swayze SC, Mc Phaul KM, Wittes JT. Replacement therapy for alpha-1 antitrypsin deficiency associated with emphysema. N Engl J Med. 1987;316(17):1055–1062. | ||
Kueppers F. The role of augmentation therapy in alpha-1 antitrypsin deficiency. Curr Med Res Opin. 2011;27:579–588. | ||
Mohanka M, Khemasuwan D, Stoller JK. A review of augmentation therapy for alpha-1 antitrypsin deficiency. Expert Opin Biol Ther. 2012;12:685–700. | ||
Gauvain C, Mornex JF, Pison C, et al. Health related quality of life in patients with alpha-1 antitrypsin deficiency: the French Experience. COPD. 2015;1:46–51. | ||
Mordwinkin NM, Louie SG. Aralast: an alpha 1-protease inhibitor for the treatment of alpha-1 antitrypsin deficiency. Expert Opin Pharmacother. 2007;8(15):2609–2614. | ||
Stoller JK, Rouhani F, Brantly M, et al. Biochemical efficacy and safety of a new pooled human alpha (1)-antitrypsin, Respitin. Chest. 2002;122(1):66–74. | ||
Respreeza EMA/554260/2016. EPAR Summary for the public. European Medicines Agency. London, 2016. | ||
Stocks JM, Brantly M, Pollock D, et al. Multicenter study: the biochemical efficacy, safety and tolerability of a new alpha-1 proteinase inhibitor, Zemaira. COPD. 2006;3(1):17–23. | ||
Blanco I, Lara B, De Serres F. Efficacy of alpha-1 antitrypsin augmentation therapy in conditions other than pulmonary emphysema. Orphanet Journal of Rare Diseases. 2011:6–14. | ||
Teschler H. Long-term experience in the treatment of alpha-1 antitrypsin deficiency: 25 years of augmentation therapy. Eur Respir Rev. 2015;24:46–51. | ||
Coan MN, Brockway WJ, Eguizabal H, et al. Preparation and properties of alpha-1 proteinase inhibitor concentrate from human plasma. Vox Sang. 1985;48(6):333–342. | ||
Burnout T, Constans J, Clerc A, Descamps J, Martinache L, Goudemand M. Biochemical and biological properties of an alpha-1 antitrypsin concentrate. Vox Sang. 1986;52(4):291–297. | ||
Stocks JM, Brantly ML, Wang-Smith L, et al. Pharmacokinetic comparability of Prolastin®-C to Prolastin® in alpha-1 antitrypsin deficiency: a raondmized study. BMC Clin Pharmacol. 2010;10:13. Doi:10.1186/1472-6904-10-13. | ||
Soy D, de la Roza C, Lara B, Esquinas C, Torres A, Miravitlles M. Alpha-1 antitrypsin deficiency: optimal therapeutic regimen based on population pharmacokinetics. Thorax. 2006;61:1059–1064. | ||
Wright G, Carver A, Cottom D. High level expression of active human alpha-1 antitrypsin in the milk of transgenic sheep. Biotechnology. 1991;9:830–834. | ||
Spencer LT, Humphries JE, Brantly ML; Transgenic Human Alpha-1 antitrypsin Study Group. Antibody response to aerosolized transgenic human alpha-1 antitrypsin. N engl J Med. 2005;352:2030–2031. | ||
Ross D, Brown T, Harper R, Pamarthi M, Nixon J, Bromirski J, et al. Production and characterization of a novel human recombinant alpha-1 antitrypsin in PERC. C6 cells. J Biotechnol. 2012;162:262–273. | ||
Hubbard RC, Brantly ML, Seller SE. Antineutrophilic elastase defenses of the lower respiratory tract in alpha-1 antitryspin deficiency directly augmented with aerosol of alpha-1 antitrypsin. Ann Intern Med. 1989;111:206–212. | ||
Siekmeier R. Lung deposition of inhaled alpha-1-proteinase inhibitor (alpha1-PI) problems and experience of alpha1-PI inhalation therapy in patients with hereditary alpha1-PI deficiency and cystic fibrosis. Eur J Med Res. 2010;15:164–174. | ||
Hubbard RC, Sellers S, Czerski D, Stephens L, Cristal RG. Biochemical efficacy and safety of monthly augmentation therapy for alpha-1 antitrypsin deficiency. JAMA. 1988;260(9):1259–1264. | ||
Stockley RA, Bayley DL, Unsal I, Dowson W. The effect of augmentation therapy on bronchial inflammation in alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2002;165:1494–1498. | ||
Ma S, Lin YY, He J, Rouhani FN, Brantly M, Turino GM. Alpha-1 antitrypsin augmentation therapy and biomarkers of elastin degradation. COPD. 2013;10:473–481. | ||
Seersholm N, Wencker M, Banik N, et al. Does alpha-1 augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha-1 antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft Zup-Therapie Von Lunge-Nerkrankungen (WATL) Alpha1-AT study group. Eur Respir J. 1997;10:2260–2263. | ||
The alpha-1-antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha-1 antitrypsin. Am J Respir Crit Care Med. 1998;158:49–59. | ||
Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha-1 antitrypsin augmentation therapy. Am J Respir Crit Care Med. 1999;160:1468–1472. | ||
Lieberman J. Augmentation therapy reduces frequency og lung infections in antitrypsin deficiency: a new hipothesis with supporting data. Chest. 2000;118:1480–1485 | ||
Wencker M, Furhmann B, Banik N, Konietzko N. Longitudinal follow-up of patients with alpha-1 antiportease inhibitor deficiency before and during therapy with IV alpha (1)-protease inhibitor. Chest. 2001;119:737–744. | ||
Tonelli AR, Rouhani F, Li N, Schreck P, Brantly ML. Alpha-1 antitrypsin augmentation therapy in deficient individuals enrolled in the Alpha-1 Foundation DNA and tissue bank. Int J COPD. 2009;4:443–452. | ||
Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J. 2009;33(6):1345–1353. | ||
Chapman KR, Stockley RA, Dawkins C, Wilkes MM, Navickis RJ. Augmentation therapy for alpha-1 antitrypsin deficiency: a meta-analysis. COPD. 2009;6:177–184. | ||
Gøtzsche PC, Johansen HK. Intravenous alpha-1 antitrypsin augmentation therapy for treating patients with alpha-1 antitrypsin deficiency and lung disease (Review). Cochrane Database Syst Rev. 2010;7:CD007851. | ||
Stockley RA, Parr DG, Piituilainen E, Stolk J, Stoel BBC, Dirksen A. Therapeutic efficacy of alpha-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respir Res. 2010;11:136. | ||
Barros-Tizón JC, Torres ML, Blanco I, Martínez MT; Investigators of the rEXA study group. Reduction of severe exacerbations and hospitalization-derived costs in alpha-1-antitrypsin-deficient patients treated with alpha-1-antitrypsin augmentation therapy. Ther Adv Respir Dis. 2012;6:67–78.9. | ||
Chapman KR, Burdon JG, Piituilainen E, et al; RAPID Trial Study Group. Intravenous augmentation treatment and lung density in severe alpha-1 antitrypsin deficiency (RAPID): a randomized, double-blind, placebo-controlled trial. Lancet. 2015;386:360–368. | ||
Gøtzsche PC, Johansen HK. Intravenous alpha-1 antitrypsin augmentation therapy for treating patients with alpha-1 antitrypsin deficiency and lung disease (review). Cochrane Database of Syst Rev. 2016;9:CD007851. | ||
McElvaney, Burdon J, Holmes M, Glanville A, Wark P, Thompson P. Long-term efficacy and safety of alpha-1 proteinase inhibitor treatment for emphysema cause by severe alpha-1 antitrypsin: an open label extension trial (RAPID-OLE). The Lancet Respiratory 2016. DOI101016/52213-2006(16)30430-1. | ||
Casas F, Hernandez JM. General treatment and intravenous treatment with alpha-1 antitrypsin. Chapter 12. Deficit de alfa-1 antitripsina: fisiopatologia, enfermedades relacionadas, diagnostico y tratamiento. Editorial Respira. ISBN 978-84-944876-8-2. | ||
Schluchter MD, Stoller JK, Barker AF, et al. Feasibility of a clinical trial of augmentation therapy for alpha(1)-antitrypsin deficiency. The Alpha 1-Antitrypsin Deficiency Registry Study Group. Am J Respir Crit Care Med. 2000;161(3 Pt 1):796–801. | ||
Dowson LJ, Guest PJ, Stockley RA. Longitudinal changes in physiological, radiological, and health status measurements in alpha (1)-antitrypsin deficiency and factors associated with decline. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1805–1809. | ||
Stolk J, Ng WH, Bakker ME, et al. Correlation between annual change in health status and computer tomography derived lung density in subjects with alpha1-antitrypsin deficiency. Thorax. 2003;58(12):1027–1030. | ||
Parr DG, Stoel BC, Stolk J, et al. Validation of computed tomographic lung densitometry for monitoring emphysema in alpha1-antitrypsin deficiency. Thorax. 2006;61(6):485–490 | ||
Hruban RH, Meziane MA, Zerhouni EA, et al. High resolution computed tomography of inflation-fixed lungs. Pathologic-radiologic correlation of centrilobular emphysema. Am Rev Respir Dis. 1987;136(4):935–940. | ||
Hayhurst MD, MacNee W, Flenley DC, et al. Diagnosis of pulmonary emphysema by computerised tomography. Lancet. 1984;2(8398):320–322. | ||
Gould GA, MacNee W, McLean A, et al. CT measurements of lung density in life can quantitate distal airspace enlargement – an essential defining feature of human emphysema. Am Rev Respir Dis. 1988;137(2):380–392. | ||
Gevenois PA, de Maertelaer V, De Vuyst P, et al. Comparison of computed density and macroscopic morphometry in pulmonary emphysema. Am J Respir Crit Care Med. 1995;152(2):653–657. | ||
Gevenois PA, De Vuyst P, de Maertelaer V, et al. Comparison of computed density and microscopic morphometry in pulmonary emphysema. Am J Respir Crit Care Med. 1996;154(1):187–192. | ||
Muller NL, Staples CA, Miller RR, et al. “Density mask”. An objective method to quantitate emphysema using computed tomography. Chest. 1988;94(4):782–787. | ||
Gould GA, Redpath AT, Ryan M, et al. Lung CT density correlates with measurements of airflow limitation and the diffusing capacity. Eur Respir J. 1991;4(2):141–146. | ||
Kinsella M, Muller NL, Abboud RT, et al. Quantitation of emphysema by computed tomography using a “density mask” program and correlation with pulmonary function tests. Chest. 1990;97(2):315–321. | ||
Dowson LJ, Newall C, Guest PJ, et al. Exercise capacity predicts health status in alpha (1)-antitrypsin deficiency. Am J Respir Crit Care Med. 2001;163(4):936–941. | ||
Johannessen A, Skorge TD, Bottai M, et al. Mortality by level of emphysema and airway wall thickness. Am J Respir Crit Care Med. 2013;187(6):602–608. | ||
Dawkins PA, Dowson LJ, Guest PJ, et al. Predictors of mortality in alpha1-antitrypsin deficiency. Thorax. 2003;58(12):1020–1026. | ||
Dowson LJ, Guest PJ, Hill SL, et al. High-resolution computed tomography scanning in alpha1-antitrypsin deficiency: relationship to lung function and health status. Eur Respir J. 2001;17(6):1097–1104. | ||
Kalender WA, Rienmuller R, Seissler W, et al. Measurement of pulmonary parenchymal attenuation: use of spirometric gating with quantitative CT. Radiology. 1990;175(1):265–268. | ||
Moroni C, Mascalchi M, Camiciottoli G, et al. Comparison of spirometric-gated and -ungated HRCT in COPD. J Comput Assist Tomogr. 2003;27(3):375–379. | ||
Hoffman EA, Reinhardt JM, Sonka M, et al. Characterization of the interstitial lung diseases via density-based and texture-based analysis of computed tomography images of lung structure and function. Acad Radiol. 2003;10(10):1104–1118. | ||
Stolk J, Dirksen A, van der Lugt AA, et al. Repeatability of lung density measurements with low-dose computed tomography in subjects with alpha-1-antitrypsin deficiency-associated emphysema. Invest Radiol. 2001;36(11):648–651. | ||
Dirksen A. Monitoring the progress of emphysema by repeat computed tomography scans with focus on noise reduction. Proc Am Thorac Soc. 2008;5(9):925–928. | ||
Stoel BC, Putter H, Bakker ME, et al. Volume correction in computed tomography densitometry for follow-up studies on pulmonary emphysema. Proc Am Thorac Soc. 2008;5(9):919–924. | ||
Shaker SB, Dirksen A, Laursen LC, et al. Volume adjustment of lung density by computed tomography scans in patients with emphysema. Acta Radiol. 2004;45(4):417–423. | ||
Parr DG, Dirksen A, Piitulainen E, et al. Exploring the optimum approach to the use of CT densitometry in a randomised placebo-controlled study of augmentation therapy in alpha 1-antitrypsin deficiency. Respir Res. 2009;10:75. | ||
Food and Drug Administration. Blood Products Advisory Committee. Clinical and Surrogate Endpoints for Evaluating Efficacy of Alpha1-Proteinase Inhibitor (Human) Augmentation Therapy. 95th Meeting Issue Summary, Gaithersburg, Maryland, USA. July 20–21; 2009. | ||
Fischer A, Stegemann J, Scheuch G, Siekmeier R. Novel devices for individualized controlled inhalation can optimized aerosol therapy in efficacy, patient care and power of clinical trails. Eur J Med Res. 2009;14:71–77. | ||
Geller DE, Kesser KC. The I-neb adaptative aerosol delivery system enhances delivery of alpha-1 antitrypsin with controlled inhalation. J Aerosol Med Pulm Drug Deliv. 2010;23:S55–S59. | ||
Sorrells S, Camprubi S, Griffin R, Chen J, Ayguasanosa J. SPARTA clinical trial design: exploring the efficacy and safety of two dose regimens of alpha-1 antitrypsin proteinase inhibitor augmentation therapy in alpha-1 antitrypsin deficiency. Respir Med. 2015;109(4):490–499. | ||
Hernandez JM, Fumero S, Alvarez A. Successful alpha-1 antitrypsin replacement therapy in a patient with alpha-1 antitrypsin deficiency and granulomatosis with polyangiitis. Rheumatology. 2013:52:755–757. | ||
Gøtzsche PC, Johansen HK. Intravenous alpha-1 antitrypsin augmentation therapy for treating patients with alpha-1 antitrypsin deficiency and lung disease (Review). Cochrane Database Syst Rev. 2010;7:CD007851. Available from: http://www.alpha1.org/Portals/0/Response-Cochrane.pdf | ||
Stoller JK, Fallat R, Schluchter MD. Augmentation Therapy with alpha-1 anitrypsin: patterns of use and adverse events. Chest 2003;123(5):1425–1434. | ||
Wencker M, Bbanik N, Jul R, Seidel R, Konietzko N. Long term treatment of alpha-1 antitrypsin deficiency related pulmonary emphysema with human alpha-1 antitrypsin. Wissenshaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WALT) alpha-1-AT-study group. Eur Respir J. 1998;11(2):428–433. | ||
Meyer FJ, Wencker M, Teschler H. Acute allergic reaction and demonstration of specific IgE antibodies against alpha-1 proteiase inhibitor. Eur Respir J. 1998;12(4):996–997. | ||
Chotirmall S, Al-Alawi M, McEnery T, McElvaney N. Alpha-1 proteinase inhibitors for the treatment of alpha-1 antitrypsin deficiency: safety, tolerability and patients outcomes. Ther Clin Risk Manag. 2015;11:143–151. | ||
Marciniuk DD, Hernandez P, Balter M, Bourbeau J, Chapman KR, Ford GT, et al; Canadian Thoracic Society COPD clinical assembly Alpha-1 Antitrypsin Deficiency Expert Working Group. Alpha-1 antitrypsin deficiency targeted testing and augmentation therapy: a Canadian Thoracic Society clnical practice guideline. Can Respir J. 2012;19-109-16. | ||
Menga G, Miravitlles M, Blanco I, Echazarreta S, Sorroche PB, Fernandez ME, et al. Normativas de diagnostic y tratamiento del deficit de alfa-1 antitripsina. RAMR. 2014;1:28–46. | ||
Casas F, Blanco I, Martinez MT, Bustamante A, Miravitlles M, Cadenas S, et al. Indications for active cases searches and intravenous alpha-1 antitrypsin treatment for patients wirh alpha-1 antitrypsin deficiency chronic pulmonary obstructive disease: an update. Arch Bronchoneumol. 2015;51(4):185–192. | ||
Lara B, Miravitlles M, Casas F. Review of national and international guidelines about diagnoses and treatment of alpha-1 antitrypsin deficiency. Deficit de alfa-1 antitripsina: fisiopatologia, enfermedades relacionadas, diagnostico y tratamiento. Editorial Respira. ISBN 978-84-944876-8-2. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.