Back to Journals » Journal of Multidisciplinary Healthcare » Volume 19
Clinical Practice of Multidisciplinary Team-Guided Comprehensive Management for Pediatric Patients with Fabry Disease: A Single-Center Case Series
Authors Liu Y, Liu X, Yang M ![]() , Wang Q, Xu W, Zeng J, Han T, Mao H, Qian S
, Wang Q, Xu W, Zeng J, Han T, Mao H, Qian S
Received 15 September 2025
Accepted for publication 8 January 2026
Published 27 January 2026 Volume 2026:19 537644
DOI https://doi.org/10.2147/JMDH.S537644
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Charles V Pollack
Yingchao Liu,1,* Xiaorong Liu,2,* Mei Yang,3 Quan Wang,1 Wenmiao Xu,1 Jiansheng Zeng,1 Tongxin Han,4 Huawei Mao,4 Suyun Qian1
1Pediatric Intensive Care Unit, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, Beijing, 100045, People’s Republic of China; 2Department of Nephrology, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, Beijing, 100045, People’s Republic of China; 3Department of Pharmacy, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, Beijing, 100045, People’s Republic of China; 4Department of Rheumatology and Immunology, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, Beijing, 100045, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Suyun Qian, Pediatric Intensive Care Unit, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, No. 56 Nan-Li-Shi Road, Beijing, 100045, People’s Republic of China, Tel +8610 59616767, Fax +8610 59718700, Email [email protected] Huawei Mao, Department of Rheumatology and Immunology, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, No. 56 Nan-Li-Shi Road, Beijing, 100045, People’s Republic of China, Tel +8610 59616316, Fax +8610 59718700, Email [email protected]
Purpose: This study aimed to illustrate the practical application and preliminary outcomes of a multidisciplinary team (MDT) approach in managing pediatric Fabry disease (FD), and provide a reference for the clinical management of this rare disease.
Methods: This single-center, prospective, observational case series was conducted at Beijing Children’s Hospital. Between March 2021 and February 2024, five pediatric patients with FD who were managed by a dedicated MDT were enrolled. The MDT operated under a model of “identification and initiation in outpatient clinics, overall management by the core team, and specialist consultation as needed.” The workflow covered a dual-path diagnostic pathway (MDT-initiated or external referral), pedigree screening, baseline assessment, individualized treatment, and long-term follow-up.
Results: Five pediatric FD probands were enrolled, with four diagnosed through the MDT-initiated pathway and one via external referral. For the four newly diagnosed patients, the MDT achieved a definitive diagnosis within 15– 30 days of its engagement, despite a prior diagnostic odyssey of 2.0– 5.9 years. Pedigree screening identified an asymptomatic sibling, enabling pre-symptomatic diagnosis. All patients commenced enzyme replacement therapy (ERT, agalsidase α) and did not develop adverse events. Through MDT coordination, they currently maintain continuous ERT at medical institutions with a travel time of 20 minutes to 2 hours. MDT-guided pain treatment, primarily with oxcarbazepine, effectively controlled neuropathic pain in most cases and improved quality of life. Psychosocial support alleviated family burdens, achieving treatment cost reimbursement rates of 60%– 85%. At one-year follow-up, symptomatic improvement and significant reductions in globotriaosylsphingosine (Lyso-GL-3) levels were observed.
Conclusion: The structured MDT approach facilitated accelerated diagnosis, early intervention, and comprehensive care in this pediatric FD cohort, yielding positive short-term outcomes and providing a practical reference for rare disease management.
Keywords: Fabry disease, rare disease, pediatrics, multidisciplinary team, clinical practice
Introduction
Fabry disease (FD) is a rare X-linked inherited disorder that was included in China’s First Batch of Rare Diseases Catalog in 2018.1 FD is caused by pathogenic variants in the GLA gene, which results in partial or complete loss of the activity of the lysosomal hydrolase α-galactosidase A (α-Gal A). This loss of activity results in the progressive accumulation of the metabolic substrate globotriaosylceramide (GL-3) and its derivative deacetyl GL-3 (Lyso-GL-3) in tissues. It then leads to multi-organ damage, which can be manifested as chronic intermittent burning pain in hands or feet, anhidrosis/hypohidrosis, cutaneous angiokeratoma, kidney damage, hypertrophic cardiomyopathy or heart failure, and stroke.2 Without early and effective intervention, the disease may progressively lead to multi-organ failure and life-threatening complications. The life expectancy of male patients could be reduced by about 15–20 years, and that of female patients could be reduced by about 6–10 years.3,4
The clinical manifestations of FD are complex, variable, and non-specific. The diagnosis and treatment process often involves multiple clinical subspecialties and is prone to misdiagnosis or delayed diagnosis. This condition results in severe complications and imposes a significant medical and economic burdens on patients, families, and society. The existing consensus recommendation is to establish a multidisciplinary team (MDT), including pediatricians, geneticists, pain specialists, and other relevant experts, to lead the management of children with FD and improve outcomes.5 In 2023, the Multidisciplinary Treatment in the Long-term Management of Fabry Disease was published in China and recommended coordinated treatment and management strategies by multiple disciplines.6
In 2021, a children’s hospital in Shanghai, China, reported that through a one-year MDT diagnosis and screening project for FD, five children with FD were diagnosed for the first time, highlighting the important role of the MDT approach in rare disease screening.7 However, the specific practical pathways and operational workflows of MDT in the comprehensive management of pediatric FD remain to be further clarified. In March 2021, the Rare Disease Center at Beijing Children’s Hospital, Capital Medical University was established. Multiple relevant disciplines formed an FD MDT to create a one-stop, comprehensive diagnosis and treatment platform for children with FD. This study aims to illustrate the practical application of the MDT approach by detailing the clinical presentations, diagnostic process, and management outcomes of the first five pediatric patients with FD under the MDT program. Results provide a reference for the management of this rare disease.
Methods
Study Design and Participants
This single-center, prospective, observational case series was approved by the Ethics Board of Beijing Children’s Hospital, Capital Medical University ([2022]-E-060-Y). From March 2021 to February 2024, patients meeting the criteria were consecutively enrolled. The inclusion criteria as follows: (1) age <18 years; (2) confirmed diagnosis of FD;1 (3) receipt of comprehensive management by the FD MDT at our hospital; and (4) signed informed consent. The exclusion criteria were (1) follow-up duration <1 year; (2) incomplete key clinical records, which precluded assessment of disease status or treatment outcomes.
MDT Framework and Workflow
Establishment of MDT
A dedicated pediatric FD MDT was established in March 2021 at Beijing Children’s Hospital, Capital Medical University, with the Pediatric Intensive Care Unit (PICU) as the designated clinical ward. The MDT operates under a model of “identification and initiation in outpatient clinics, overall management by the core team, and specialist consultation as needed” to optimize resource use. The MDT conducted regular FD-focused training for its members.
Composition of MDT
The MDT consisted of three components: (1) outpatient identification team, which is composed of physicians from rheumatology and immunology, cardiology, nephrology, neurology, gastroenterology, dermatology, and ophthalmology and is responsible for identifying children with high-risk features of FD during outpatient settings and initiating the diagnostic process; (2) core team who is responsible for overall management and comprises PICU physicians, clinical pharmacists, clinical geneticists, nursing staff, and social workers; (3) consultation specialists, which include experts from nephrology, rheumatology and immunology, neurology, gastroenterology, dermatology, ophthalmology, cardiology, psychology, pathology, and clinical nutrition, to provide consultations as needed.
Diagnostic Process
Diagnosis was established through two pathways. (1) For MDT-initiated diagnosis, the process started when physicians in the outpatient identification team identified children with high-risk FD features (eg, burning pain in hands or feet, cutaneous angiokeratoma, anhidrosis/hypohidrosis, corneal verticillate, unexplained chronic kidney disease, unexplained hypertrophic myocardiopathy or heart failure, unexplained early-onset stroke, unexplained abdominal pain or intermittent diarrhea, and unexplained hearing loss). All suspected cases were screened using the dried blood spots (DBS) triple-test, which analyzed α-Gal A activity, Lyso-GL-3 concentration, and GLA gene sequencing. Diagnosis was confirmed according to the Expert Consensus for Diagnosis and Treatment of Fabry Disease in China (2021).1 (2) For external referral, patients with a previously confirmed FD diagnosis were referred to our center. They were initially received by the outpatient identification team and then directly admitted to the designated clinical ward (PICU) for ongoing management by the core team.
Pedigree Screening
Following the diagnosis of the proband, a standardized pedigree screening was initiated. A clinical geneticist constructed the pedigree chart and performed genetic risk assessments for all consenting first-degree relatives (parents, siblings). Relatives who met the screening criteria were advised to undergo DBS triple-test. If a pathogenic variant was identified, then screening was recommended to other at-risk relatives.
Baseline Assessment and Treatment
Before treatment, a comprehensive baseline assessment was conducted. This assessment evaluated multi-organ involvement (kidney, heart, nervous system/brain, etc.), Mainz Severity Score Index (MSSI) evaluation, Brief Pain Inventory (BPI), and laboratory parameters. Based on the findings, an enzyme replacement therapy (ERT) regimen was initiated under the guidance of the clinical pharmacist, administered and monitored by nursing staff. Consultations with relevant specialists were integrated into the care plan as clinically indicated. Social workers provided psychological support, resource linkage services, and assistance with medical aid application.
Follow-up
After discharge, long-term follow-up was conducted by the PICU physician and social worker via telephone and WeChat to monitor disease progression and treatment response and offered continuous support. The FD MDT workflow is summarized in Figure 1.
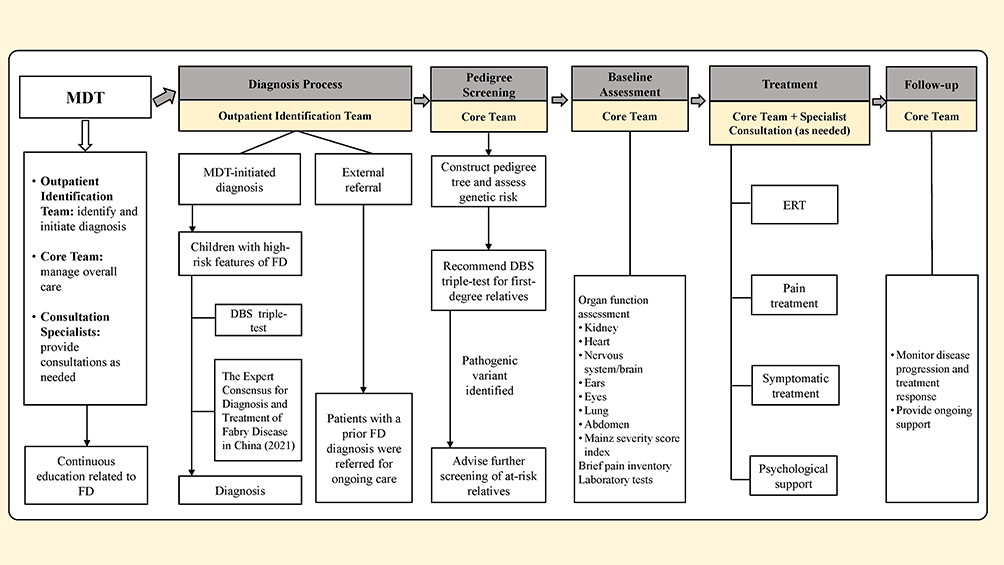 |
Figure 1 The Workflow Diagram of the Fabry disease multidisciplinary team. Abbreviations: DBS, dried blood spots; ERT, enzyme replacement therapy; FD, Fabry disease; MDT, multidisciplinary team. |
Collection of Clinical Data
Clinical data were collected from electronic medical records and included demographic data, such as age, time from symptom onset to diagnosis, main symptoms; auxiliary examinations, such as α-Gal A activity, Lyso-GL-3 level, genetic testing, fundoscopic examination, hearing test, echocardiogram, and brain magnetic resonance imaging; diagnosis and treatment data, such as drug dose, interval, and adverse drug reactions; and follow-up data, such as current treatment location, pain profile, and treatment cost.
Results
Diagnostic Findings and Time to Diagnosis
Five pediatric probands with FD were enrolled in this study, four of whom were diagnosed through the MDT-initiated pathway and one through external referral. The age at symptom onset ranged from 5.0 to 8.6 years, with the presenting symptom being cutaneous angiokeratoma in case 4 and bilateral toe burning pain (with or without finger involvement) in the four other cases. All the five probands presented with bilateral acral burning pain and hypohidrosis. Four cases exhibited either short or relatively short stature, three had cutaneous angiokeratoma, two had thickened lips, one presented with intermittent abdominal pain, and one presented with mild high-frequency bilateral hearing loss (Table 1). Prior to definitive diagnosis, all five children had visited multiple other hospitals. Cases 1 and 5 had previously been misdiagnosed with “juvenile idiopathic arthritis” and “joint pain,” respectively, and had received treatment with “immunomodulatory drugs.”
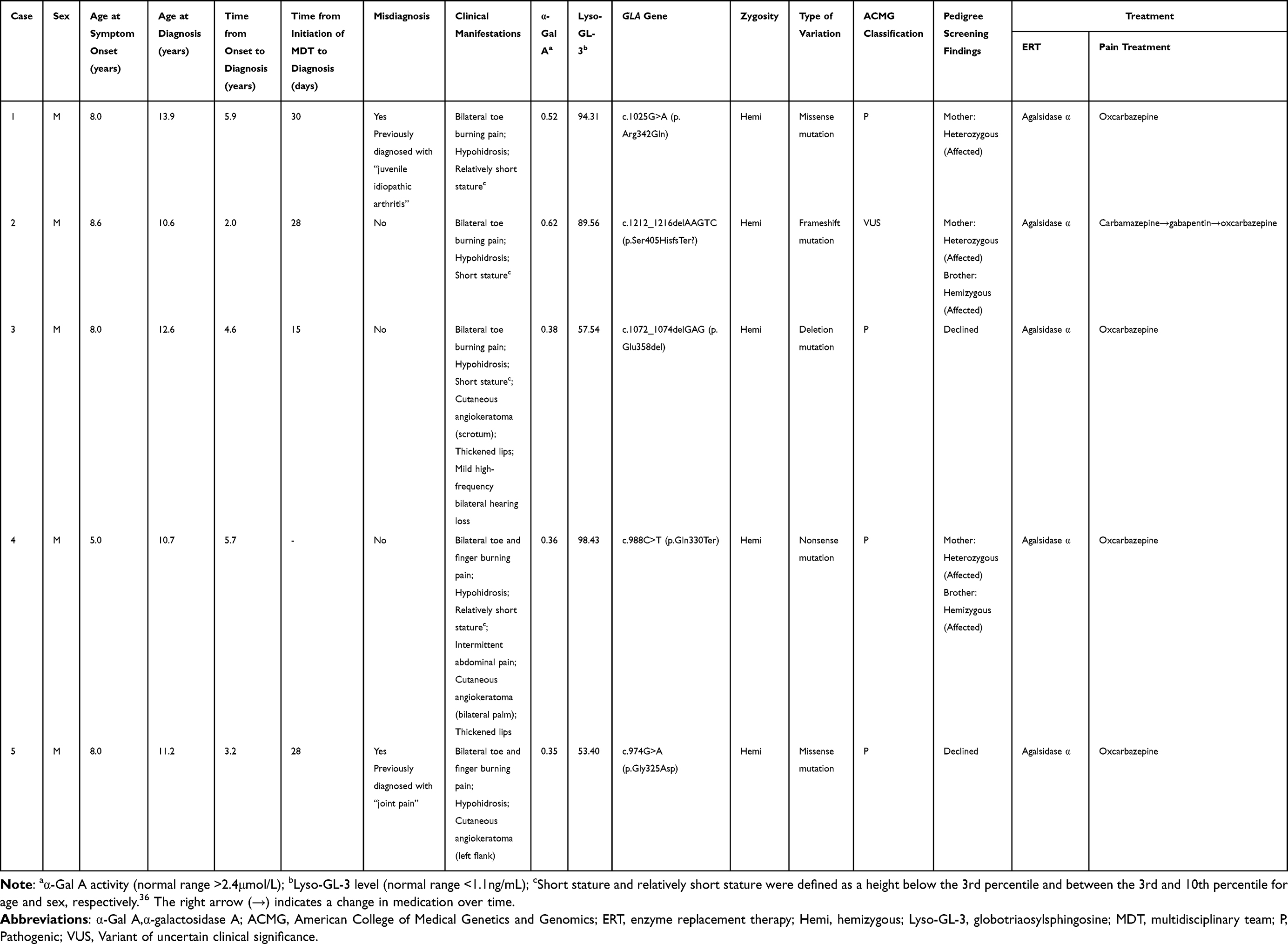 |
Table 1 Clinical Characteristics, Genetic Features, and Treatment of Five Probands with Fabry Disease |
Cases 1, 2, 3, and 5 were enrolled through the MDT-initiated pathway, and their diagnostic process was initiated in the rheumatology and immunology outpatient clinic. The DBS results revealed significantly reduced α-Gal A activity and pathogenic GLA gene variants in cases 1, 3, and 5, confirming the definitive diagnosis of FD. Case 2 harbored a GLA frameshift variant (c.1212_1216delAAGTC, p.Ser405HisfsTer?) classified as a variant of uncertain significance. The clinical geneticists in the MDT established the definitive diagnosis through comprehensive evaluation of the following: (i) significantly reduced α-Gal A activity and elevated Lyso-GL-3 levels and (ii) identification of the same pathogenic variant in an affected heterozygous mother and a hemizygous affected brother (Table 1). The four cases experienced a prolonged diagnostic odyssey of 2.0 to 5.9 years from symptom onset to diagnosis; by contrast, the MDT process achieved a definitive diagnosis within 15 to 30 days.
Pedigree Screening Results
The pedigree screening identified five affected relatives across the families of cases 1, 2, and 4. Notably, the asymptomatic younger brother of case 4 harbored the same hemizygous pathogenic GLA variant. His significantly reduced α-Gal A activity, thereby confirming the diagnosis of FD. The relatives of cases 3 and 5 declined pedigree screening (Table 1).
Baseline Assessment Results
The baseline assessment of organ function for the five children indicated varying degrees of organ involvement, with MSSI scores ranging from 6 to 14, all classified as mild (Table 2, Supplementary Table 1). Case 1 exhibited signs of “depression” upon admission. After the psychological assessment conducted by the clinical psychologist, the patient was confirmed to have depression and anxiety.
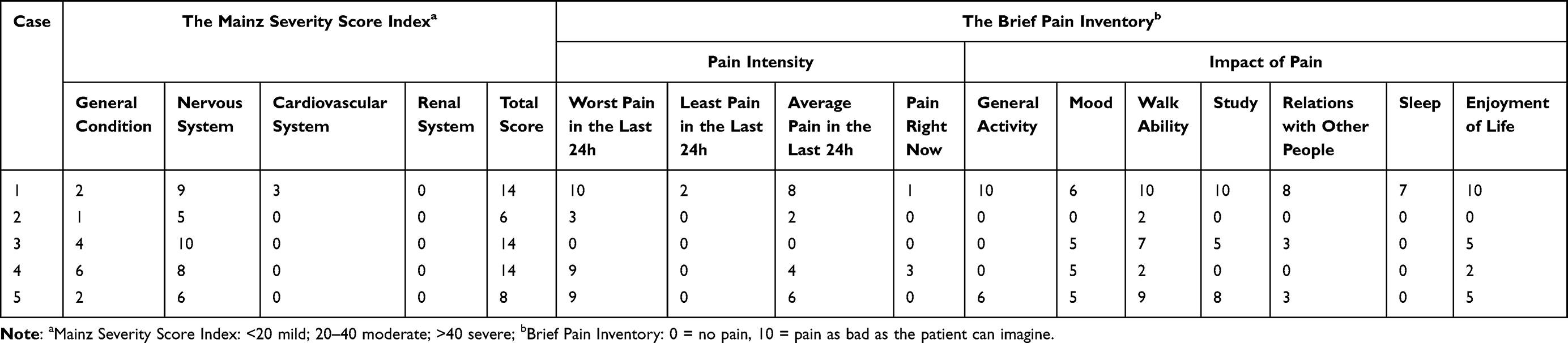 |
Table 2 The Mainz Severity Score Index and Brief Pain Inventory at Admission |
Pain was assessed using the BPI. Four cases reported pain episodes within 24 hours, with average pain intensity scores ranging from 2 to 8 points, and the most severe case reaching 10 points. Pain affected the walking ability of all the five children, with scores ranging from 2 to 10 points. Additionally, mood and enjoyment were affected in four children, study and relations with other people were impacted in three, and sleep was disturbed in one child (Table 2).
Treatment and Supportive Care
ERT
Under the guidance of the clinical pharmacist in the MDT, agalsidase α (0.2 mg/kg intravenous infusion, every two weeks) was administered for ERT. During the treatment, the nursing team closely monitored vital signs. Following one to two initial infusions, cases 1 through 4 transitioned to continue ERT at their local hospitals, while case 5 remained under our care. No infusion-related adverse reactions (eg, rash, angioedema, or hypotension) were observed in any of the five patients.
Pain Treatment
Pain treatment was individualized. During initial hospitalization, case 1 presented with persistent burning pain in bilateral toes (BPI score: 10), which significantly limited ambulation. Following the recommendation of the clinical pharmacist, oral oxcarbazepine was administered. The pain decreased substantially within approximately 30 minutes and resolved completely after one day. Case 2 had been prescribed carbamazepine prior to admission. After the first ERT and discharge, the patient experienced increased pain frequency and prolonged duration. The parents increased the dosage of carbamazepine on their own, but it was ineffective. After consultation, the medication was switched to gabapentin, which also failed to control the pain. A subsequent switch to oxcarbazepine resulted in significant pain reduction. On hospital day 4, case 3 experienced bilateral toe pain (BPI score: 4), accompanied by activity limitations. The pain was alleviated after administration of oxcarbazepine. Case 4 had no pain attacks during the hospital stay. Case 5 presented with persistent burning pain in bilateral fingers and toes accompanied by activity limitations (BPI score: 9). After taking oxcarbazepine, the pain was relieved, and the patient was able to climb stairs and perform simple physical activities.
Social and Psychological Support
Due to recurrent pain episodes, case 1 developed depression and anxiety, leading to one year of school absence. His parents also experienced significant anxiety regarding disease prognosis and treatment expenses. Social workers in the MDT communicated with his parents through interviews, phone calls, and other methods to guide them in reframing their perception of lifelong treatment from an “unfortunate fate” to a manageable condition with accessible medical solutions. Furthermore, they assisted the parents in learning healthcare policies, facilitated applications for financial aid programs, and arranged charity-funded free accommodation during hospitalization, empowering the families to shift their mindset and regain confidence.
Follow-up
Follow-up was conducted by the PICU physicians and social workers. At the time of discharge, an individualized follow-up plan was developed for each patient based on the baseline assessment results. One year after discharge, growth and sweating condition improved in cases 1, 2, and 4, with acral neuralgia alleviated in cases 1, 2, 4, and 5, enabling them to return to normal school attendance. Case 3 continued to experience relatively frequent pain attacks and was currently on a break from school, but the severity of the pain decreased. The Lyso-GL-3 levels were significantly reduced in all five children compared with their baseline measurements (Table 3).
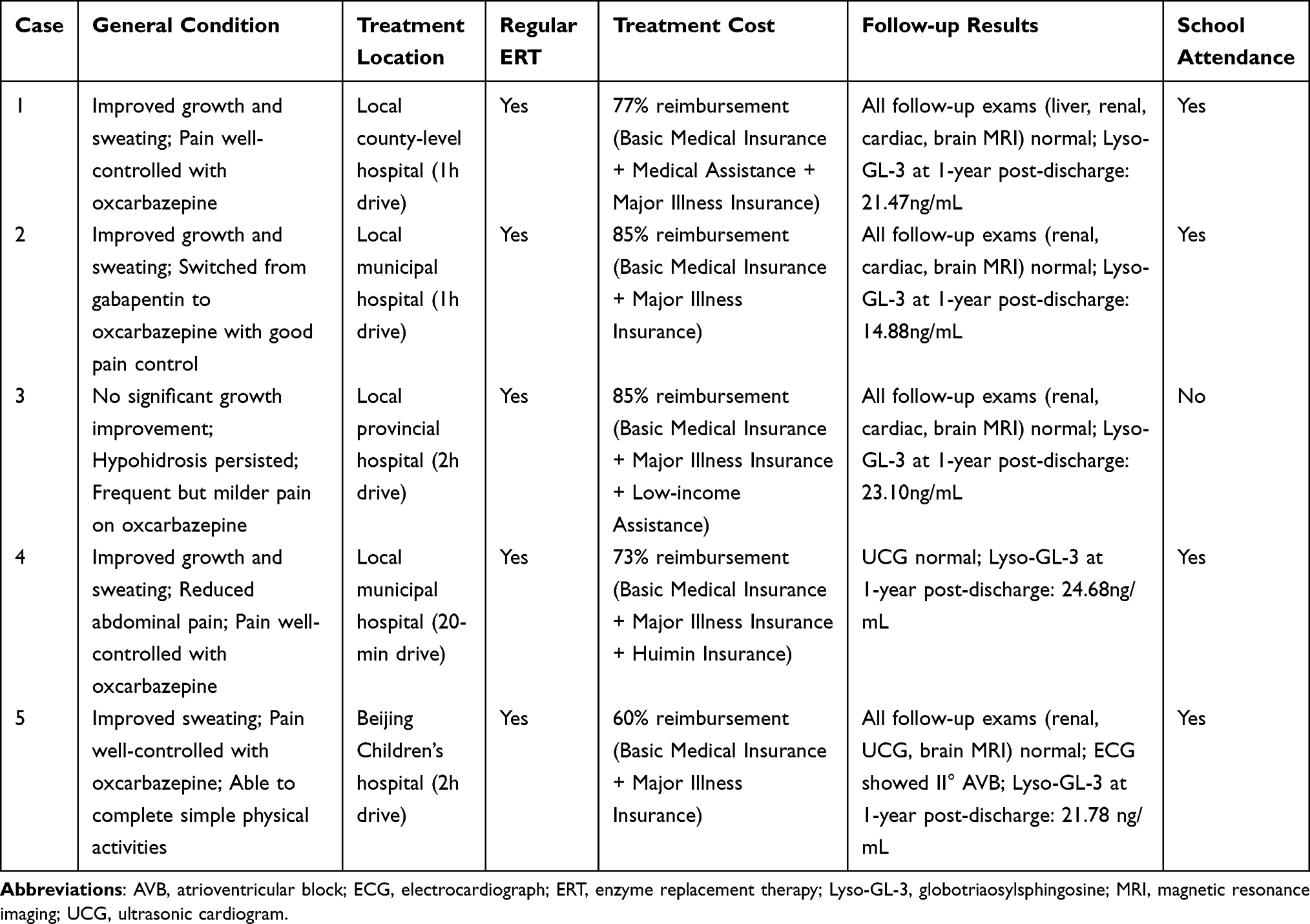 |
Table 3 Summary of Follow-up for Five Patients with Fabry Disease |
To ensure uninterrupted care, the MDT proactively coordinated post-discharge ERT for cases 1–4. For Case 1, the MDT’s sustained connection facilitated a transition from a distant municipal hospital (3.5-hour travel) to a closer county-level facility (1-hour travel) within the first year, effectively reducing the distance barrier to long-term treatment adherence. Case 5 remained under direct care at our center. Currently, all the five patients continue to receive ERT regularly at medical institutions within a travel time of 20 minutes to 2 hours; the comprehensive reimbursement rate for treatment costs ranges from 60% to 85% (Table 3).
Furthermore, social workers also provided ongoing psychological support to case 1 and his father to facilitated their return to normal life. The child’s father has begun working away from home. Notably, the mother of case 1, who was identified through pedigree screening, initiated ERT in August 2023 after experiencing a sudden cerebral infarction. The younger brother of case 4 is also scheduled to initiate ERT in the near future.
Discussion
FD often manifests symptoms during early childhood, and such symptoms progressively worsen over time. Therefore, early identification and systematic management are essential to improve long-term disease outcomes. The Fabry Registry reported that the median age of symptom onset was 6 years in males and 9 years in females, with neuropathic pain, gastrointestinal, and skin symptoms being the most frequent symptoms.8 The clinical presentations of our cohort were consistent with these findings. In recent years, the proportion of FD cases diagnosed by pediatricians, geneticists, general practitioners, and internists considerably increased. This proportion has risen from 18.9% before 2006 to 39.6% between 2007 and 2013.9 Given these findings, establishing a pediatric MDT involving multiple disciplines is a recommended approach for the diagnosis and treatment of FD. Our study contributes a detailed operational framework and preliminary outcomes of the MDT approach in a Chinese pediatric setting.
Impact of MDT on Diagnosis
FD is highly heterogeneous and exhibits significant variability in age of onset, affected organs, clinical manifestations, and disease severity. The limited awareness and diagnostic capabilities of clinicians regarding FD often result in delayed detection or misdiagnoses. The lack of widespread availability of specific diagnostic methods for FD, such as α-Gal A activity assays, plasma GL-3 and Lyso-GL-3 level testing, and genetic testing, further contributes to diagnostic delays. An analysis of Fabry Outcome Survey (FOS) revealed that the time from symptom onset to final diagnosis was 13.7±12.9 years for males and 16.3±14.7 years for females, with 25% of the patients having been misdiagnosed.10 In China, Yu et al11 reported that the time from symptom onset to diagnosis for children with classic FD was 13.50±10.08 years. In the present study, the five children took 2.0 to 5.9 years from symptom onset to final diagnosis, with cases 1 and 5 having been misdiagnosed with “juvenile idiopathic arthritis” and “joint pain,” respectively. To address these challenges, we established a pediatric FD MDT. In our practice, the MDT provided systematic training for its members on the diagnosis and treatment of the disease. Furthermore, the MDT integrated relevant medical resources, promoted the implementation of specific diagnostic tests for FD, optimized the diagnostic and treatment process, and bridged the key links in diagnosis and treatment. For the four newly diagnosed children, the “MDT-initiated” pathway achieved a definitive diagnosis within 15 to 30 days. The MDT approach facilitates a significantly accelerated diagnostic timeline to mitigate diagnostic delays.
Beyond accelerating initial diagnosis, the MDT also plays a pivotal role in identifying at-risk family members. Pedigree screening led by the MDT helps identify potential patients and facilitates early intervention.12 Laney et al13 reported an average of five family members diagnosed with FD for every proband. In the present study, the younger brother of case 4 was diagnosed through pedigree screening during the asymptomatic stage, achieving “pre-symptomatic diagnosis” and providing a critical window for early intervention. Expert consensus, both internationally and in China, indicate that asymptomatic FD boys with pathogenic variant may benefit from preventive ERT.1,5,14 This child is scheduled to initiate ERT, and such a “pre-symptomatic management” approach may positively influence his prognosis. However, pedigree screening still faces barriers such as financial burden and disease stigmatization, as evidenced in our results, where screening was not completed due to family refusal in case 3 and 5. In the future, the MDT approach should expand its scope to actively incorporate public education and social support to improve the acceptance and effectiveness of pedigree screening.
Impact of MDT on Disease Severity Evaluation
Upon diagnosis, the MDT utilized the MSSI to systematically assess disease severity and guide the following individualized management. The MSSI, a clinical scoring system specific for FD, has been extensively validated in adult populations.15–17 Growing evidence also supports its applicability in pediatric populations. A multicenter study demonstrated that MSSI scores in children with FD tend to increase with age.18 Consistent with our findings, Guo et al19 reported five pediatric cases with MSSI scores all falling within the mild range (0–16 points), which primarily involved general symptoms and neurological manifestations. While MSSI effectively identifies non-specific symptoms in children and facilitates longitudinal monitoring, its clinical discrimination remains limited due to inadequate weighting of core symptoms such as pain and fatigue, coupled with the rarity of high-scoring end-stage organ damage in pediatric populations. Future development of pediatric-specific FD assessment tools is needed to more accurately capture early symptoms and quality of life changes.
Impact of MDT on ERT
ERT is the specific treatment for FD. Adults with FD have a higher incidence of major clinical events, including renal, cardiovascular, and cerebrovascular events, compared with pediatric patients.20 A recent FOS analysis found that initiating ERT in childhood or early adulthood attenuated the progression of renal and/or cardiac disease compared to initiation after 30 years of age.21 Consistent with these findings, international and Chinese consensus guidelines recommend immediate initiation of ERT in pediatric patients upon the emergence of FD-specific symptoms because early treatment is associated with great potential for benefit.1,5,14 In China, two ERT drugs, namely, agalsidase α and agalsidase β, have been approved and marketed. Both drugs are comparable in terms of improving symptoms in children with FD, that is, they reduce the incidence of cardiovascular, cerebrovascular, and renal complications and extend survival time.22,23 Early initiation of ERT offers benefits for children and has no significant serious adverse events.21,24–26 In this context, the core role of the MDT is to formulate individualized and sustainable treatment plans. To this end, physicians and clinical pharmacists evaluated key factors including drug accessibility, compliance, and cost, ultimately selecting agalsidase α. This drug is readily available on the Beijing municipal drug procurement platform and has been included in the Chinese basic medical insurance catalog to ease the economic burden for patients. Moreover, the MDT facilitated connections to local hospitals for these patients to ensure, continuity of care. Our findings demonstrated the viability of this approach. During the infusion of agalsidase α, none of the five children experienced any adverse events. At one-year follow-up, all patients showed clinical improvement to varying degrees, accompanied by a significant reduction in Lyso-GL-3 levels from baseline. Furthermore, the doctors involved introduced parents to other therapeutic options, such as oral chaperone therapy for amenable mutations and potentially curative gene therapies, even though these are not yet clinically available in China, to equip families with comprehensive information and foster a sense of hope. Consequently, the value of the MDT in ERT lies in its comprehensive approach, ensuring not only guideline-concordant treatment initiation but also the development of a practical, sustainable, and long-term care strategy for pediatric rare diseases.
Impact of MDT on Pain Treatment
Neuropathic pain is one of the earliest and most common symptoms in patients with FD. Li et al27 reported that neuropathic pain in boys typically occurs between the ages of 3 and 9, mostly presenting as episodic burning or stabbing pain in the extremities (fingertips, palms, toes, and soles), with variable durations. These episodes can be triggered by physical activity and changes in temperature. Some patients may experience systemic, disabling episodes of severe pain, known as “pain crises.” Miao et al28 found that the incidence of neuropathic pain was as high as 91.4%. Hopkin et al8 reported that in children with FD, neuropathic pain accounted for 50.6% of all clinical manifestations, making it the most common symptom. In this study, all five children experienced acral neuralgia, and cases 1 and 3 had to take a break from school due to severe pain. Effective and aggressive pain management for patients with FD can significantly improve their quality of life.
Regarding the selection of analgesics, the MDT engaged in a thorough discussion. The precise mechanism underlying neuropathic pain in FD remains unclear. Recent experimental studies suggested that some of the spontaneous pain type with FD may be explained by the upregulation of sodium-ion channels (Nav1.8) and transient receptor potential cation channel subfamily V member 1(TRPV1) or an increase in lyso-GL-3-dependent calcium (Ca2+) influx.29–31 These findings imply that ion channel blockers could potentially alleviate pain. Carbamazepine is recommended as a first-line treatment for neuropathic pain in relevant Chinese clinical practice guidelines.1,32 However, carbamazepine is associated with numerous adverse reactions, particularly in Asians carrying the human leukocyte antigen allele (HLA-B*1502), who are at risk of severe and even fatal skin reactions. Therefore, pharmacogenetic testing is necessary before initiating carbamazepine therapy. Given these considerations, clinical pharmacists recommend switching to oxcarbazepine, which has comparable efficacy but a better safety profile than carbamazepine. Structurally similar to carbamazepine, oxcarbazepine offers equivalent pain relief with a significantly lower incidence of rash. Following this treatment, neuropathic pain in cases 1, 2, 4, and 5 was effectively controlled, so they were able to attend school and participate in simple physical activities. Case 3 experienced frequent pain attacks, but the severity of the episodes was reduced. However, current guidelines lack specific recommendations on when and how to discontinue medication for children with well-controlled pain. Further studies involving a larger cohort of children with FD are needed to provide robust evidence for optimizing pain management strategies.
Psychological Support and Social Assistance Provided by MDT
Depression and diminished quality of life are significant complications in patients with FD, with an incidence rate ranging from 15% to 62%.33–35 In our study, case 1 exhibited signs of depression and anxiety during the initial psychological assessment at admission. His father was similarly anxious, primarily due to concerns about the disease prognosis, financial burden, and societal judgment. After admission, social workers proactively provided psychological support to the child and his father and assisted them in navigating the challenges associated with disease treatment. Focusing on establishing a correct understanding of the disease, social workers communicated with the child and his father through face-to-face talks and phone calls to assist the family regain their normal lives. Social workers address a critical gap in traditional care for rare diseases, which often focuses predominantly on physiological management while overlooking psychological well-being. They can integrate additional humanistic concepts into the comprehensive management of rare diseases. Additionally, social workers leveraged their strengths to help the patients and their family access social resources to alleviate their burden. This finding was particularly evident in interpreting medical insurance policies and applying for relevant funds; as reported in the results, the MDT approach led to a high reimbursement rate (60% to 85%) and helped the family build confidence to continue treatment.
Limitations
This case-series has the following limitations. First, the single-center design and relatively small sample size may restrict the broad applicability of the findings. Second, the relatively short follow-up period limited a full assessment of the long-term prognosis of FD and the sustained effectiveness of the MDT approach. Therefore, future studies should employ a prospective controlled design to systematically compare key outcomes between MDT-managed and conventionally managed groups, to quantify its true value.
Conclusions
This case series demonstrates that implementing a structured MDT approach was associated with improved management outcomes in pediatric FD, including accelerated diagnosis, early intervention via pedigree screening, and comprehensive care delivery. This integrated approach alleviated symptoms, improved treatment access, provided psychosocial support, and yielded positive short-term outcomes, thereby serving as a practical framework for rare disease management.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article and its Supplementary Information File.
Ethical Approval
This prospective, observational study involving human participants was in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The Ethics Board of Beijing Children’s Hospital, Capital Medical University ([2022]-E-060-Y) approved this study. Written informed consent was obtained from a parent or legal guardian of all pediatric participants prior to their enrollment in the study.
Consent for Publication
All authors approved the final version of the manuscript.
All authors consent to publication.
Acknowledgments
The authors thank all the patients and their parents who participated in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
All authors declare no conflicts of interest in this work.
References
1. Chinese Fabry Disease Expert Panel. Expert consensus for diagnosis and treatment of Fabry disease in China (2021). Zhonghua Nei Ke Za Zhi. 2021;60(4):321–13. doi:10.3760/cma.j.cn112138-20201218-01028
2. Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124(3):189–203. doi:10.1016/j.ymgme.2018.06.004
3. Nowicki M, Bazan-Socha S, Blazejewska-Hyzorek B, et al. Enzyme replacement therapy in Fabry disease in Poland: a position statement. Pol Arch Intern Med. 2020;130(1):91–97. doi:10.20452/pamw.15117
4. Schiffmann R. Fabry disease. Handb Clin Neurol. 2015;132:231–248. doi:10.1016/B978-0-444-62702-5.00017-2
5. Germain DP, Fouilhoux A, Decramer S, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet. 2019;96(2):107–117. doi:10.1111/cge.13546
6. Chinese National Expert Collaborative Group of Fabry Disease; China Medical Education Association Clinical Nephrology Committee. Multidisciplinary treatment in the long-term management of Fabry disease. Zhonghua Nei Ke Za Zhi. 2023;62(8):949–955. doi:10.3760/cma.j.cn112138-20230218-00095
7. Shen Q, Liu J, Chen J, et al. Multidisciplinary approach to screening and management of children with Fabry disease: practice at a Tertiary Children’s Hospital in China. Orphanet J Rare Dis. 2021;16(1):509. doi:10.1186/s13023-021-02136-1
8. Hopkin RJ, Bissler J, Banikazemi M, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008;64(5):550–555. doi:10.1203/PDR.0b013e318183f132
9. Reisin R, Perrin A, Garcia-Pavia P. Time delays in the diagnosis and treatment of Fabry disease. Int J Clin Pract. 2017;71(1):e12914. doi:10.1111/ijcp.12914
10. Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34(3):236–242. doi:10.1111/j.1365-2362.2004.01309.x
11. Yu C, Zhou Y, Li Z, et al. Clinical features and treatment effect of enzyme replacement therapy in patients with Fabry disease. Chin J Nephrol. 2022;38(6):497–503. doi:10.3760/cma.j.cn441217-20211104-00069
12. Germain DP, Moiseev S, Suarez-Obando F, et al. The benefits and challenges of family genetic testing in rare genetic diseases - lessons from Fabry disease. Mol Gene Genom Med. 2021;9(5). doi:10.1002/mgg3.1666
13. Laney DA, Fernhoff PM. Diagnosis of Fabry disease via analysis of family history. J Gene Counsel. 2008;17(1):79–83. doi:10.1007/s10897-007-9128-x
14. Vaisbich MH, Andrade LGM, Silva CAB, et al. Recommendations for the diagnosis and management of Fabry disease in pediatric patients: a document from the rare diseases committee of the Brazilian Society of Nephrology (Comdora-SBN). Journal brasileiro de nefrologia. 2022;44(2):268–280. doi:10.1590/2175-8239-jbn-2021-0216
15. Whybra C, Kampmann C, Krummenauer F, et al. The Mainz severity score index: a new instrument for quantifying the Anderson-Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin Genet. 2004;65(4):299–307. doi:10.1111/j.1399-0004.2004.00219.x
16. Altarescu G, Chicco G, Whybra C, et al. Correlation between interleukin-6 promoter and C-reactive protein (CRP) polymorphisms and CRP levels with the Mainz Severity Score Index for Fabry disease. J Inherit Metab Dis. 2008;31(1):117–123. doi:10.1007/s10545-007-0716-6
17. McCarron EP, Chinnadurai R, Meyer J, et al. Real-world clinical outcomes in adult patients with Fabry disease: a 20-year retrospective observational cohort study from a single centre. Mol Gene Metabol Rep. 2025:43. doi:10.1016/j.ymgmr.2025.101229
18. Ries M, Ramaswami U, Parini R, et al. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr. 2003;162(11):767–772. doi:10.1007/s00431-003-1299-3
19. Gou P, Leng J, Cheng XR, et al. Clinical evaluation, accurate diagnosis and treatment of four pedigrees with Fabry’s disease. Front Pediatrics. 2023:11. doi:10.3389/fped.2023.1057014
20. Eng CM, Fletcher J, Wilcox WR, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30(2):184–192. doi:10.1007/s10545-007-0521-2
21. Parini R, Pintos-Morell G, Hennermann JB, et al. Analysis of renal and cardiac outcomes in male participants in the fabry outcome survey starting agalsidase alfa enzyme replacement therapy before and after 18 years of age. Drug Des Devel Ther. 2020;14:2149–2158. doi:10.2147/DDDT.S249433
22. Liu Y, Li Y, Li P, et al. Effectiveness and safety of enzyme replacement therapy in the treatment of Fabry disease: a Chinese monocentric real-world study. Orphanet J Rare Dis. 2024;19(1):422. doi:10.1186/s13023-024-03441-1
23. Spada M, Baron R, Elliott PM, et al. The effect of enzyme replacement therapy on clinical outcomes in paediatric patients with Fabry disease - A systematic literature review by a European panel of experts. Mol Genet Metab. 2019;126(3):212–223. doi:10.1016/j.ymgmr.2019.100454
24. van der Veen SJ, Körver S, Hirsch A, et al. Early start of enzyme replacement therapy in pediatric male patients with classical Fabry disease is associated with attenuated disease progression. Mol Genet Metab. 2022;135(2):163–169. doi:10.1016/j.ymgme.2021.12.004
25. Khan A, Sirrs SM, Bichet DG, et al. The safety of agalsidase alfa enzyme replacement therapy in Canadian patients with Fabry disease following implementation of a bioreactor process. Drugs R D. 2021;21(4):385–397. doi:10.1007/s40268-021-00361-4
26. Germain DP, Charrow J, Desnick RJ, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. 2015;52(5):353–358. doi:10.1136/jmedgenet-2014-102797
27. Li Q, Wang J, Tian M, et al. Clinical features and enzyme replacement therapy in 10 children with Fabry disease. Front Pediatr. 2023;11:1084336. doi:10.3389/fped.2023.1084336
28. Miao Y, Zhao Y, Liu J, et al. Characteristics of neuralgia in patients with Fabry disease in China. Chin J Neurol. 2022;55(1):15–20. doi:10.3760/cma.j.cn113694-20210430-00311
29. Bennett DL, Clark AJ, Huang J, et al. The role of voltage-gated sodium channels in pain signaling. Physiol Rev. 2019;99(2):1079–1151. doi:10.1152/physrev.00052.2017
30. Trimmer JS. Ion channels and pain: important steps towards validating a new therapeutic target for neuropathic pain. Exp Neurol. 2014;254:190–194. doi:10.1016/j.expneurol.2014.01.019
31. Toyoda H. Ion channels involved in spontaneous pain. Sci Reposit. 2018;1:2–7. doi:10.31487/j.NNB.2018.02.001
32. Zhu Q, Fan B, Zhang D, et al. Consensus of Chinese experts on the diagnosis and treatment of peripheral neuropathic pain. Chin J Pain Med. 2020;26(5):321–328. doi:10.3969/j.issn.1006-9852.2020.05.001
33. Bolsover FE, Murphy E, Cipolotti L, et al. Cognitive dysfunction and depression in Fabry disease: a systematic review. J Inherit Metab Dis. 2014;37(2):177–187. doi:10.1007/s10545-013-9643-x
34. Arends M, Korver S, Hughes DA, et al. Phenotype, disease severity and pain are major determinants of quality of life in Fabry disease: results from a large multicenter cohort study. J Inherit Metab Dis. 2018;41(1):141–149. doi:10.1007/s10545-017-0095-6
35. Ali N, Gillespie S, Laney D. Treatment of depression in adults with Fabry disease. JIMD Rep. 2018;38:13–21. doi:10.1007/8904_2017_21
36. Li H, Ji CY, Zong XN, et al. Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years. Zhonghua Er Ke Za Zhi. 2009;47(7):487–492. doi:10.3760/cma.j.issn.0578-1310.2009.07.003
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Implementation and Outcomes of a Standardized Multidisciplinary Treatment Outpatient Model in Two Pediatric Tertiary Hospitals
Wang M, Ye C, Gao X, Qian Y, Feng T, Shi Y
Journal of Multidisciplinary Healthcare 2026, 19:599215
Published Date: 9 April 2026