")
Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 6
Clinical potential of ataluren in the treatment of Duchenne muscular dystrophy
Authors Namgoong J, Bertoni C
Received 7 February 2016
Accepted for publication 14 April 2016
Published 13 May 2016 Volume 2016:6 Pages 37—48
DOI https://doi.org/10.2147/DNND.S71808
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Thomas Müller
John Hyun Namgoong, Carmen Bertoni
Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, CA, USA
Abstract: Duchenne muscular dystrophy (DMD) is an autosomal dominant, X-linked neuromuscular disorder caused by mutations in dystrophin, one of the largest genes known to date. Dystrophin gene mutations are generally transmitted from the mother to male offspring and can occur throughout the coding length of the gene. The majority of the methodologies aimed at treating the disorder have focused on restoring a shorter, although partially functional, dystrophin protein. The approach has the potential of converting a severe DMD phenotype into a milder form of the disease known as Becker muscular dystrophy. Others have focused on ameliorating the disease by targeting secondary pathologies such as inflammation or loss of regeneration. Of great potential is the development of strategies that are capable of restoring full-length dystrophin expression due to their ability to produce a normal, fully functional protein. Among these strategies, the use of read-through compounds (RTCs) that could be administered orally represents an ideal option. Gentamicin has been previously tested in clinical trials for DMD with limited or no success, and its use in the clinic has been dismissed due to issues of toxicity and lack of clear benefits to patients. More recently, new RTCs have been identified and tested in animal models for DMD. This review will focus on one of those RTCs known as ataluren that has now completed Phase III clinical studies for DMD and at providing an overview of the different stages that have led to its clinical development for the disease. The impact that this new drug may have on DMD and its future perspectives will also be described, with an emphasis on the importance of further assessing the clinical benefits of this molecule in patients as it becomes available on the market in different countries.
Keywords: nonsense mutations, premature termination codons, PTC, nonsense-mediated decay, tRNA suppressors, read-through, aminoglycosides, RTC13, PTC124, Translarna, ataluren
Introduction
Neuromuscular diseases are a group of heterogeneous disorders that affect muscle, neuromuscular junctions, and motor neurons. To date, over 40 genes have been linked to several forms of these disorders, and genetic defects can be either acquired or inherited. The majority of these diseases manifest early in life and lead to progressive deterioration of functions. Mutations in the dystrophin gene are associated with two distinct pathologies: Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD).1,2
The dystrophin gene is one of the largest genes identified to date, spanning approximately 2.5 Mb on chromosome X. The incidence of DMD is approximately 1 in every 3,500 males, while in BMD, the incidence is much lower and has been estimated to be 1 in every 10,000 patients. Mutations in DMD can occur throughout the length of the gene and are prevalently characterized by large deletions and single point mutations. Duplications, insertions, and small deletions have also been reported, although at a lower frequency.
Among the diseases caused by mutations in the dystrophin gene, DMD is the most severe and usually manifests within the first 3–5 years of the patient’s life. It is caused by mutations of the dystrophin gene that disrupt the reading frame of the mRNA coding sequence, which leads to complete absence of dystrophin expression in skeletal and cardiac muscles. The disease is characterized by progressive muscle weakness that affects the trunk first and then the lower and upper extremities, ultimately leading to wheelchair dependency by the age of 14. In BMD patients, dystrophin gene mutations are generally associated with large deletions of the gene, which result in the expression of a shorter, but still in-frame transcript that leads to the production of a partially functional dystrophin protein. A few cases have also been reported that are characterized by nonsense mutations that result in significant levels of altered exon splicing of the dystrophin mRNA and that lead to reduced levels of dystrophin protein or to the expression of shorter forms of the protein.3–6 Although varying from patient to patient, the prognosis in BMD is less severe than that of DMD, and affected individuals generally live well into their middle age or later. The most common causes of death in DMD and BMD patients are respiratory and heart failure.
Therapeutic potentials of approaches to DMD
Treatment options for DMD have been widely explored over the past 30 years. Corticosteroids are considered standard care for DMD patients and have demonstrated evident benefits to patients, but are associated with strong side effects.7–10 Several approaches aimed at restoring dystrophin expression have already reached the clinic with promising results. They range from gene replacement through the use of viral11–15 and nonviral approaches16–18 to manipulation of splicing through the use of antisense oligonucleotides that act by redirecting splicing of the dystrophin mRNA and that allow the expression of shorter BMD-like dystrophin isoforms.19–30 Other strategies are focused on compensating for the loss of dystrophin by upregulating utrophin, a homologue of dystrophin,31–36 with promising results in humans.37,38 Additional strategies for DMD aim at ameliorating the pathology by increasing muscle strength,39–41 reducing muscle fibrosis,42–44 and decreasing inflammation.45–50 Although promising, these strategies can only improve the quality of life of patients and delay disease progression.
To date, the assessment of the benefits achieved by a therapy over the risks associated with the use of a new drug has been based, primarily, on the opinions of experts in the field and on the reviews of the clinical data obtained pre- and postapproval of new drugs. Similarly, the benefit–risk assessment in DMD is considered to be a complex issue, and the information regarding potential treatments for the disease available to the US Food and Drug Administration (FDA) and other regulatory agencies worldwide is very limited. The identification of new approaches that are now reaching the clinic has evidenced the need to address this very important problem and to revise laws and regulations that control the entry into market of new drugs for DMD. The efforts undertaken thus far by caregivers, advocates, academic researchers, and pharmaceutical drug developers have been pivotal in addressing some of the key issues that surround the evaluation of new therapeutics, especially in the context of clinical expectations to be achieved for DMD. Currently, it is clear that a cure for DMD, defined as a treatment that would completely restore health in patients, is still out of reach. This is primarily due to the complexity of the disease, the vast area of muscle tissue that needs to be treated, and the lack of approaches that can efficiently target each individual fiber within the large number of muscles that compose the human body. However, the whole scientific community agrees that approaches that could significantly delay the progression of the disease would represent a tremendous achievement. This opinion is also shared by patients and caregivers as stated in recently published surveys.51–53 Importantly, these surveys have clearly demonstrated the willingness of patients and caregivers to accept significant risks and side effects in return for slowing or stabilizing the progression of the disease even in the absence of survival benefits, an increased risk of death, or additional serious disability. The FDA is currently considering including DMD into the Patient-Focused Drug Development Initiative,54 which would ultimately allow families, researchers, and sponsors to ease some of the regulatory requirements needed for approval.
Pharmacological approaches targeting nonsense mutations
In recent years, great emphasis has been placed on the discovery of approaches, especially pharmacological treatments that could be used to restore normal, full-length dystrophin due to their potential to reverse the course and clinical outcome of the disease. Read-through (RT) of nonsense mutations represents an ideal approach to DMD due to its ability to bypass the premature stop codon and to act on virtually any region of the dystrophin gene independently of the location in which the mutation resides (Figure 1). Furthermore, according to the Human Gene Mutation Database,55 nonsense mutations leading to premature termination codons (PTCs) are responsible for up to 30% of inherited genetic disorders and account for approximately 12% of all mutations listed in the database.56 This highlights the broad spectrum of application that any approach aimed at suppressing these types of mutations may have in medicine. Among those, cystic fibrosis (CF), DMD, ataxia telangiectasia (A-T), hemophilia, retinal degeneration, and several forms of lysosomal storage diseases are, perhaps, the most known and those for which data from animal or human cells models are available. Other forms of genetic disorders are also amenable to suppression of nonsense mutations as described in detail in a recent review.57 The majority are rare or ultra-rare disorders and, although difficult to determine accurately, recent analyses suggest that about 30 million people in the United States (US) and circa 300 million people worldwide would benefit from drugs that suppress nonsense mutations.58 It has been estimated that the market size for an RT drug would reach $1 billion by the year 2019, and so it is highly attractive to companies.59
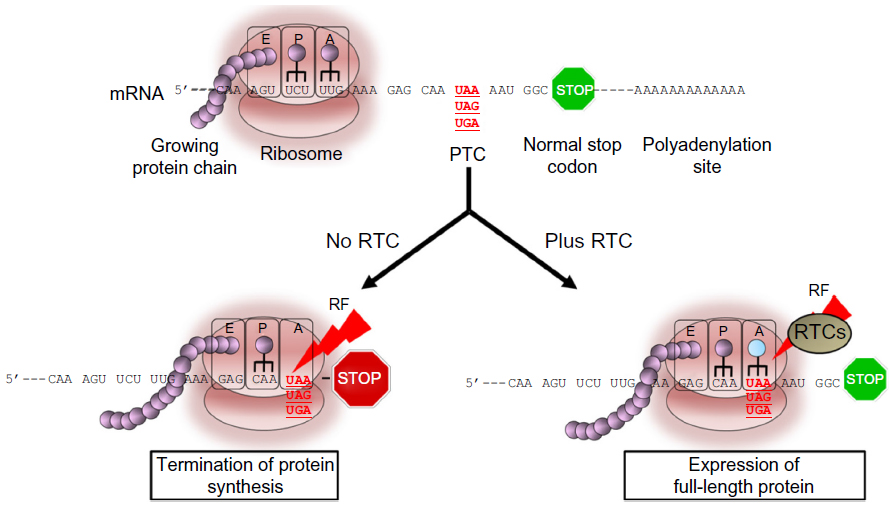 | Figure 1 Read-through activity in eukaryotic cells mediated by read-through compounds (RTCs). |
Aminoglycosides and nonaminoglycoside antibiotics for the treatment of DMD
The ability of certain antibiotics to suppress PTCs in eukaryotic cells has been known since the early 1990s.60–65 Several pivotal studies have demonstrated that antibiotics like gentamicin and G418 can interfere with the region of the ribosome that monitors codon–anticodon interactions with near-cognate aminoacyl-transfer RNA (tRNA) or suppressor tRNAs (Figure 1). This interaction leads to the substitution of the PTC with a different amino acid. In prokaryotic cells, like bacteria, the affinity of the aminoglycoside with the 16S rRNA subunit is very strong, inducing a conformational change that is similar to that occurring upon cognate tRNA binding. The interference leads to extensive misreading at both sense and nonsense codons, which ultimately results in aberrant expression profiles and cell death. In eukaryotic cells, the difference in structure in the 18S rRNA subunit, the homologous of the bacteria 16S rRNA, renders the affinity of aminoglycoside much weaker, resulting in a significantly lower frequency of misincorporation at PTC sites. Importantly, the substitution of the stop codon with an amino acid in eukaryotic cells appears to be restricted to PTC as read-through of normal stop codons has never been seen in these cells with any of the antibiotics tested thus far.66–69 This specificity and selectivity is likely to be due to the presence of a complex transcription machinery composed of different proteins and regulatory factors that control normal termination of protein synthesis and that ensures that the protein is terminated at its normal stop codon.
The type of stop codon being targeted influences the efficiency and fidelity of the read-through activity of aminoglycosides. The UAA (ochre) codon is the most difficult to suppress, followed by the UAG (amber), and finally, the UGA (opal) termination codons.70 Similarly, suppression of the PTC appears to be influenced by the specific nucleic acid immediately following the stop codon71 as well as the sequence surrounding it.72
Studies conducted to demonstrate the efficacy of aminoglycoside compounds to restore dystrophin expression have utilized the mdx mouse model for DMD. In this naturally occurring strain, a G-to-T transversion in exon 23 of the dystrophin gene converts a GAA codon into a UAA, thus resulting in premature termination of protein synthesis and absence of dystrophin expression in skeletal muscle.73 The muscle pathology in mdx is similar to that of DMD patients and presents with increased susceptibility to muscle damage, large areas of degeneration, and inflammation, but a life span that is close to that of normal mice. These characteristics render this mouse particularly suitable in studies aimed at determining the efficacy of approaches capable of restoring dystrophin expression, especially those targeting nonsense mutations.
Barton-Davies et al63 were the first ones to demonstrate the ability of gentamicin to target and restore full-length dystrophin expression in mdx mice both in vitro and in vivo. Mice dosed systemically for 14 days showed improved muscle strength and increased membrane stability, suggesting that the administration of the compound for short periods of time was sufficient to ameliorate muscle pathology. Results have been confirmed independently in mdx mice dosed for up to 12 weeks, further prompting the testing of this compound in DMD patients.74
Clinical trials in DMD using gentamicin have shown controversial results, with little or no dystrophin expression detected following administration of the drug to patients.75–77 Ultimately, its use in the clinic was dismissed due to the long-term side effects, particularly the nephrotoxicity and ototoxicity, associated with its use, especially over prolonged periods of time. Nonetheless, these studies have served as proof of concept for the possibility of using small molecules to suppress PTCs in patients with DMD. More recently, new derivatives of aminoglycosides with reduced toxicity have been synthesized and evaluated in an in vitro luciferase-based assay for their ability to read-through nonsense mutations in the DMD gene.78 However, their ability to restore dystrophin expression in vivo in animal models still remains unknown.
Several other antibiotics with RT ability have been identified in the recent years and evaluated for their ability to suppress nonsense mutations in multiple in vitro and in vivo PTC models as, reviewed by others.57 Negamycin is the only nonaminoglycoside antibiotic to have been evaluated as a possible candidate for DMD.79 A 4-week study in mdx receiving the compound systemically showed clear expression of dystrophin in tibialis anterior as well as heart muscles (the only two muscles analyzed) isolated from treated mice, suggesting that negamycin may have important therapeutic value for the treatment of this disorder. Structure–activity relationship studies of this compound have recently been conducted to further improve its efficacy.80 However, a direct comparison between the activity of these new compounds to that of other antibiotics or known RTCs have not been performed yet, and therefore, their potential as valid therapeutic candidates for DMD or other genetic disorders caused by nonsense mutations has yet to be determined.
Among the compounds that have shown great promise for the treatment of DMD is RTC13, a novel nonaminoglycoside RTC identified using a protein transcription/translation–enzyme-linked immunosorbent assay, using A-T as a genetic disease model. RTC13 was shown to efficiently restore full-length dystrophin in mdx in vitro as well as in vivo.81,82 Furthermore, new compounds were identified using SRA studies, several of which are currently under investigation for their ability to suppress PTC in animal models for the disease.83 These compounds are likely to further enrich the pool of RTCs that can be moved forward into clinical testing for the treatment of DMD and other diseases alike.
Discovery and evaluation of ataluren (PTC124)
Two luciferase-based high-throughput screens of nearly 800,000 low-molecular-weight compounds were performed by PTC Therapeutics and used to identify nonaminoglycoside compounds capable of promoting ribosomal read-through of PTCs.84 Comprehensive medicinal chemistry studies have led to the identification of PTC124, also known as ataluren, which was shown to efficiently restore dystrophin expression in human cells isolated from a DMD patient and the mdx mouse.84 Importantly, muscle strength was significantly restored following systemic administration of ataluren in mdx mice for up to 4 weeks of continuous dosing, further emphasizing the therapeutic relevance of the compound for the treatment of DMD.
However, shortly after the publication of these findings, the validity of the screen utilized for the identification of PTC124 was challenged by reports demonstrating the ability of the compound to bind the luciferase protein. This binding was shown to increase the stability of the protein, resulting in a significant increase in luciferase activity that was independent of its ability to read-through PTCs.85,86 These results have fueled debates on whether the compound had any activity at all, which was in part supported by an independent study conducted using different reporter systems, all of which showed no detectable levels of read-through activity mediated by PTC124.87 Moreover, the inability of PTC124 to suppress nonsense mutations in several mouse and human models of relevant diseases has further contributed to the critiques and skepticisms surrounding the efficacy of this molecule for the treatment of nonsense mutations.88–90 These findings are in contrast to those reported by many other groups that have shown the activity of the compound in several models, including reports demonstrating low, although detectable, rescue of protein expression in vitro and in vivo.78,81,82,89,91–96
The differences in the ability of ataluren to suppress premature stop codons known to be associated with specific genetic disorders may be dependent on several factors. The sensitivity of the assays being utilized to assess the efficacy of the compound may have been, at least in part, responsible for the apparent lack of activity detected in some models. Furthermore, factors like mRNA stability and its susceptibility to degradation by the nonsense-mediated decay pathway are likely to have played a crucial role in the differences in activity observed. Indeed, it is well recognized that these factors can vary largely depending on the gene being analyzed, restricting the activity of an RTC to a limited window of opportunity in time during which the compound can interact with the rRNA. Other factors, like protein stability and normal rate of turnover of the protein being restored by the RTC, are crucial aspects that impact its abundance into a cell. In fact, proteins with a long half-life are more likely to accumulate in a cell following treatment with an RTC compared to products that have a rapid turnover and, therefore, should be easier to detect using standard biochemical assays. Thus, it is plausible to assume that effects detected following treatment may vary widely from model to model and that certain genetic defects may be more amenable to treatments with RTCs than others. Nonetheless, the results obtained thus far have clearly evidenced limitations in the efficacy of ataluren and have demonstrated the need of identifying new RTCs with improved activity. Despite those limitations, the encouraging data obtained in the mdx84 and animal models for CF91 were highly encouraging and have warranted further evaluation of the potential of ataluren for the treatment of those disorders and its advancement into clinical testing.
Clinical development of ataluren
Preclinical safety studies in rats and dogs that received PTC124 orally for 28 days showed no adverse effects on neurological, pulmonary, or cardiovascular functions, not even at high doses.91 Phase I single- and multiple-dose studies in healthy volunteers were used to determine the pharmacokinetics and safety of ataluren and to identify the ideal dosing schedule necessary to achieve optimal target plasma concentrations for subsequent studies in CF and DMD patients.97 Studies were conducted in a total of 62 subjects, age 18–30 years, who were randomized into two groups. Single-dose escalating studies were performed in 18 subjects who received PTC124 at a concentration of 3, 10, 30, 100, 150, and 200 mg/kg. Side effects included headache, dizziness, nausea, vomiting, diarrhea, and/or abdominal pain and were reported in all subjects receiving PTC124 at the doses of 150 and 200 mg/kg. Side effects progressively decreased in a concentration-dependent manner in healthy volunteers who received the lower doses. Serum chemistry results showed an increase in alanine aminotransferase (ALT) and aspartate aminotransferase (AST), suggesting possible liver toxicity of the drug, at least in the higher doses utilized.
The multiple-dose study conducted in Phase I trial was performed in 30 healthy volunteers. Subjects were randomized into four groups, each receiving the compound every 12 hours at doses of 10, 20, 30, and 50 mg/kg, respectively, and for up to 7 days. Increases in serum transaminase values, primarily ALT and AST, were noted in approximately 30%–50% of the subjects who received the compound, independently of the doses being administered, confirming the presence of possible liver toxicity of the drug. Importantly, analyses on peripheral blood mononuclear cells and plasma failed to show signs of read-through of normal stop codons, validating the results previously obtained using in vitro systems and those achieved in animal models.84
The safety data obtained in healthy volunteers prompted the initiation of Phase IIa clinical trials in CF and DMD patients. The initial results of the two clinical trials conducted by independent groups in CF patients were slightly discordant. In fact, one study reported statistically significant improvement in epithelial electrophysiological abnormalities caused by the absence of expression of the CF transmembrane conductance regulator in some of the patients treated,98 while the other showed no significant effects.57 In DMD patients, the Phase IIa clinical trial was conducted over a 28-day period of active administration of the compound. Subjects were randomized into three groups that received ataluren three times a day, following breakfast, lunch, and dinner at a dose of respectively 4, 4, 8 mg/kg/d (group 1: low dose), 10, 10, 20 mg/kg/d (group 2: medium dose), and 20, 20, 40 mg/kg/d (group 3: high dose). No side effects were noted at any of the doses administered, although the plasma levels of ataluren were lower than those detected in adults in Phase I studies. Expression of dystrophin was observed in some of the muscle biopsies isolated from boys treated with the low and medium doses, but not in any of the samples isolated from patients who received the high dose. Following treatment, serum creatinine kinase (CK), a marker of muscle integrity, AST, and ALT levels, which are known to be elevated in DMD patients as a result of the disease, decreased significantly, but changes in muscle strength and timed functions were small and not statistically significant.99
A Phase IIb trial was conducted in 2008 to evaluate the safety and efficacy of ataluren in DMD/BMD boys with nonsense mutations.100 A total of 174 patients, aged 9–20 years, were randomized into three groups of approximately 60 patients per group that received either placebo, ataluren at a dose of 10, 10, 20 mg/kg, or ataluren at doses of 20, 20, 40 mg/kg orally three times daily after meals and for up to 48 weeks. The primary end point was change in 6-minute walk test (6MWT), a widely used method to assess efficacy of therapeutic interventions in clinical trials for DMD boys. This test has been used to quantify ambulatory function in DMD boys who are not yet wheelchair dependent and relies on measuring the distance in meters covered during a period of 6 minutes. The 6MWT is strictly dependent on the state of progression of the disease, and therefore, values can vary from patient to patient, especially in relation to age.101–103 However, because a clear decline in motor function can be detected over a period of several weeks, each patient can be used as his own control, and changes in motor function observed following treatment with a potential drug or other therapeutic interventions can be compared to that obtained at baseline, prior to treatment. As such, this test is currently considered the gold standard in the field for all the clinical trials in DMD and is generally used as a primary outcome measure for all studies aimed at determining efficacy of a potential therapy in this population. Importantly, it has been established that in DMD, as well as other patients affected by neuromuscular disorders, a 30 m difference in 6MWT should be considered the minimal clinically important difference to allow for an intervention to be considered therapeutically relevant. Similar ranges have also been considered meaningful for several other drugs that have received approval for marketing in multiple inherited conditions.104–107 Secondary outcome measures included decreases in CK levels, expression of dystrophin in biopsies obtained from biceps muscles, heart rate, and additional motor function tests.100
The initial results released at the conclusion of the Phase IIb study were disappointing, as they showed no statistically significant differences in the 6MWT among the patients who received ataluren compared to DMD boys treated with placebo. Even more disappointing was the clear presence of a bell-shaped or U-shaped curve response in patients who underwent active treatment with the drug. In particular, patients treated with the higher dose showed a decline in walking distance comparable to that achieved in the placebo group. The post hoc analysis was able to show significant effects among different subgroups of patients who received the lower dose, but not in patients treated with the higher dose.100 On average, at the end of the study, subjects who received ataluren at a dose of 10, 10, 20 mg/kg were shown to be able to cover more distance than boys who received placebo. Of those, patients who were able to walk more than 350 m at baseline and who received the lower dose of ataluren showed the greatest difference, with an average of 68.2 more meters covered over placebo-dosed patients. All other secondary outcome measures failed to provide significant improvements in patients receiving active treatment compared to those in the placebo-control group. Similarly, no dystrophin expression was detected in any of the muscle biopsies, further limiting the evidence of efficacy of the drug.
The reasons for the apparent lack of efficacy in patients who received the higher dose are not clear and have not been investigated in detail. However, several hypotheses that could explain the inhibitory effects observed in patients receiving the higher dose have been suggested. Among those, the more plausible appears to be related to possible interference of the RTC with normal processes of protein translation and protein synthesis as a result of off-target effects. Importantly, the effects observed do not appear to be confined to ataluren alone, since a dose-dependent inhibition of read-through activity has been observed in vitro as well as in vivo using a number of different RTCs including gentamicin.108–110
The ability of ataluren to delay disease progression in DMD patients was assessed in a Phase III study, which has recently concluded (PTC Therapeutics press release).111 The study enrolled over 220 patients in 53 sites across 18 countries. Most of the subjects who were enrolled in the Phase IIb study also participated in the trial. Patients between the ages of 7 and 16 years were randomized into two groups of approximately 110 subjects per group. Each group received either placebo or ataluren at a dose identical to that previously shown to have meaningful effects in Phase IIb trials and consisting of three daily doses of 10, 10, and 20 mg/kg of drug administered after breakfast, lunch, and dinner, respectively. The study was conducted for a period of 48 weeks and was subsequently extended to an open-label extension study in which all subjects were allowed to receive ataluren at the dose of 10, 10, and 20 mg/kg as described above for an additional 50 weeks. The primary end point was change from baseline in the 6MWT. The results of the initial double-blind study have been recently released and have confirmed those obtained in the Phase IIb study.100 Overall, in the whole population that received ataluren, the benefit in the 6MWT was only of 15 m, which was not statistically significant compared to the group that received placebo. Analyses of data from subgroups of patients demonstrated a significant delay in loss of ambulation in subjects who, at baseline, were able to walk over 300 m in the 6MWT, consistent with the results obtained in Phase IIb studies. Secondary outcome measures included additional tests of muscle function, CK levels, and cardiac evaluation. Whether the benefits obtained in some of the patients will be shown to have been sustained throughout the duration of the extension study and to what extent the effects achieved have been beneficial to patients remain unknown as a detailed analysis of all the end points is currently in progress. Importantly, the study also included additional tertiary end points that were introduced to assess their sensitivity and ability to detect improvement in muscle or motor function following treatment. If proven sufficiently sensitive, these new tests may provide new and improved methods to assess the efficacy of therapeutic interventions in DMD patients and could become an integral part of future clinical trials for these patients.
Approval of ataluren in DMD/BMD
Requesting an approval for the use of any drug worldwide requires the filing of different applications in each country for which approval is sought. The US and Europe (EU) are the two main regulatory agencies in the world that control how drugs are moved into the market. In the US, the progression to drug approval following the filing of an investigational new drug application and testing in human subjects can be a lengthy and time-consuming process. The results of clinical trials need to be compiled in a new drug application (NDA), which is subjected to extensive scrutiny by the FDA. In the EU, approval of new drugs is usually requested through the European Medicines Agency (EMA), a centralized system that makes a determination for all the states that are members of the European community. The data presented to both agencies need to demonstrate not only the safety of the drug for which approval is requested but also, and foremost, its efficacy.
Because DMD and BMD are both rare and life-threatening conditions for which unmet needs for treatment exist, they are considered orphan diseases and, therefore, companies or sponsors that are working on the development of treatments for these disorders are eligible to receive incentives from governments, including the US through the FDA, under the Orphan Drug Act, and the EMA. Incentives include scientific advice and protocol assistance to the applicant during the development of the medicine, as well as more frequent consultations on various issues related to the requirements needed to be met prior to receiving approval. Most importantly, orphan designation confers the drug under investigation to qualify for fast track status, which uses pathways that are intended to speed up the drug approval process. When completed successfully and based on the recommendations and decisions of specific designated committees, the process could ultimately lead to what is known in the US as an “accelerated approval”, or in EU as a “conditional approval”.
Despite the incentives, the process of requesting and obtaining approval is among the most tedious and one that has proven to be difficult for ataluren. PTC Therapeutics submitted an NDA to the FDA in 2011, but approval was refused based, primarily, on the lack of statistical significance obtained in the Phase IIb trial. A formal dispute resolution request was filed immediately after by the company requesting to be allowed to resubmit the NDA with the updated meta-analysis conducted on the specific subgroups of patients and which showed significant effects in the population that received the lower dose. Despite the new results, the FDA reaffirmed the appropriateness of its earlier decision and rejected the possibility of filing a second NDA, but allowed for the decision to be reconsidered following the completion of the Phase III clinical trial. Currently, the company is finalizing the data analysis of the trial and is expected to file an NDA for marketing approval from the FDA within the year.
Similarly, the request to approve ataluren in EU has seen a series of initial rejections. In January 2014, the Committee for Medicinal Products for Human Use (CHMP) at the EMA that oversees applications seeking approval adopted a negative opinion, recommending the refusal of the granting of the conditional marketing authorization for ataluren for the treatment of DMD and BMD. Once again, the principal reason was based on the apparent lack of efficacy of the results obtained in Phase IIb studies. Additional concerns included weaknesses identified in the pharmacology data, particularly a lack of relevant data on the pharmacodynamic effects of ataluren in humans, reinforcing the uncertainties raised on its mechanism of action and the dose–response relationship seen in Phase IIb trials. After appeal, the CHMP Committee, although still considered the results not to be sufficiently robust due to the variability in the primary efficacy data, opted to accept the conclusions made by the company and obtained from the post hoc analyses supporting the efficacy of the drug in specific subgroups of patients. The CHMP concluded that the results derived from the Phase IIb studies may be considered clinically relevant and recommended conditional marketing authorization for ataluren in August 2014. The approval represents a huge milestone for the neuromuscular field since this new compound has become the first drug to be approved for the treatment of diseases caused by mutations in the dystrophin gene. Under the terms of the conditional approval, authorization will be granted for 1 year and renewal is subjected to yearly revision of all the clinical safety data available postapproval, as well as those generated from the Phase III studies. The CHMP opinion on ataluren has been sent to the European Commission for adoption of a decision on a marketing authorization for all EU. Germany has been the first European country to provide ataluren to qualifying DMD and BMD patients.
Currently, PTC Therapeutics is expected to submit the results of the study to the EMA in hopes of receiving final approval for the drug in EU. Ataluren will be available to patients under the trade name of Translarna.
Impact of administering Translarna to BMD and DMD patients on societal burden
According to the most recent analyses performed in the US and EU, the costs of standards of care for a patient with DMD is significantly higher than the mean per-capita health expenditure and, although they differ from country to country, they range between US$100,000 and US$120,000 (or International dollars) per year. These costs reflect the expenses paid by health care providers, as well as out-of-pocket expenses paid by each patient’s family.112–114 The approval and commercialization of ataluren will bring the costs of standard of care for qualifying patients to approximately $400,000 per year for each of the patients receiving the drug. In fact, the current list price of Translarna is about US$3,000 per gram of drug, which translates to approximately US$300,000 per year of treatment for each DMD patient. The list price reflects the needs of the company or companies that own the rights for the sale of the drug and their investors to recuperate the funds invested to develop the product and to receive revenues from its selling. Although high, these costs are justified by the limited number of patients who qualify for treatment and are within those estimated for other drugs that are currently approaching the market for the same disease. Nonetheless, these increases in health care costs have already generated concerns and criticisms by politicians in the US and in EU. The National Health System in the United Kingdom (UK) has refused to provide funding of Translarna for DMD, leading to a huge setback for the British families who were awaiting the availability of the drug in Britain. The decision is extremely disappointing considering that only between 100 and 200 patients in the UK are eligible for treatment with Translarna, and therefore, the overall increase in health care costs to the British Government would only be minimal. Following several debates between the company, family advocates, and academicians, the British Government is now considering revising its position if the drug can be provided at a lower cost. An alternative approach to contain the costs of health care for DMD patients with nonsense mutation is to allow the use of a generic drug. However, such a drug will not be available for at least several more years due to the patent that protects the rights of manufacturing the compound, rendering this option highly unlikely. It will be interesting now to see whether the request made to lower the price will be accepted and what other alternatives will be proposed to ensure that patients who can benefit from the drug receive the best standard of care available. However, it needs to be emphasized that limiting the revenues obtained for new drugs that target rare diseases may prove to be a mistake, as this could ultimately discourage companies from pursuing the development of new pharmaceutical products targeting those disorders.
Concluding remarks
The identification and commercialization of ataluren has offered new hope for the treatment of many disorders caused by nonsense mutations and, in particular, DMD. The efforts made by private foundations, patients and their caregivers, and other nonprofit organizations have clearly demonstrated the impact that the community can have on the advancement of strategies to treat rare disorders like DMD. These efforts, combined with the incentives offered by government agencies to pharmaceutical companies, have enabled the establishment of strong research and development activities within industry sectors. Ataluren is the result of just one of those efforts. Importantly, its approval by the EMA, and possibly in the near future by the FDA, will prompt its testing in other genetic disorders caused by nonsense mutations, thus allowing for the establishment of its beneficial effects in other diseases. The impact that this small molecule has on the Duchenne community goes beyond its perspective therapeutic applicability. From a patient’s perspective and that of their caregivers, having the first approved drug developed to specifically target the disease represents a huge conquest and pay-off for their hard work and dedication demonstrated to generate awareness for this rare disorder. This is likely to be just the first of many steps forward that the patients and their families will witness in the near future toward the optimization of an effective therapy for the disease. From a scientific perspective, the use of a drug specifically designed to restore full-length dystrophin expression will enable to better understand the impact that a therapeutic application may have on disease progression and to correlate low, although detectable effects, with improvements on both quality of life and life expectancy of qualifying individuals. Furthermore, its use in patients may enable the establishment of some of the key parameters needed to consider a therapeutic application clinically relevant. For instance, it may enable the correlation of the amount of muscle mass preserved in patients who undergo treatment for prolonged periods of time with the overall delay in loss of muscle function detected in those patients and its impact on secondary pathologies that arise as a result of the disease. Finally, even if the levels of dystrophin protein being restored by ataluren remain low, they may be sufficient to reduce phenotypic variances often observed among patients, allowing the identification of new factors or genetic modifiers that play active roles in the disease process.115,116
In conclusion, although the results obtained in clinical trials clearly demonstrate that ataluren may not be able to reverse the disease pathology in patients, its use in the clinic is likely to have an impact on disease progression. DMD boys who undergo active treatment at very early stages, immediately followed their diagnosis, are likely to benefit the most by RTCs. However, it is too early to be able to predict the extent of the benefits achieved on both life expectancy and its quality. Side effects will have to be evaluated in detail over prolonged periods of administration and throughout the lifetime of the patients to ensure its safety. Ultimately, developing new compounds with improved read-through activity is critical to maximize beneficial effects to patients. Nonetheless, the introduction of ataluren in the market represents an important milestone in the field that needs to be regarded as a tremendous achievement.
Acknowledgments
The authors thank Kaylee M Yan for careful reading of the manuscript. The work was supported by a grant from the Muscular Dystrophy Association (184494), the National Institute of Health (1R21NS076761), and the Department of Defense Congressionally Directed Medical Research Programs (MD120021) to CB.
Disclosure
The authors report no conflicts of interest in this work.
References
Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. | |
Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2:90–95. | |
Shiga N, Takeshima Y, Sakamoto H, et al. Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. J Clin Invest. 1997;100:2204–2210. | |
Tuffery-Giraud S, Saquet C, Thorel D, et al. Mutation spectrum leading to an attenuated phenotype in dystrophinopathies. Eur J Hum Genet. 2005;13:1254–1260. | |
Flanigan KM, Dunn DM, von NA, et al. Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene. Hum Mutat. 2011;32:299–308. | |
Flanigan KM, Dunn D, Larsen CA, Medne L, Bonnemann CB, Weiss RB. Becker muscular dystrophy due to an inversion of exons 23 and 24 of the DMD gene. Muscle Nerve. 2011;44:822–825. | |
Falzarano MS, Scotton C, Passarelli C, Ferlini A. Duchenne muscular dystrophy: from diagnosis to therapy. Molecules. 2015;20:18168–18184. | |
Drachman DB, Toyka KV, Myer E. Prednisone in Duchenne muscular dystrophy. Lancet 1974;2:1409–1412. | |
Mendell JR, Moxley RT, Griggs RC, et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. 1989;320:1592–1597. | |
Escolar DM, Hache LP, Clemens PR, et al. Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy. Neurology. 2011;77:444–452. | |
Scott JM, Li S, Harper SQ, et al. Viral vectors for gene transfer of micro-, mini-, or full-length dystrophin. Neuromuscul Disord. 2002; 12(Suppl 1):S23–S29. | |
Gregorevic P, Blankinship MJ, Allen JM, et al. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 2004;10:828–834. | |
Konieczny P, Swiderski K, Chamberlain JS. Gene and cell-mediated therapies for muscular dystrophy. Muscle Nerve. 2013;47:649–663. | |
Yue Y, Ghosh A, Long C, et al. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol Ther. 2008;16:1944–1952. | |
Lai Y, Yue Y, Liu M, et al. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol. 2005;23:1435–1439. | |
Acsadi G, Dickson G, Love DR, et al. Human dystrophin expression in mdx mice after intramuscular injection of DNA constructs. Nature. 1991;352:815–818. | |
Bertoni C, Jarrahian S, Wheeler TM, et al. Enhancement of plasmid-mediated gene therapy for muscular dystrophy by directed plasmid integration. Proc Natl Acad Sci U S A. 2006;103:419–424. | |
Romero NB, Braun S, Benveniste O, et al. Phase I study of dystrophin plasmid-based gene therapy in Duchenne/Becker muscular dystrophy. Hum Gene Ther. 2004;15:1065–1076. | |
Alter J, Lou F, Rabinowitz A, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. 2006;12:175–177. | |
Lu QL, Mann CJ, Lou F, et al. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med. 2003;9:1009–1014. | |
De Angelis FG, Sthandier O, Berarducci B, et al. Chimeric snRNA molecules carrying antisense sequences against the splice junctions of exon 51 of the dystrophin pre-mRNA induce exon skipping and restoration of a dystrophin synthesis in Delta 48-50 DMD cells. Proc Natl Acad Sci U S A. 2002;99:9456–9461. | |
van Deutekom JC, Bremmer-Bout M, Janson AA, et al. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum Mol Genet. 2001;10:1547–1554. | |
Wilton SD, Fall AM, Harding PL, McClorey G, Coleman C, Fletcher S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol Ther. 2007;15:1288–1296. | |
Mendell JR, Campbell K, Rodino-Klapac L, et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med. 2010;363:1429–1437. | |
Rodino-Klapac LR, Montgomery CL, Bremer WG, et al. Persistent expression of FLAG-tagged micro dystrophin in nonhuman primates following intramuscular and vascular delivery. Mol Ther. 2010;18:109–117. | |
Voit T, Topaloglu H, Straub V, et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014;13:987–996. | |
Hammond SM, Wood MJA. PRO-051, an antisense oligonucleotide for the potential treatment of Duchenne muscular dystrophy. Curr Opin Mol Ther. 2010;12:478–486. | |
Cirak S, Feng L, Anthony K, et al. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol Ther. 2012;20:462–467. | |
Cirak S, rechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. | |
Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74:637–647. | |
Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349–353. | |
Tinsley J, Deconinck N, Fisher R, et al. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;4:1441–1444. | |
Gilbert R, Nalbanoglu J, Tinsley JM, Massie B, Davies KE, Karpati G. Efficient utrophin expression following adenovirus gene transfer in dystrophic muscle. Biochem Biophys Res Commun. 1998;242:244–247. | |
Wakefield PM, Tinsley JM, Wood MJ, Gilbert R, Karpati G, Davies KE. Prevention of the dystrophic phenotype in dystrophin/utrophin-deficient muscle following adenovirus-mediated transfer of a utrophin minigene. Gene Ther. 2000;7:201–204. | |
Krag TO, Bogdanovich S, Jensen CJ, et al. Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc Natl Acad Sci U S A. 2004;101:13856–13860. | |
Tinsley JM, Fairclough RJ, Storer R, et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One. 2011;6:e19189. | |
Tinsley J, Robinson N, Wilson FX, Horne G, Fairclough RJ, Davies K. Future clinical and biomarker development for SMTC1100, the first utrophin modulator to enter clinical trials for Duchenne Muscular Dystrophy (DMD). Neuromuscular Disord. 2013;23:813. | |
Bertoni C. Therapy development for neuromuscular diseases: translating hope into promise. Future Neurol. 2013;8:399–401. | |
Bogdanovich S, Krag TO, Barton ER, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. | |
Wagner KR, McPherron AC, Winik N, Lee SJ. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–836. | |
Haidet AM, Rizo L, Handy C, et al. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci U S A. 2008;105:4318–4322. | |
Cohn RD, van EC, Habashi JP, et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–210. | |
Bish LT, Yarchoan M, Sleeper MM, et al. Chronic losartan administration reduces mortality and preserves cardiac but not skeletal muscle function in dystrophic mice. PLoS One. 2011;6:e20856. | |
Spurney CF, Sali A, Guerron AD, et al. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J Cardiovasc Pharmacol Ther. 2011;16:87–95. | |
Messina S, Bitto A, Aguennouz M, et al. Nuclear factor kappa-B blockade reduces skeletal muscle degeneration and enhances muscle function in Mdx mice. Exp Neurol. 2006;198:234–241. | |
Grounds MD, Torrisi J. Anti-TNFalpha (Remicade) therapy protects dystrophic skeletal muscle from necrosis. FASEB J. 2004;18:676–682. | |
Pierno S, Nico B, Burdi R, et al. Role of tumour necrosis factor alpha, but not of cyclo-oxygenase-2-derived eicosanoids, on functional and morphological indices of dystrophic progression in mdx mice: a pharmacological approach. Neuropathol Appl Neurobiol. 2007;33:344–359. | |
Hodgetts S, Radley H, Davies M, Grounds MD. Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFalpha function with Etanercept in mdx mice. Neuromuscul Disord. 2006;16:591–602. | |
Piers AT, Lavin T, Radley-Crabb HG, Bakker AJ, Grounds MD, Pinniger GJ. Blockade of TNF in vivo using cV1q antibody reduces contractile dysfunction of skeletal muscle in response to eccentric exercise in dystrophic mdx and normal mice. Neuromuscul Disord. 2011;21:132–141. | |
Peterson JM, Kline W, Canan BD, et al. Peptide-based inhibition of NF-kappaB rescues diaphragm muscle contractile dysfunction in a murine model of Duchenne muscular dystrophy. Mol Med. 2011;17:508–515. | |
Peay HL, Tibben A, Fisher T, Brenna E, Biesecker BB. Expectations and experiences of investigators and parents involved in a clinical trial for Duchenne/Becker muscular dystrophy. Clin Trials. 2014;11:77–85. | |
Peay HL, Hollin I, Fischer R, Bridges JF. A community-engaged approach to quantifying caregiver preferences for the benefits and risks of emerging therapies for Duchenne muscular dystrophy. Clin Ther. 2014;36:624–637. | |
Franson TR, Peay H. Benefit-risk assessments in rare disorders: the case for therapeutic development in Duchenne muscular dystrophy as the prototype for new approaches. Available from: http://www parentprojectmd org/site/DocServer/br_paper_v11__2_ pdf;jsessionid=2C381495CB3753608053FD8DD624B686 app247d?docID=14503. Accessed April 16, 2016. | |
US Food and Drug Administration (FDA). Enhancing benefit-risk assessment in regulatory decision-making. Available from: http://wwwfda gov/forindustry/userfees/prescriptiondruguserfee/ucm326192 htm 2014. Accessed March 1, 2014. | |
Human Gene Mutation Database. Available from: http://www.hgmd.org www.hgmd.org. Accessed March 1, 2016. | |
Mort M, Ivanov D, Cooper DN, Chuzhanova NA. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29:1037–1047. | |
Lee HLR, Dougherty JP. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol Ther. 2012;136:227–266. | |
Keeling KM, Xue X, Gunn G, Bedwell DM. Therapeutics based on stop codon readthrough. Annu Rev Genomics Hum Genet. 2014;15:371–394. | |
Mullard A. EMA reconsiders ‘read-through’ drug against Duchenne muscular dystrophy following appeal. Nat Biotech. 2014;32:706. | |
Allen L-A, Raetz CRH. Partial phenotypic suppression of a peroxisome-deficient animal cell mutant treated with aminoglycoside G418. J Biol Chem. 1992;267:13191–13199. | |
Bedwell DM, Kaenjak A, Benos DJ, et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med. 1997;3:1280–1284. | |
Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–469. | |
Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104:375–381. | |
Keeling KM, Brooks DA, Hopwood JJ, Li P, Thompson JN, Bedwell DM. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum Mol Genet. 2001;10:291–299. | |
Du M, Jones JR, Lanier J, et al. Aminoglycoside suppression of a premature stop mutation in a Cftr-/- mouse carrying a human CFTR-G542X transgene. J Mol Med. 2002;80:595–604. | |
Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Annu Rev Biochem. 2005;74:129–177. | |
Ogle JM, Brodersen DE, Clemons WM Jr, Tarry MJ, Carter AP, Ramakrishnan V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001;292:897–902. | |
Purohit P, Stern S. Interactions of a small RNA with antibiotic and RNA ligands of the 30S subunit. Nature. 1994;370:659–662. | |
Wilhelm JM, Jessop JJ, Pettitt SE. Aminoglycoside antibiotics and eukaryotic protein synthesis: stimulation of errors in the translation of natural messengers in extracts of cultured human cells. Biochemistry. 1978;17:1149–1153. | |
Sun J, Chen M, Xu J, Luo J. Relationships among stop codon usage bias, its context, isochores, and gene expression level in various eukaryotes. J Mol Evol. 2005;61:437–444. | |
Bonetti B, Fu L, Moon J, Bedwell DM. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J Mol Biol. 1995;251:334–345. | |
Tate WP, Poole ES, Horsfield JA, et al. Translational termination efficiency in both bacteria and mammals is regulated by the base following the stop codon. Biochem Cell Biol. 1995;73:1095–1103. | |
Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. | |
De LA, Nico B, Rolland JF, et al. Gentamicin treatment in exercised mdx mice: Identification of dystrophin-sensitive pathways and evaluation of efficacy in work-loaded dystrophic muscle. Neurobiol Dis. 2008;32:243–253. | |
Politano L, Nigro G, Nigro V, et al. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003;22:15–21. | |
Wagner KR, Hamed S, Hadley DW, et al. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol. 2001;49:706–711. | |
Malik V, Rodino-Klapac LR, Viollet L, Mendell JR. Aminoglycoside-induced mutation suppression (stop codon readthrough) as a therapeutic strategy for Duchenne muscular dystrophy. Ther Adv Neurol Disord. 2010;3:379–389. | |
Goldmann T, Overlack N, Moller F, et al. A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol Med. 2012;4:1186–1199. | |
Arakawa M, Shiozuka M, Nakayama Y, et al. Negamycin restores dystrophin expression in skeletal and cardiac muscles of mdx mice. J Biochem (Tokyo). 2003;134:751–758. | |
Hamada K, Taguchi A, Kotake M, et al. Structure-activity relationship studies of 3-epi-deoxynegamycin derivatives as potent readthrough drug candidates. ACS Med Chem Lett. 2015;6:689–694. | |
Du L, Damoiseaux R, Nahas S, et al. Nonaminoglycoside compounds induce readthrough of nonsense mutations. J Exp Med. 2009; 206:2285–2297. | |
Kayali R, Ku JM, Khitrov G, Jung ME, Prikhodko O, Bertoni C. Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum Mol Genet. 2012;21:4007–4020. | |
Jung ME, Ku JM, Du L, Hu H, Gatti RA. Synthesis and evaluation of compounds that induce readthrough of premature termination codons. Bioorg Med Chem Lett. 2011;21:5842–5848. | |
Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. | |
Auld DS, Thorne N, Maguire WF, Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci U S A. 2009;106:3585–3590. | |
Auld DS, Lovell S, Thorne N, et al. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc Natl Acad Sci U S A. 2010;107:4878–4883. | |
McElroy SP, Nomura T, Torrie LS, et al. A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol. 2013;11:e1001593. | |
Dranchak PK, Di PE, Snowden A, et al. Nonsense suppressor therapies rescue peroxisome lipid metabolism and assembly in cells from patients with specific PEX gene mutations. J Cell Biochem. 2011;112:1250–1258. | |
Koopmann TT, Verkerk AO, Bezzina CR, de Bakker JM, Wilde AA. The chemical compound PTC124 does not affect cellular electrophysiology of cardiac ventricular myocytes. Cardiovasc Drugs Ther. 2012;26:41–45. | |
Brumm H, Muhlhaus J, Bolze F, et al. Rescue of melanocortin 4 receptor (MC4R) nonsense mutations by aminoglycoside-mediated read-through. Obesity (Silver Spring). 2012;20:1074–1081. | |
Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci U S A. 2008;105:2064–2069. | |
Sarkar C, Zhang Z, Mukherjee AB. Stop codon read-through with PTC124 induces palmitoyl-protein thioesterase-1 activity, reduces thioester load and suppresses apoptosis in cultured cells from INCL patients. Mol Genet Metab. 2011;104:338–345. | |
Tan L, Narayan SB, Chen J, Meyers GD, Bennett MJ. PTC124 improves readthrough and increases enzymatic activity of the CPT1A R160X nonsense mutation. J Inherit Metab Dis. 2011;34:443–447. | |
Goldmann T, Overlack N, Wolfrum U, Nagel-Wolfrum K. PTC124-mediated translational readthrough of a nonsense mutation causing Usher syndrome type 1C. Hum Gene Ther. 2011;22:537–547. | |
Yu H, Liu X, Huang J, Zhang Y, Hu R, Pu J. Comparison of read-through effects of aminoglycosides and PTC124 on rescuing nonsense mutations of HERG gene associated with long QT syndrome. Int J Mol Med. 2014;33:729–735. | |
Fang YW, Yang SS, Chau T, et al. Therapeutic effect of prenatal alkalization and PTC124 in Na(+)/HCO3(-) cotransporter 1 p.W516* knock-in mice. Gene Ther. 2015;22:374–381. | |
Hirawat S, Welch EM, Elfring GL, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol. 2007;47:430–444. | |
Kerem E, Hirawat S, Armoni S, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372:719–727. | |
Bonnemann C, Finkel R, Wong C, et al. G.P.3.05 Phase 2 study of PTC124 for nonsense mutation suppression therapy of Duchenne muscular dystrophy (DMD) [abstract]. Neuromuscul Disord. 2007;17:783. | |
Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50:477–487. | |
ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166:111–117. | |
Butland RJ, Pang J, Gross ER, Woodcock AA, Geddes DM. Two-, six-, and 12-minute walking tests in respiratory disease. Br Med J (Clin Res Ed). 1982;284:1607–1608. | |
McDonald CM, Henricson EK, Han JJ, et al. The 6-minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve. 2010;41:500–510. | |
Lynn S, Aartsma-Rus A, Bushby K, et al. Measuring clinical effectiveness of medicinal products for the treatment of Duchenne muscular dystrophy. Neuromuscul Disord. 2015;25:96–105. | |
McDonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve. 2013;48:343–356. | |
McDonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other clinical endpoints in Duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. 2013;48:357–368. | |
Henricson E, Abresch R, Han JJ, et al. The 6-minute walk test and person-reported outcomes in boys with Duchenne muscular dystrophy and typically developing controls: longitudinal comparisons and clinically-meaningful changes over one year. PLoS Curr. 2013;5. | |
Owen SC, Doak AK, Ganesh AN, et al. Colloidal drug formulations can explain “bell-shaped” concentration-response curves. ACS Chem Biol. 2014;9:777–784. | |
Anwer K, Earle KA, Shi M, et al. Synergistic effect of formulated plasmid and needle-free injection for genetic vaccines. Pharm Res. 1999;16:889–895. | |
Li M, Andersson-Lendahl M, Sejersen T, Arner A. Muscle dysfunction and structural defects of dystrophin-null sapje mutant zebrafish larvae are rescued by ataluren treatment. FASEB J. 2014;28:1593–1599. | |
PTC Therapeutics press release. Available from: http://ir.ptcbio.com/releasedetail.cfm?releaseid=936905. Accessed March 1, 2016. | |
Ouyang L, Grosse SD, Kenneson A. Health care utilization and expenditures for children and young adults with muscular dystrophy in a privately insured population. J Child Neurol. 2008;23:883–888. | |
Larkindale J, Yang W, Hogan PF, et al. Cost of illness for neuromuscular diseases in the United States. Muscle Nerve. 2014;49:431–438. | |
Landfeldt E, Lindgren P, Bell CF, et al. The burden of Duchenne muscular dystrophy: an international, cross-sectional study. Neurology. 2014;83:529–536. | |
Flanigan KM, Ceco E, Lamar KM, et al. LTBP4 genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Ann Neurol. 2013;73:481–488. | |
Bello L, Kesari A, Gordish-Dressman H, et al. Genetic modifiers of ambulation in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study. Ann Neurol. 2015;77:684–696. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.