")
Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 6
Clinical management of tuberous sclerosis complex over the lifetime of a patient
Received 7 May 2014
Accepted for publication 20 August 2014
Published 26 August 2015 Volume 2015:6 Pages 139—146
DOI https://doi.org/10.2147/PHMT.S67342
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Laurens Holmes, Jr
Michael Frost,1 John Hulbert2
1Minnesota Epilepsy Group, PA, St Paul, MN, USA; 2Urologic Physicians, PA, Edina, MN, USA
Abstract: Tuberous sclerosis complex (TSC) is a genetic disorder characterized by nonmalignant tumors (hamartomas) that can occur in various organ systems, including the brain, kidneys, lungs, skin, eyes, and heart. Clinical manifestations of TSC can occur at any age, thereby making the diagnosis difficult. No typical disease presentation is known, and the clinical presentation usually differs between pediatric and adult patients. Furthermore, variable penetrance of the genetic mutation causes a range of disease severity from very mild to severe, and affected individuals can go undetected for years because many of the clinical manifestations of TSC lack specificity. Once a diagnosis is made, TSC management strategies should be tailored to address the symptoms and risks most relevant to the age of the patient. Improved understanding of the genetic basis of TSC and of the central issue of mTOR overactivation has led to use of pharmacotherapies such as the mTOR inhibitors everolimus and sirolimus in the treatment of TSC disease. In Phase II and III studies, everolimus has demonstrated efficacy and safety in the treatment of both brain (subependymal giant cell astrocytoma) and renal (angiomyolipoma) manifestations associated with TSC. It is important to bear in mind that TSC is a lifelong condition, and for those diagnosed as children, a continuum of care will be needed as they transition from pediatric to adult health services. Clearly identifying the likely differences among diagnosis, monitoring, and management of pediatric and adult patients with TSC is an important step in enabling efficiencies to be maximized without compromising the care provided to patients.
Keywords: mammalian target of rapamycin inhibitors, mTOR treatment, treatment strategies
Introduction
Tuberous sclerosis complex (TSC) is a genetic disorder with an incidence of approximately 1 in 5,000–6,000 live births and a prevalence between 1 million and 1.5 million people worldwide.1,2 It is characterized by nonmalignant tumors (hamartomas) that occur in various organ systems, including brain, kidneys, lungs, skin, eyes, and heart.1,3,4 TSC is passed on in an autosomal dominant pattern,1,5 and although the hereditary nature of TSC was recognized early in the twentieth century,5 it was not until the 1990s that the genes responsible were finally identified.5,6 Approximately 70%–85% of individuals who have a definitive diagnosis of TSC have an identifiable mutation in one of two genes, TSC1 or TSC2.7,8 These genes encode for two cellular proteins – hamartin and tuberin, respectively5,6,9 – that are involved in the PI3K/Akt signaling pathway (reviewed by Huang and Manning).9 Hamartin and tuberin form a heterodimer that inhibits the mammalian target of rapamycin (mTOR) protein kinase, which is a key regulator of several cellular functions, including nutrient sensing, protein synthesis, and cellular growth and proliferation.9 Mutation of the TSC1 or TSC2 gene leads to functional loss of the hamartin/tuberin dimer, which, in turn, results in constitutive activation of the mTOR complex 1 (mTORC1) and uncontrolled cellular growth and proliferation.10 There is evidence that mutations in the TSC2 gene may result in more severe disease in multiple organs than mutations in the TSC1 gene.8 Improved understanding of the genetic basis of TSC and of the central issue of mTOR overactivation has led to the development of new pharmacotherapies directly targeting the affected pathways and has considerably changed the options available for managing the disease.
Clinical manifestations of TSC can occur at any age, thereby making the diagnosis difficult. No typical disease presentation is known, and the clinical presentation usually differs between pediatric and adult patients. Furthermore, variable penetrance of the genetic mutation causes a range of disease severity from very mild to severe, and in affected individuals, the condition can go undetected for years because many of the clinical manifestations of TSC lack specificity. The diagnosis of a patient with TSC is dependent on the presence of a constellation of symptoms, or on a TSC1 or TSC2 pathogenic mutation.11,12 Once the diagnosis is made, TSC management strategies should be tailored to address the symptoms and risks most relevant to the age of the patient. It is important to bear in mind that TSC is a lifelong condition, and for those diagnosed as children, a continuum of care will be needed as they transition from pediatric to adult health services.13 Details regarding common clinical manifestations of TSC over a patient’s lifetime are discussed below. In addition, the role of mTOR inhibitors and other management strategies currently utilized to treat these manifestations are discussed with consideration of age-appropriate therapy.
TSC manifestations over a patient’s lifetime
TSC gene penetrance is approximately 100%; however, clinical manifestations of the disease may appear at different ages (Table 1) and severity can change over the lifetime of a patient.4,14–16 For example, angiomyolipoma lesion size and renal complications have been shown to increase with age.17 In addition, symptoms can vary between family members with TSC, and it is important to recognize the different manifestations likely to be seen among pediatric, adolescent, and adult patients.
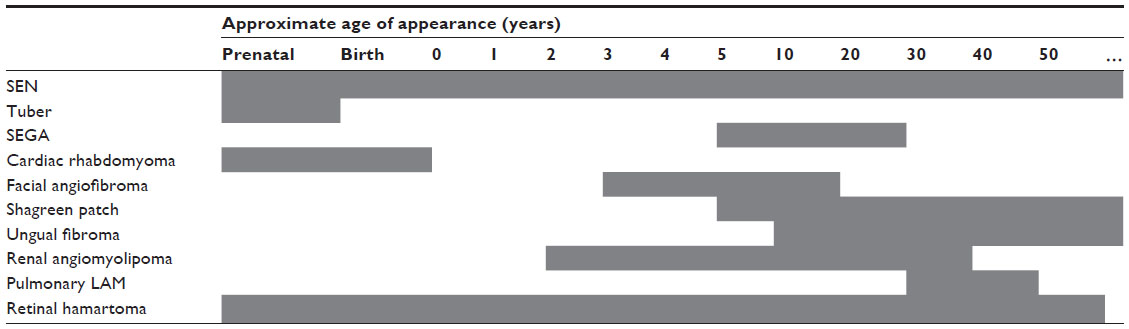 | Table 1 Age of TSC manifestation appearance3,4,14 |
Cardiac rhabdomyoma is one of the earliest manifestations of TSC, developing typically around week 22 to 28 of gestation.18 This tumor occurs in up to approximately 50%–70% of patients with TSC, primarily during the fetal and early neonatal periods.3,19–21 In a large database collected by the Tuberous Sclerosis Alliance on 1,187 patients, 461 patients had cardiac rhabdomyoma, of which 42 cases were diagnosed by fetal ultrasound.22 In a retrospective chart review by Staley et al, patients diagnosed during the first week of life presented with ≥1 cardiac rhabdomyoma.23 Most babies diagnosed with a cardiac rhabdomyoma will go on to be diagnosed with TSC.19,20 These lesions are usually asymptomatic and tend to regress spontaneously with increasing age;20,21 however, in a small percentage of patients, they cause symptoms such as outflow obstruction, valvular dysfunction, or arrhythmia.19–21,23,24 Among 16 patients with prenatal cardiac rhabdomyoma (identified between weeks 17 and 33), two patients were identified as the result of fetal arrhythmia.23 Patients may be treated for cardiac rhabdomyoma; everolimus may be effective in causing regression.24
Abnormalities of the central nervous system associated with TSC typically manifest in childhood and can lead to problematic neurological and psychological issues, including epilepsy, cognitive deficits, and autism.2 Central nervous system anatomical abnormalities include subependymal giant cell astrocytomas (SEGAs), subependymal nodules (SENs), cortical tubers, and changes to the microarchitecture of the brain.1,12,25 Cortical tubers are a hallmark of TSC;2 they often develop before birth and occur in more than 80% of patients.1 Identified SENs and SEGAs develop from childhood through adolescence and have a similar histology, although SEGAs are differentiated by their larger size and serial growth. SEGAs occur in 5%–20% of patients with TSC; their usual location in the region of the foramen of Monro can eventually block the flow of cerebrospinal fluid, leading to acute or subacute hydrocephalus with associated morbidity and mortality.2
Cortical tubers and associated microanatomical abnormalities in the central nervous system have been linked to the occurrence of seizures,26–28 which are experienced by a large percentage of patients with TSC.4,29 TSC-associated epilepsy is likely to appear during the first year of life; it is often severe, progressive, and resistant to treatment and is more likely to impact cognitive abilities than non-TSC–related epilepsy.29–31
Skin abnormalities are a common early manifestation of TSC.4 In childhood, the most common skin complications are hypomelanotic macules (occurring in more than 90% of patients with TSC).32,33 Shagreen patches and facial angiofibromas also usually develop during childhood. Angiofibromas typically appear from 3–4 years of age and progressively worsen as a patient gets older; shagreen patches are usually apparent before 10 years of age and increase in number as a patient ages.33–35 Ungual fibromas (Koenen tumors) may develop from adolescence into early adulthood.4,33,34 Skin lesions can negatively impact a patient’s quality of life, confidence, and psychological well-being. On occasion, the lesions may bleed and become infected.33
Retinal hamartomas are another manifestation seen early in childhood.36 They are usually asymptomatic and are not progressive, but they may obstruct vision in some patients.4
Renal manifestations of TSC are common, occurring in more than 80% of patients with TSC.8,37–39 Renal angiomyolipoma is the most common lesion encountered, but cystic renal disease also occurs frequently;8,37,39,40 renal cancer has a similar rate of occurrence as the general population, but on average it occurs 25 years sooner in those with TSC, than the general population.41–43
Angiomyolipoma occurs in up to 80% of patients with TSC, typically starting in the first decade of life, with a median age at diagnosis of 10–11 years.8,37–40,44 Renal disease seems more severe with TSC2 mutation and in association with constitutional deletions involving TSC2 and PKD1.8,45 Contiguous gene syndrome may result in renal insufficiency (although only 1%–2% of patients with TSC have severe renal insufficiency).44,45 Overall, however, the morbidity and mortality reported with renal lesions associated with TSC are of great significance; renal manifestations are a common cause of death in children and the most common cause of death in adults with TSC.46
Lymphangioleiomyomatosis (LAM) is a pulmonary disorder that typically presents in early adulthood, with a mean age of symptom onset of 30–35 years.2,47–50 It occurs almost exclusively in women,47,49–51 although rare cases have been reported in men.52,53 It is characterized by diffuse infiltration of the lungs by smooth muscle cells and gradual replacement of the pulmonary parenchyma with cysts. Patients usually present with progressive dyspnea on exertion or recurrent pneumothorax.2,48–51,54 The incidence (based on radiologic surveys) in women with TSC is in the range of 26%–48%.54,55 To clearly differentiate between TSC-associated LAM and spontaneous LAM (sLAM), TSC diagnostic criteria were recently amended and now require the presence of additional TSC features when both LAM and angiomyolipomata are present.12 Diagnosis of LAM may be aided by detection of vascular endothelial growth factor (VEGF)-D, an angiogenic biomarker that is elevated in patients with LAM.56,57
Several other organs may also be involved in TSC, although more rarely or with less associated morbidity. Hamartoma can occur in bones, jaw, and liver, and dental pitting has been reported more frequently in older patients with TSC than in the general population.58–60 Nonrenal hamartomas and the presence of >3 dental enamel pits are considered minor features of TSC that should be sought during diagnostic workup.12 Angiomyolipoma may occur outside of the kidneys but less commonly than in the kidneys.61,62 Because of their relative specificity to TSC, the presence of ≥2 angiomyolipomata, regardless of whether they are renal, is considered a major feature of TSC.12
Recent progress with targeted mTOR treatment
Improved understanding of the genetic basis of TSC and of the central issue of mTOR overactivation has led to development of new pharmacotherapies that enable new management strategies for TSC. The mTOR inhibitors everolimus and sirolimus (rapamycin) have been investigated for the treatment of SEGA and angiomyolipoma in patients with TSC, as well as for other manifestations, including seizures, LAM, and skin lesions. Everolimus has demonstrated efficacy in treating both SEGA and angiomyolipoma associated with TSC in Phase II and III studies.63–65 Everolimus (Afinitor®) has been approved by the US Food and Drug Administration (FDA) for use in pediatric and adult patients with TSC who have SEGA that requires therapeutic intervention but cannot be curatively resected, and in adults with renal angiomyolipoma and TSC not requiring immediate surgery.66 Although it is not FDA approved for these treatments, sirolimus has demonstrated efficacy in a few case reports and a small (n≤25) open-label study in patients with TSC-associated angiomyolipoma, LAM, and SEGA.67–69
Current management strategies in TSC – continuum of care
It is important to recognize the differences between pediatric and adult patients with TSC with respect to diagnosis, monitoring, and appropriate management. TSC is a multifaceted disorder that can manifest in different ways throughout the life of the individual, and a multidisciplinary approach to the clinical management of TSC is required.13
Unless symptoms requiring attention are noted, such as arrhythmia or obstruction, minimal monitoring or management is recommended in neonates identified with rhabdomyoma.13 Pediatricians and general practitioners need to be aware that cardiac lesions generally do not worsen into adulthood, and follow-up should continue until the lesions decrease in size.13 Therefore, repeated testing and specialist referrals may not be needed over time, although cardiologists are useful for diagnosis and for consultation if symptomatic lesions are noted.
Neurological manifestations should be managed from birth to adulthood.13 Prevention of early-onset seizures, if possible, may improve a patient’s quality of life over the long term by reducing the morbidity often associated with epilepsy. In patients with TSC who have infantile spasms, vigabatrin has become the first-line therapy of choice, whereas in the non-TSC setting, adrenocorticotropic hormone (ACTH) is the first-line therapy of choice.70,71 Additionally, there is some evidence to suggest mTOR inhibitors may ameliorate or prevent seizures in TSC as demonstrated in two Phase I/II clinical studies evaluating everolimus in patients with SEGA, and a case series evaluating both sirolimus and everolimus in patients with SEGA;63,72–74 however, more robust studies are needed in this area. Other options for poorly controlled or refractory epilepsy include consumption of a ketogenic diet, surgical excision of tubers, vagus nerve stimulation, and corpus callosotomy.75 Treating epilepsy early may reduce the negative impact of early-onset seizures on later neurological functioning.70
Teaching parents to recognize seizures is a key element in achieving early diagnosis.13,70 Identification of electroencephalographic (EEG) seizures through the use of EEG monitoring in young infants may allow very early detection of abnormalities and initiation of preventive therapy.13,70 Additional significant neurological manifestations of TSC include neurobehavioral and cognitive difficulties such as mental retardation, autism, and learning difficulty, for which patients should be comprehensively assessed at diagnosis.13 Individuals with TSC are often of normal or near normal intellect and may exhibit obsessive-compulsive tendencies, generalized and social anxiety, shifting attention, and problems with multistep reasoning, or they may have profound mental retardation.2 A retrospective analysis examining psychiatric and behavioral disorders in patients with TSC identified 62 of 157 evaluable patients with confirmed psychiatric comorbidity.76 Psychiatric conditions included behavioral disorders (intermittent explosive disorder, oppositional defiant disorder, and self-injurious behavioral disorder), autism spectrum disorders, anxiety disorders, attention-deficit/hyperactivity disorder, and mood and thought disorders.76 On average, each patient was diagnosed with two psychiatric conditions (mean 2.42±1.27 diagnoses/patient).76 Management of psychiatric conditions includes the use of antipsychotics, antidepressants, and anticonvulsants with mood-stabilizing properties and is often associated with overall improvement or stabilization of psychiatric symptoms.76
Monitoring for the development or progression of potentially life-threatening conditions in the central nervous system such as SEGA is also warranted.13 Patients should undergo regular brain imaging at least until adulthood.13 Symptomatic or enlarging SEGAs should be surgically excised when possible.13,77 For growing but otherwise asymptomatic SEGA, or for SEGA requiring intervention that is not amenable to resection, everolimus therapy can be considered to reduce SEGA volume.13,66
To monitor eye health in patients with identified ophthalmologic lesions, annual ophthalmologic evaluation is recommended.13 As with cardiac lesions, health providers should be aware that retinal lesions usually do not get worse as the patient grows up, so repeated evaluations every 6 months are not needed. Similarly, more frequent assessment in children receiving vigabatrin for seizures (to monitor for potential ophthalmologic toxicity, as recommended by the FDA) is now considered of limited benefit in the absence of specific clinical concerns.13
Renal angiomyolipoma requires close monitoring and careful management in both childhood and adult life.13 Although it is often asymptomatic, the nature of angiomyolipoma with fragile aneurysmal blood vessels surrounded by soft adipose tissue can lead to spontaneous and severe renal hemorrhage78 and can be life threatening—a situation that is more frequent as the size increases to >4 cm.79 Furthermore, some angiomyolipomata have low fat content and are difficult to differentiate radiologically from malignant renal tumors. It is regrettable that in the past, far too frequently in both instances, this has resulted in nephrectomy, which in a condition that is bilateral and almost always benign should be the last resort. Fortunately, nowadays, both therapeutic embolization and, for those for whom embolization is not possible, nephron-sparing, minimally invasive surgical techniques (partial nephrectomy or ablative therapy), allied with greater understanding of TSC by physicians, patients, families, guardians, and caregivers, have enabled avoidance of nephrectomy, except as a last resort.13
Atypical (ie, low fat content) angiomyolipoma has presented a particular diagnostic challenge because of the radiological difficulty associated with differentiating it from renal cell cancer.80 However, greater understanding of TSC by radiologists, attention to the growth rate of the lesion, understanding that the incidence of atypical angiomyolipoma/renal cell cancer in TSC is 50:1, and safer biopsy techniques with HMB45 staining (specific for angiomyolipoma) should greatly aid diagnosis.80,81 A few angiomyolipomata appear to metastasize despite benign pathology; this may be a feature of multicentric origin or of actual metastasis.82,83 Treatment with everolimus can now be considered for asymptomatic, growing angiomyolipomata greater than 3 cm in diameter, as long as emergency intervention is not required;13 everolimus has demonstrated significantly greater angiomyolipoma response rates compared with placebo in Phase III clinical trials.64–66 Additionally, time to angiomyolipoma progression was statistically significantly longer in patients taking everolimus compared with those taking placebo.65
Identification and management of LAM are required almost exclusively for women in adulthood, usually from the third to fourth decade onward. mTOR inhibitors are recommended for the treatment of TSC-associated LAM in patients with moderate-to-severe lung disease or rapid progression; for patients with respiratory failure, lung transplantation is considered.13 The efficacy and safety of mTOR inhibitors such as everolimus and sirolimus have been examined in clinical trials, but the numbers of patients included have been low.65,84 Ongoing and future studies will enhance our understanding of the clinical benefits of mTOR inhibitors in the treatment of LAM. In the Phase III trial of everolimus in patients with angiomyolipoma associated with TSC or sLAM, 28% of patients in the everolimus arm and 18% in the placebo arm had LAM.65 By week 24, lung function had deteriorated less in patients receiving everolimus than in those given placebo, but differences were small (diffusion capacity for carbon monoxide [DLCO] −3% vs −8%; forced expiratory volume in 1 second [FEV1] −1% vs −4%) and interpretation was difficult in light of the small patient numbers and the short duration of the study.65 One open-label trial of sirolimus in patients with angiomyolipoma associated with TSC or sLAM reported some improvement from baseline in respiratory measures,69 although another reported no effect on pulmonary function.85
Future direction
Limited resources are available for research because of lack of funding or other priorities; therefore, it will become increasingly important to target treatment of TSC to ensure that it is as effective as possible. Early diagnosis and proactive treatment strategies initiated before the development of symptoms are becoming increasingly important.12 Use of everolimus as neoadjuvant therapy to reduce tumor volume as a means of improving surgical outcomes should be considered. Antiepileptic drug treatment administered before the development of clinical seizures was effective in reducing the onset of seizures and seizure severity and was associated with improved mental outcomes.86 Infants received vigabatrin (100–150 mg/kg/day) within 1 week after active epileptic discharges appeared on EEG (ie, preventive treatment), or the same dosage regimen was started within 1 week after the onset of clinical seizures (standard treatment).86 At 24 months of age, a significantly higher percentage of patients with epilepsy who received preventive treatment were seizure-free compared with patients with epilepsy who received standard treatment (P<0.0003). Mean IQ scores were significantly higher (P<0.05) and the percentage of patients with intellectual disability was significantly lower in the preventive treatment group (P=0.031).86 No patients in the preventive treatment group developed moderate, severe, or profound intellectual disability at 24 months of age.86
Clearly identifying the likely differences between diagnosis, monitoring, and management of pediatric and adult patients with TSC is an important step in enabling efficiencies to be maximized without compromising care provided to patients. The aim should be to enable an effective continuum of care for patients with TSC and their families as they transition from childhood into adulthood. Specialized TSC transition clinics that treat both children and adults regardless of organ system involvement could improve continuity of care for all patients with TSC, but particularly the more severely affected.13 Parents are the most effective advocates for young children with TSC, but as children mature into adolescence and adulthood, the need for comprehensive specialized care in one place is increased. All TSC facilities need to have the capability of addressing both medical requirements and psychosocial issues affecting individuals and their families.13 Avoiding unnecessary testing (although, at present, only 1–2% of patients with TSC develop end stage renal disease) can further reduce costs without compromising care.13
Acknowledgments
Editorial assistance was provided by ApotheCom (funded by Novartis Pharmaceuticals Corporation).
Disclosure
Michael Frost has no conflicts of interest to disclose. John Hulbert serves on the speakers’ bureau for Novartis Pharmaceuticals Corporation, for which he may receive remuneration, and is a member of the National Scientific Advisory Board of the Tuberous Sclerosis Alliance, for which he receives no compensation.
References
Baskin HJ Jr. The pathogenesis and imaging of the tuberous sclerosis complex. Pediatr Radiol. 2008;38(9):936–952. | |
Franz DN, Bissler JJ, McCormack FX. Tuberous sclerosis complex: neurological, renal and pulmonary manifestations. Neuropediatrics. 2010;41(5):199–208. | |
Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345–1356. | |
Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–668. | |
European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75(7):1305–1315. | |
van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277(5327):805–808. | |
Sancak O, Nellist M, Goedbloed M, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype – phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet. 2005;13(6):731–741. | |
Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68(1):64–80. | |
Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412(2):179–190. | |
Krymskaya VP. Tumour suppressors hamartin and tuberin: intracellular signalling. Cell Signal. 2003;15(8):729–739. | |
Krueger D, Northrup H. TSC Clinical Consensus Conference Summary of Updates: Diagnosis, Treatment and Surveillance Guidelines. Silver Springs, MD: TSC; 2012. | |
Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):243–254. | |
Krueger DA, Northrup H; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):255–265. | |
Hallett L, Foster T, Liu Z, Blieden M, Valentim J. Burden of disease and unmet needs in tuberous sclerosis complex with neurological manifestations: systematic review. Curr Med Res Opin. 2011;27(8):1571–1583. | |
Anlauf M, Garbrecht N, Bauersfeld J, et al. Hereditary neuroendocrine tumors of the gastroenteropancreatic system. Virchows Arch. 2007; 451 Suppl 1:S29–S38. | |
Napolioni V, Curatolo P. Genetics and molecular biology of tuberous sclerosis complex. Curr Genomics. 2008;9(7):475–487. | |
Tsai JD, Wei CC, Chen SM, Lue KH, Sheu JN. Association between the growth rate of renal cysts/angiomyolipomas and age in the patients with tuberous sclerosis complex. Int Urol Nephrol. 2014;46(9):1685–1690. | |
Franz DN. Non-neurologic manifestations of tuberous sclerosis complex. J Child Neurol. 2004;19(9):690–698. | |
Bader RS, Chitayat D, Kelly E, et al. Fetal rhabdomyoma: prenatal diagnosis, clinical outcome, and incidence of associated tuberous sclerosis complex. J Pediatr. 2003;143(5):620–624. | |
Smythe JF, Dyck JD, Smallhorn JF, Freedom RM. Natural history of cardiac rhabdomyoma in infancy and childhood. Am J Cardiol. 1990;66(17):1247–1249. | |
Jóźwiak S, Kawalec W, DŁuzewska J, Daszkowska J, Mirkowicz-Malek M, Michalowicz R. Cardiac tumours in tuberous sclerosis: their incidence and course. Eur J Pediatr. 1994;153(3):155–157. | |
Tuberous Sclerosis Alliance [webpage on the Internet]. TSC national history database project. Available from: http://www.tsalliance.org/pages.aspx?content[=]558. Accessed May 6, 2014. | |
Staley BA, Vail EA, Thiele EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics. 2011;127(1):e117–e125. | |
Tiberio D, Franz DN, Phillips JR. Regression of a cardiac rhabdomyoma in a patient receiving everolimus. Pediatrics. 2011;127(5):e1335–e1337. | |
O’Callaghan FJ, Martyn CN, Renowden S, Noakes M, Presdee D, Osborne JP. Subependymal nodules, giant cell astrocytomas and the tuberous sclerosis complex: a population-based study. Arch Dis Child. 2008;93(9):751–754. | |
Koh S, Jayakar P, Dunoyer C, et al. Epilepsy surgery in children with tuberous sclerosis complex: presurgical evaluation and outcome. Epilepsia. 2000;41(9):1206–1213. | |
Weiner HL, Carlson C, Ridgway EB, et al. Epilepsy surgery in young children with tuberous sclerosis: results of a novel approach. Pediatrics. 2006;117(5):1494–1502. | |
Cusmai R, Chiron C, Curatolo P, Dulac O, Tran-Dinh S. Topographic comparative study of magnetic resonance imaging and electroencephalography in 34 children with tuberous sclerosis. Epilepsia. 1990;31(6):747–755. | |
Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51(7):1236–1241. | |
Curatolo P, Bombardieri R, Cerminara C. Current management for epilepsy in tuberous sclerosis complex. Curr Opin Neurol. 2006;19(2):119–123. | |
Thiele EA. Managing epilepsy in tuberous sclerosis complex. J Child Neurol. 2004;19(9):680–686. | |
Sooriakumaran P, Gibbs P, Coughlin G, et al. Angiomyolipomata: challenges, solutions, and future prospects based on over 100 cases treated. BJU Int. 2010;105(1):101–106. | |
Webb DW, Clarke A, Fryer A, Osborne JP. The cutaneous features of tuberous sclerosis: a population study. Br J Dermatol. 1996;135(1):1–5. | |
Józwiak S, Schwartz RA, Janniger CK, Bielicka-Cymerman J. Usefulness of diagnostic criteria of tuberous sclerosis complex in pediatric patients. J Child Neurol. 2000;15(10):652–659. | |
Hall MR, Kovach BT, Miller JL. Unilateral facial angiofibromas without other evidence of tuberous sclerosis: case report and review of the literature. Cutis. 2007;80(4):284–288. | |
Budde K, Gaedeke J. Tuberous sclerosis complex-associated angiomyolipomas: focus on mTOR inhibition. Am J Kidney Dis. 2012;59(2):276–283. | |
Ewalt DH, Sheffield E, Sparagana SP, Delgado MR, Roach ES. Renal lesion growth in children with tuberous sclerosis complex. J Urol. 1998;160(1):141–145. | |
Rakowski SK, Winterkorn EB, Paul E, Steele DJ, Halpern EF, Thiele EA. Renal manifestations of tuberous sclerosis complex: incidence, prognosis, and predictive factors. Kidney Int. 2006;70(10):1777–1782. | |
Casper KA, Donnelly LF, Chen B, Bissler JJ. Tuberous sclerosis complex: renal imaging findings. Radiology. 2002;225(2):451–456. | |
Cook JA, Oliver K, Mueller RF, Sampson J. A cross sectional study of renal involvement in tuberous sclerosis. J Med Genet. 1996;33(6):480–484. | |
Washecka R, Hanna M. Malignant renal tumors in tuberous sclerosis. Urology. 1991;37(4):340–343. | |
Al-Saleem T, Wessner LL, Scheithauer BW, et al. Malignant tumors of the kidney, brain, and soft tissues in children and young adults with the tuberous sclerosis complex. Cancer. 1998;83(10):2208–2216. | |
Bjornsson J, Short MP, Kwiatkowski DJ, Henske EP. Tuberous sclerosis-associated renal cell carcinoma. Clinical, pathological, and genetic features. Am J Pathol. 1996;149(4):1201–1208. | |
Castagnetti M, Vezzu B, Laverda A, Zampieri S, Rigamonti W. Urological counseling and followup in pediatric tuberous sclerosis complex. J Urol. 2007;178(5):2155–2159. | |
Sampson JR, Maheshwar MM, Aspinwall R, et al. Renal cystic disease in tuberous sclerosis: role of the polycystic kidney disease 1 gene. Am J Hum Genet. 1997;61(4):843–851. | |
Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc. 1991;66(8):792–796. | |
Urban T, Lazor R, Lacronique J, et al. Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM”O”P). Medicine (Baltimore). 1999;78(5):321–337. | |
Taylor JR, Ryu J, Colby TV, Raffin TA. Lymphangioleiomyomatosis. Clinical course in 32 patients. N Engl J Med. 1990;323(18):1254–1260. | |
Kitaichi M, Nishimura K, Itoh H, Izumi T. Pulmonary lymphangioleiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med. 1995; 151(2 Pt 1):527–533. | |
Castro M, Shepherd CW, Gomez MR, Lie JT, Ryu JH. Pulmonary tuberous sclerosis. Chest. 1995;107(1):189–195. | |
Ryu JH, Moss J, Beck GJ, et al; NHLBI LAM Registry Group. The NHLBI lymphangioleiomyomatosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med. 2006;173(1):105–111. | |
Aubry MC, Myers JL, Ryu JH, et al. Pulmonary lymphangioleiomyomatosis in a man. Am J Respir Crit Care Med. 2000;162(2 Pt 1):749–752. | |
Dwyer JM, Hickie JB, Garvan J. Pulmonary tuberous sclerosis. Report of three patients and a review of the literature. Q J Med. 1971;40(157):115–125. | |
Costello LC, Hartman TE, Ryu JH. High frequency of pulmonary lymphangioleiomyomatosis in women with tuberous sclerosis complex. Mayo Clin Proc. 2000;75(6):591–594. | |
Franz DN, Brody A, Meyer C, et al. Mutational and radiographic analysis of pulmonary disease consistent with lymphangioleiomyomatosis and micronodular pneumocyte hyperplasia in women with tuberous sclerosis. Am J Respir Crit Care Med. 2001;164(4):661–668. | |
Xu KF, Zhang P, Tian X, et al. The role of vascular endothelial growth factor-D in diagnosis of lymphangioleiomyomatosis (LAM). Respir Med. 2013;107(2):263–268. | |
Young LR, Vandyke R, Gulleman PM, et al. Serum vascular endothelial growth factor-D prospectively distinguishes lymphangioleiomyomatosis from other diseases. Chest. 2010;138(3):674–681. | |
Sampson JR, Attwood D, al Mughery AS, Reid JS. Pitted enamel hypoplasia in tuberous sclerosis. Clin Genet. 1992;42(1):50–52. | |
Mlynarczyk G. Enamel pitting: a common symptom of tuberous sclerosis. Oral Surg Oral Med Oral Pathol. 1991;71(1):63–67. | |
Lygidakis NA, Lindenbaum RH. Pitted enamel hypoplasia in tuberous sclerosis patients and first-degree relatives. Clin Genet. 1987;32(4):216–221. | |
Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998;13(12):624–628. | |
Cheung H, Ambrose RE, Lee PO. Liver hamartomas in tuberous sclerosis. Clin Radiol. 1993;47(6):421–423. | |
Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363(19):1801–1811. | |
Franz DN, Belousova E, Sparagna S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicenter, randomized, placebo-controlled phase 3 trial. Lancet. 2013;381(9861):125–132. | |
Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:817–824. | |
Afinitor (everolimus) tablets for oral administration [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2012. | |
Wienecke R, Fackler I, Linsenmaier U, Mayer K, Licht T, Kretzler M. Antitumoral activity of rapamycin in renal angiomyolipoma associated with tuberous sclerosis complex. Am J Kidney Dis. 2006;48(3):e27–e29. | |
Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59(3):490–498. | |
Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358(2):140–151. | |
Curatolo P, Jóźwiak S, Nabbout R; TSC Consensus Meeting for SEGA and Epilepsy Management. Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol. 2012;16(6):582–586. | |
Go CY, Mackay MT, Weiss SK, et al; Child Neurology Society; American Academy of Neurology. Evidence-based guideline update: medical treatment of infantile spasms. Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2012;78(24):1974–1980. | |
Krueger DA, Care MM, Agricola K, Tudor C, Mays M, Franz DN. Everolimus long-term safety and efficacy in subependymal giant-cell astrocytoma. Neurology. 2013;80(6):574–580. | |
Krueger DA, Wilfong AA, Holland-Bouley K, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74(5):679–687. | |
Cardamone M, Flanagan D, Mowat D, Kennedy SE, Chopra M, Lawson JA. Mammalian target of rapamycin inhibitors for intractable epilepsy and subependymal giant cell astrocytomas in tuberous sclerosis complex. J Pediatr. 2014;164(5):1195–1200. | |
Connolly MB, Hendson G, Steinbok P. Tuberous sclerosis complex: a review of the management of epilepsy with emphasis on surgical aspects. Childs Nerv Syst. 2006;22(8):896–908. | |
Chung TK, Lynch ER, Fiser CJ, et al. Psychiatric comorbidity and treatment response in patients with tuberous sclerosis complex. Ann Clin Psychiatry. 2011;23(4):263–269. | |
de Ribaupierre S, Dorfmuller G, Bulteau C, et al. Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: when should we operate? Neurosurgery. 2007;60(1):83–89. | |
Kellner DS, Ercolani MC, Isom-Batz G, Javit DJ, Armenakas NA. Renal angiomyolipoma presenting with massive retroperitoneal hemorrhage. Hosp Physician. 2004;40(12):34–36. | |
Oesterling JE, Fishman EK, Goldman SM, Marshall FF. The management of renal angiomyolipoma. J Urol. 1986;135(6):1121–1124. | |
Patel U, Simpson E, Kingswood JC, Saggar-Malik AK. Tuberose sclerosis complex: analysis of growth rates aids differentiation of renal cell carcinoma from atypical or minimal-fat-containing angiomyolipoma. Clin Radiol. 2005;60(6):665–673; discussion 663–664. | |
Pea M, Bonetti F, Zamboni G, et al. Melanocyte-marker-HMB-45 is regularly expressed in angiomyolipoma of the kidney. Pathology. 1991;23(3):185–188. | |
Hulbert JC, Graf R. Involvement of the spleen by renal angiomyolipoma: metastasis or multicentricity? J Urol. 1983;130(2):328–329. | |
Ferry JA, Malt RA, Young RH. Renal angiomyolipoma with sarcomatous transformation and pulmonary metastases. Am J Surg Pathol. 1991;15(11):1083–1088. | |
McCormack FX, Inoue Y, Moss J, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364(17):1595–1606. | |
Davies DM, de Vries PJ, Johnson SR, et al. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: a phase 2 trial. Clin Cancer Res. 2011;17(12):4071–4081. | |
Jóźwiak S, Kotulska K, Domanska-PakieŁa D, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol. 2011;15(5):424–431. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.