Back to Journals » Therapeutics and Clinical Risk Management » Volume 17
Clinical Management of Relapsed/Refractory Hemophagocytic Lymphohistiocytosis in Adult Patients: A Review of Current Strategies and Emerging Therapies
Authors Yildiz H ![]() , Bailly S, Van Den Neste E, Yombi JC
, Bailly S, Van Den Neste E, Yombi JC
Received 27 January 2021
Accepted for publication 25 March 2021
Published 14 April 2021 Volume 2021:17 Pages 293—304
DOI https://doi.org/10.2147/TCRM.S195538
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Halil Yildiz,1 Sarah Bailly,2 Eric Van Den Neste,2 Jean Cyr Yombi1
1Departement of Internal Medicine and Infectious Diseases, Cliniques Universitaires Saint Luc, Bruxelles, Belgique; 2Departement of Hematology, Cliniques Universitaires Saint Luc, Bruxelles, Belgique
Correspondence: Halil Yildiz
Departement of Internal Medicine and Infectious Diseases, Cliniques Universitaires Saint Luc, 10 Avenue Hippocrate, Bruxelles, 1200, Belgique
Email [email protected]
Introduction: Haemophagocytic lymphohistiocytosis (HLH) is a severe disorder with high mortality. The aim of this review is to update clinical management of relapsed/refractory HLH in adults, with a focus on current and new therapies.
Methods: We searched relevant articles in Embase and PUBMED with the MESH term “hemophagocytic lymphohistiocytosis; refractory; relapsing; adult.”
Results: One hundred eight papers were found; of these, 22 were retained for this review. The treatment of HLH in adult is based on the HLH-94 regimen. The response rate is lower than in pediatric patients, and 20– 30% are refractory to this therapy. DEP regimen and allogenic hematopoietic stem cell transplantation (HSCT) are associated with complete response and partial response in 27% and 49.2%, respectively. However, many patients fail to achieve a stable condition before HSCT, and mortality is higher in them. New drugs have been developed, such as emapalumab, ruxolitinib, and alemtuzumab, and they may be used as bridges to the curative HSCT. They are relatively well tolerated and have few or mild side effects. With these agents, the rate of partial response ranges from 14.2% to 100%, while the rate of complete response is highly variable according to study and medication used. The number of patients who achieved HSCT ranged from 44.8% to 77%, with a survival rate of 55.9% to 100%. However, the populations in these studies are mainly composed of mixed-age patients (pediatric and adult patients), and studies including only adult patients are scarce.
Conclusion: Relapsed or refractory HLH in adult patients is associated with poor outcome, and consolidation with HSCT may be required in some cases. Mortality related to HSCT is mainly due to active HLH disease before HSCT and post HSCT complications. New drugs, such as empalumab, ruxolitinib, and alemtuzumab are interesting since these agents may be used as bridges to HSCT with increases in the numbers of patients proceeding to HSCT and survival rate.
Keywords: haemophagocytic lymphohistiocytosis, refractory, doxorubicin, etoposide, methylprednisolone regimen, hematopoietic stem cell transplantation, emapalumab
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a severe disorder with high cytokine release and activation of macrophages, cytotoxic T lymphocytes, and NK cells, which can lead to multiple organ dysfunction and death.1–4 The classification of HLH into a primary form (mostly seen in children and secondary to genetic defects in several genes) and a secondary form (acquired, mostly seen in adults and secondary to infectious, malignant, and autoimmune diseases), is useful and easy to understand. However, this classification does not represent correctly the complexity of the genetic defects in HLH, in which biallelic severe mutations lead to earlier onset HLH, and mild mutations lead to later onset HLH.5–10 Moreover, monoallelic mutations or gene polymorphisms in genes associated with familial HLH have been found in patients with infectious, rheumatologic, and malignant diseases related HLH.11,12 For these reasons, it should be better to classify patients according to familial HLH (genetic); malignancy-related HLH; rheumatologic disease-related HLH; drug-induced HLH (such as immune check point inhibitor); and immune-compromised HLH. Patients without genetic mutations or with mutations of unknown significance should be named « HLH disease-NOS ». Since familial HLH can also be triggered by infections, HLH occurring with infectious diseases should also be classified as «HLH disease NOS».10 Macrophage activation syndrome (MAS) is the preferred term when HLH is secondary to autoimmune or autoinflammatory disease.
In adult HLH, the reported mortality rate varies widely, from 26.5% to 74.8%.3 Infectious, autoimmune, and malignancy diseases should be excluded, and disease-specific treatment must be given as soon as possible. Corticosteroids combined or not with etoposide is the cornerstone of treatment, but new therapies are emerging and are under investigation, especially in relapsed/refractory cases.4
The objective of this review is to update clinical management of relapsed/refractory HLH in adults, with a focus on current and new therapies. Primary HLH will be the term used for patients with genetic mutations. “MAS” will be used for HLH secondary to rheumatologic diseases, and “secondary HLH” will be used for HLH related to other diseases (infection, malignancy).
Methods
We searched published articles in Pubmed and EMBASE with the term « hemophagocytic lymphohistiocytosis; refractory; relapsed; adult ». Papers not related to relapsed/refractory HLH and those concerning pediatric patients were excluded. One hundred eight papers were found. Of these, 86 articles were excluded for the following reasons: pediatric patients [n=12], case reports [n=22], not related to relapsed/refractory HLH treatment [n=40], or duplicate manuscripts [12]. Finally, 22 papers were retained for this review.
Pathogenesis of HLH
Hereditary HLH is characterised by hyperactivation of cytotoxic T-lymphocytes (CTLs), NK-cell dysfunction, and overproduction of interferon gamma (IFN-γ). In normal situations, CTLs are able to kill antigen-presenting dendritic cells using the perforin-dependent pathway. In HLH, these dendritic cells are persistent and responsible for abnormal activation of CTLs.13 Other cells that play an interesting role are regulatory T cells (Tregs) and NK-cells. Tregs are normally responsible for elimination of CTLs by using perforin and granzyme-dependent pathways. In primary HLH, activated CTLs express high levels of CD25 in their membranes, leading to IL-2 consumption and downregulation of Tregs.1 NK-cell dysfunction leads to uncontrol activation and proliferation of CTLs. Activated CTLs produce high levels of IFN-γ, which then stimulate macrophages and lead to high production of cytokines such as IL1β, IL6 and TNFα. Recently, a novel cytokine (IL33) has been described. IL33 seems to induce CTL activation and IFN-γ production by using the MyD88 pathway.14
HLH in adult patients (nonhereditary) is mainly triggered by infection, malignancy, and autoimmune/autoinflammatory disease but the pathogenesis is complex and not fully understood since genetic defect are also found in adult patients. These patients may have reduced NK cell and CTL function, as in familial HLH. The responsible mechanism seems to be secondary to acquired low expression of perforin. The high cytokine levels, classically encountered in HLH, are thought to be responsible for decreased NK cell and CTL cytotoxicity. Interestingly, many infectious disease such as Epstein-Barr virus (EBV), H5N1, and HSV-1 and tumour cells are also able to suppress CTLs and NK cell cytotoxicity.15,16 More recently, Carvelli et al showed that most patients with adult HLH have lymphopenia but normal NK cell function and reduced levels of IFN-γ, which means that there are other explanations for disease occurrence, such as chronic activation of Toll-like receptors, high exposure to IL6, and increased production and exposure to IL18.12 Carvelli et al analyzed genes implicated in HLH and did not find biallelic mutations, but they identified a variant of uncertain significance in 50% of patients.12
Genetic Alterations in Adult HLH
In primary HLH, 9 genes are associated: PRF1, UNC13D, STX11, STXBP2, RAB27A, LYST, SH2D1A, BIRC4, and NLRC4.4–10
The gene mutation in type 1 familial HLH (FHL type 1) is unknown. Mutations in PRF1 are responsible for a defect in perforin. Approximately 30–40% of patients with primary HLH carry this mutation, which is responsible for FHL type 2. Up to 9% of the population carry a polymorphism in this gene, so genetic analysis should be interpreted carefully and consider the clinical characteristics of the patients.
Mutations of UNC13D and STX11 are responsible for FHL type 3 and 4, respectively, and account for 30% and 5% of FHL. These mutations are responsible for a defect in the cytotoxic exocytosis pathway.
Mutations in STXBP2 are classified as FHL type 5 and account for 5–20% of FHL. Mutations in this gene are responsible for defects in the cytotoxic exocytosis pathway, which is slightly different clinically from other FHL since patients may present with enteropathy and sensorineural hearing loss. Other diseases, such as X-linked severe combined immunodeficiency syndrome (X-SCID, XLP1 Duncan disease [mutation in SH2D1A gene]), Chediak-Higashi syndrome (mutation in LYST gene), Griscelli type 2 (mutation in RAB27A gene), BIRC4 and XLP2 syndrome (NLRC4) have been associated with HLH. Griscelli type 2 and Chediak-Higashi diseases are associated with defects in the cytotoxic exocytosis pathway and clinically present with albinism. Duncan disease (Purtilo syndrome) is an X-linked lymphoproliferative disease characterized by severe immune reaction secondary to EBV infection, with bone marrow failure, hepatitis, and lymphoma. XLP2 syndrome is also an X-linked lymphoproliferative disease secondary to mutations in XIAP (BIRC4) associated with defects in the inflammasome pathway such as BIRC4.
In secondary HLH, it has been suggested that there are no genetic mutations, although mutations may be found in adult patients. Heterozygous mutations or polymorphisms have been found in genes associated with primary HLH, such as PRF-1, UNC13D, STX11, STXBP2, RAB27A, SH2D1A, LYST, and NLRC4.7,11,17–20 PRF-1 polymorphism is found in 9% of the general population, which raises concern about the real implication of this gene polymorphism, but probably it is responsible for dysfunction in T-cell cytotoxicity. Carvelli et al, recently found that the PRF-1 A91V variant was present in 50% of adult patients with secondary HLH,12 which is in line with findings of others.6,17,18
For these reasons, Brisse et al proposed a model for HLH in which several factors (genetic, infection, malignancy, autoimmune disease, inflammatory disease, immunodeficiency) are combined and, when a threshold is reached, an uncontrolled inflammatory reaction occurs.10
Causes of Adult HLH
Infections (such as EBV, cytomegalovirus, HIV, and Mycobacterium tuberculosis), lymphoma, and autoimmune/autoinflammatory diseases are the triggers that are most frequently associated with adult HLH. If a trigger is not found, HLH is considered idiopathic. Table 1 lists the main causes that have been reported.8,10,21
 |
Table 1 Etiology of Secondary HLH in Adult Patients |
Infection
Viral Infection
The most frequent viral infections are primary or reactivation of EBV (43%) and cytomegalovirus (9%) infection. Other viral infections, such as HIV, Dengue, influenza, herpes simplex virus, varicella zoster, parvovirus B19, and hepatitis have been associated with HLH (Table 1).8,10,21 In patients with HIV and HLH, attention should be given to the possible association of malignancy and opportunistic infections.22
Bacterial Infection
The most frequent bacterial infection associated with HLH is Mycobacterium tuberculosis, with more than 70 cases published and a high mortality rate in untreated patients.8,10,21 Other bacterial infections have been associated with HLH, such as rickettsia, brucella, coxiella, ehrlichia (Table 1).
Fungal and Parasitic Infection
Rarely, fungal and parasitic infections associated with secondary HLH have been described, but mainly in patients with immunodeficiency.8,10,21 The most frequent fungal infections are histoplasma, candida, and cryptococcus. Parasitic infections, such as leishmania, and plasmodium infection, should be ruled out in patients who have traveled in developing countries. Fungal and parasite infection associated with HLH are listed in Table 1.
Malignancy
The most frequent malignant diseases described with HLH are lymphomas, but HLH secondary to solid cancers also have been described. The frequency seems to be 1% among patients with haematological diseases and 20% among patients with large B-cell intravascular lymphoma and T-cell lymphoma.23
Autoimmune/Autoinflammatory Diseases
When HLH is secondary to autoimmune or autoinflammatory diseases, macrophage activation syndrome (MAS) is the preferred term.3,8,10,21,24 The most frequent autoimmune/autoinflammatory diseases associated with HLH are systemic lupus erythematosus (prevalence 4%) and Still’s disease (12%). Table 1 lists autoimmune and autoinflammatory diseases associated with HLH.
Drugs
Drug-related HLH is rare and is summarized in Table 1.8 Immune checkpoint inhibitors are commonly used for treatment of solid cancers, and a few cases of HLH secondary to nivolumab, pembrolizumab, ipilimumab, and nivolumab have been described.25,26 The mechanism is not understood, but it seems that immune checkpoint inhibitors can favor differentiation of T regulatory cells and induction of T cell tolerance by upregulation of the indoleamine-pyrrole 2,3-dioxygenase pathway.27,28
Others: Post-Organ Transplantation, Pregnancy
HLH has been described also with pregnancy and haematologic or solid-organ transplantation.3,21,29,30 The frequency of HLH seems to be higher with haemopoietic stem-cell transplantation than with umbilical cord blood transplantation and autologous/allogenic transplantation.21,29 HLH has been described after solid-organ transplantation such as kidney and liver transplantation and seems to be mainly related to opportunistic infection (herpedae virus infection, tuberculosis, toxoplasmosis, bartonella and Pneumocystis jiroveci infection) and post-transplant lymphoproliferative disease.30
Diagnosis of HLH in Adults
HLH-2004 criteria and the HLH-probability calculator (HScore) are the most useful criteria. According to the HLH-2004 diagnostic criteria, individuals must have a genetic analysis compatible with HLH or meet ≥ 5 of 8 diagnostic criteria. However, the HLH-2004 criteria have been mostly validated in studies including children, with adult patients less represented.4 The HScore has been developed retrospectively in adult patients and is easy to calculate using the link http://saintantoine.aphp.fr.score/).31 Table 2 shows the main characteristics of these two scores. A retrospective study compared the HScore and HLH-2004 criteria in both pediatric and adult patients and found that the HScore has a better diagnostic sensitivity and specificity than do the HLH-2004 criteria.32
 |
Table 2 HLH-2004 Diagnostic Criteria and HScore for the Diagnosis of HLH |
Distinguishing HLH from sepsis may be difficult. Chaturvedi et al described the usefulness of flow cytometry to differentiate between HLH from sepsis. They showed that patients with HLH exhibit high level of activated T cells (CD 38 high/HLADR+) with activation of CD8+ T cells, and that a threshold of >7% CD 38 high/HLADR+ cells among CD8+ T cells has a strong positive and negative predictive value for distinguishing HLH from sepsis.33
Identifying patients with genetic HLH (primary HLH) is challenging, and NK-cell function test has been used for several years.34 More recently, perforin expression and CD107a upregulation tests have been compared to NK-cell function test; perforin expression and CD107a expression tests were more accurate in identifying patients with genetic HLH.35 These tests should be proposed for patients with a familial history of HLH, partial albinism, or relapsing HLH; in young patients with adult EBV-HLH or lymphoproliferation; or in idiopathic HLH.4 However, these tests (NK function test, perforin expression, CD107a) and, even genetic tests, are not easily available and are only performed in specialized centers. Recently, Merli et al described the use of an easy and widely available test, QuantiFeron-TB Gold (QFT-G) to detect patients with primary HLH.36 As described before, IFN-γ play a key role in the pathogenesis of primary HLH (1, 10, 37). Merli et al showed that QFT-G test results were intermediate in all children with primary HLH. This was explained by the high serum IFN-γ levels, which characterized patients with primary HLH.36 Even though some diseases, such as SLE and dermatomyositis may also present with high serum IFN-γ levels, this simple test may be a good diagnostic test to identify patients in whom a genetic mutation is suspected.
Treatment of Adult HLH
Systemic corticosteroids (typically dexamethasone) are the cornerstone of treatment. When more aggressive therapy is necessary, etoposide-based HLH-94 and HLH-2004 protocols are commonly used.4 In the HLH-94 regimen, corticosteroids, etoposide, cyclosporine A, and intrathecal therapy (methotrexate) are used. Etoposide should be given once a week with or without a reduced dose (150 mg/m2 or 50–100 mg/m2). Reduced doses of etoposide should be considered for adult patients who have comorbidities in whom chemotherapy may cause organ damage.38 Intrathecal drug infusion therapy is recommended only in cases of progressive neurological manifestations or if there is no improvement in the cerebrospinal fluid findings. Cyclosporine A is used after 8 weeks in HLH-94, which differs from the HLH-2004 protocol, were cyclosporine A was administered at week 1.4 HLH-94 remains the standard of care because HLH-2004 failed in improving the outcome. Many patients with secondary HLH required <8 weeks of etoposide and the need to continue etoposide therapy should be evaluated weekly.4 Patients with active disease after 8 weeks of etoposide should be given maintenance treatment according to the HLH-94 protocol. Consolidation with HSCT should be proposed in appropriately selected patients if a familial form is identified and in relapsed, refractory cases.39 Patients treated with HLH-94 and HSCT treatment had a 5-year survival rate of 54%; thus, a consolidation treatment should be given when necessary, as in primary HLH, relapsing HLH, and high-risk hematological malignancies.
In malignant disease-associated HLH (Mal-HLH), corticosteroids are used to treat inflammation. If the HLH is not highly active and not associated with organ damage, malignancy-specific treatment should be proposed. Otherwise, reduced dose of etoposide (50–100 mg/m2) should be considered.40 Since lymphomas are the main cause of Mal-HLH, a CHOP-like protocol (CHOEP [cyclophosphamide, doxorubicin, vincristine, etoposide, prednisone] or DA-EPOCH [dose-adapted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin]) can be used.41 More recently, Guangqiang et al showed that the use of DEP regimen (doxorubicin, etoposide, methylprednisolone), as first-line therapy, was superior to HLH-94 regimen in patients with lymphoma-associated HLH.42 In younger HLH patients with EBV-associated lymphoma, genetic testing can be useful for excluding HLH-associated mutations since this may be an indication to perform HSCT. HLH sometimes develops during chemotherapy, making the diagnosis difficult since cytopenia and liver function abnormalities may be secondary to the chemotherapy. Corticosteroids (dexamethasone, prednisolone) are possible therapeutic options, but etoposide use should be avoided since bone marrow reconstitution is important in that context. Moreover, since these patients are neutropenic, infection should always be excluded before giving any HLH treatment.43
In infectious disease-associated HLH, viral infections such as EBV, cytomegalovirus, and HIV are the most common triggers. In severe EBV-HLH, glucocorticoids combined with rituximab should be the preferred treatment. In HIV-associated HLH (HIV-HLH), physicians must carefully excluded lymphomas and opportunistic infections, since these conditions are frequent in people living with HIV/AIDS.22 The prognosis of HIV-HLH has improved with the use of highly active antiretroviral treatment, and corticosteroids should be given only in hyperinflammatory cases.4 In HLH due to infectious agents affecting the mononuclear phagocyte system such as rickettsia, brucella and coxiella, treatment with corticosteroids should be avoided since these pathogens respond well to disease-specific treatment.4
In cases due to rheumatological diseases (macrophage activation syndrome, MAS-HLH), corticosteroids are the preferred treatment, followed by an anti-IL-1 or anti-IL-6 drug when the immediate response is insufficient.4
Assessment of Response
Despite treatment of adult HLH with the HLH-94 regimen, the response rate is lower than in pediatric patients, in whom 70–80% have at least a partial response to dexamethasone and etoposide. However, 20–30% are refractory to this therapy, and guidelines for second-line therapy are lacking. There is no clear definition of refractory HLH, but the absence of response (partial and/or complete) at 2–3 weeks, after standard HLH therapy, is often suggestive of refractory HLH.4,37,44 A complete response is characterized by disappearance of all symptoms and normalization of HLH biological signs, such as ferritin, triglycerides, hemoglobin, neutrophil counts, glutamate pyruvate transaminase, and sCD25 [if available]). However, there might be a lag in clinical and biomarker responses, such as the decline of ferritin level, which might take much longer despite treatment. A partial response is generally defined as an improvement of at least ≥2 symptoms and laboratory markers within 2 weeks after initiation of treatment.37,44,45 Since HLH patients frequently are receiving transfusion (red blood cells, platelets), these biomarkers may be difficult to interpret, and other indicators may be used, such as bilirubin, albumin, and fibrinogen.37,44,45
When dealing with relapsed/refractory HLH, especially idiopathic HLH, it is important that a secondary cause has not been missed. Since infections and malignancies are the main causes of secondary HLH, a thoraco-abdominal CT scan and/or 18 fluorodeoxyglucose-positron-computer tomography (18FDG-PET/CT) may be useful. The usefulness of 18F-FDG-PET/CT has been demonstrated in recent studies, and it seems that PET/CT is a helpful diagnostic tool for identifying the trigger (infectious or malignancy disease) and helping clinicians identify the hypermetabolic areas where biopsies may be performed to establish a diagnosis.46,47
Hematopoietic Stem Cell Transplantation (HSCT)
Consolidation with HSCT should be proposed in appropriately selected patients if a familial form (primary HLH) is identified and/or in relapsed, refractory cases. In primary HLH, allogeneic HSCT is the only definitive cure. Henter et al described the outcome of children undergoing HSCT, finding that the 5-year survival was 54%.48 Mortality in patients undergoing HSCT is mainly transplant-related (pulmonary complications, graft-versus-host disease, and infection). Matched related donors and matched unrelated donors are associated with better survival after HSCT, whereas mismatched unrelated donors and haploidentical donors are associated with poorer survival, although the results are still acceptable and the use of such donors should be proposed when matched donors are not available. Active HLH disease at the time of HSCT seems to portend poor outcomes and is associated with higher mortality.49 For that reason, HLH-94 induction therapy is followed by continuation therapy (alternating weekly pulses of VP-16 and dexamethasone combined with cyclosporin A). Patients should also have conditioning regimen (busulfan-containing myeloablative conditioning or reduced intensity regimen [alemtuzumab, fludarabine, melphalan]) and graft vs host disease prophylaxis before HSCT is performed.4 A reduced-intensity regimen seems to be associated with lower transplant-related mortality but also has higher rates of graft failure and relapse.50
If HSCT is indicated in primary HLH, it does not mean that every patient with genetic mutation should undergo HSCT. In fact, the finding of a heterozygous or homozygous A91V variant in the PRF1 gene is not an indication for HSCT, except if the variant is associated with other severe mutations.
There is a lack of prospective trials in adult HLH patients, and most studies have been done in children. Use of HSCT should be considered in case of recurrent/refractory disease. As previously described, EBV- and lymphoma-associated HLH are the major types of HLH in adult patients, and they may be associated with relapsing/refractory disease. HSCT may be performed in these patients even with haploidentical donors.51,52
Treatment of Relapsed/Refractory Adult HLH
No standardized treatment exists for relapsed or refractory hemophagocytic lymphohistiocytosis.
Chemotherapeutic Agents and HSCT
The DEP regimen and HSCT have been effective as salvage therapy for adult refractory HLH.4 Wang et al found, in a multicenter study with the DEP regimen, that complete response was achieved in 27% (17/63) of patients and partial response in 49.2% (31/63).44 Wang et al investigated the efficacy of a modified DEP regimen in combination with PEG-aspargase (L-DEP) in 28 patients with refractory EBV-HLH.53 Complete and partial response was achieved in 32.1% and 53.5%, respectively, and thirteen patients then received HSCT. The major side effects of the L-DEP regimen were high serum amylase concentrations, abnormal liver tests, and coagulation disorders. However, the mortality rate was higher in patients who did not reach a stable condition before HSCT was performed. Mortality associated with HSCT is mainly due to post-HSCT complications (graft failure, veno-occlusive disease, and pulmonary complications) and active HLH disease before HSCT.4,54
Non-Chemotherapeutic Agents
There is a need for effective salvage therapies that can control the disease and be a bridge to the curative HSCT.4,54 New drugs such as emapalumab, ruxolitinib, nivolumab, and alemtuzumab have been developed and may be therapeutic options.
Emapalumab is an antibody that competitively blocks free interferon gamma and binds also to IFN-γ bound to its receptor, thus avoiding signal transduction. The safety and efficacy of emapalumab were confirmed in a trial with 34 patients (mainly pediatric patients) with treatment-naïve or relapsed/refractory HLH.55 Patients with active Mycobacterium, Shigella, Histoplasma, Leishmania, Salmonella, or Campylobacter infection were excluded (since these infections are associated with IFN-γ neutralization), as well as were patients with rheumatologic or malignant disease. Emapalumab has been approved by the United States Food and Drug Administration since November 2018 for treatment of adult or pediatric primary HLH that is refractory, relapsing, or intolerant to conventional therapy. Emapalumab is given intravenously at a dose of 1 mg/kg combined with dexamethasone (5–10 mg/m2). Emapalumab is given twice per week, and the dose can be raised to 10 mg/kg according to patients’ tolerance and clinical evolution. A latent tuberculosis infection should always be excluded with the interferon-gamma release-assay prior to giving emapalumab infusion, and monitoring for EBV and cytomegalovirus infections by blood PCR should be performed every 2 weeks. Patients should receive prophylaxis also for herpes zoster and Pneumocystis jiroveci with acyclovir and trimethoprim-sulfamethoxazole, respectively.56 However, targeting a single cytokine (interferon gamma) with emapalumab may not be sufficient, and targetting multiple cytokines simultaneously may be tried.57
Ruxolitinib is a Janus Kinase inhibitor that selectively targets JAK1 and 2 pathways activated by several cytokines such as interferon gamma, interleukin 2, and interleukin 6, which are elevated in HLH.56 The first case of ruxolitinib used in an adult HLH patient was described by Sin et al in a 38-year-old woman with EBV-related HLH.58 Wang et al studied the effectiveness of ruxolitinib in pediatric and adult patients and showed that the overall response rate was as high as 73.5%, with a complete and partial response rate of 14.7% and 58.8%, respectively.59 The first study that included exclusively adult patients was published by Ahmed et al and included 5 adult patients with secondary HLH. Ruxolitinib was given orally at a dose of 15 mg twice daily during 28-day cycle or until progression of HLH or occurrence of severe toxicity.60 Ruxolitinib was associated with a good clinical response and low side effects, and it was easily managed in the clinic; only one patient developed grade 4 neutropenia. More data concerning longer follow-up with 7 more patients has just been published; an overall response rate of 77% and complete and partial response rates of 38.4% were found.61 A larger study was a multicenter prospective study which investigated the efficacy of ruxolitinib combined with a DEP regimen (DEP-Ru) in relapsed/refractory HLH; 54 patients were included, of whom 28 had EBV-related HLH, 5 had MAS-HLH, 3 had Mal-HLH, 6 had familial HLH, 1 had pregnancy-HLH, 1 had drug-HLH, and 2 had infection.62 Complete and partial response was achieved in 15.1% and 58.5%, respectively. The overall response rate was 73.6%. DEP-Ru seems to have higher efficacy in MAS-HLH and idiopathic HLH than in EBV- or lymphoma-associated HLH.62
Nivolumab, a PD-1 inhibitor, approved for the treatment of several cancers has been effective in EBV+ gastric cancer and NK/T lymphoma.63 Liu et al described retrospectively the outcome of 7 patients treated with nivolumab for relapsing/refractory EBV-associated HLH.64 All 7 patients tolerated the treatment, and complete clinical response was achieved in 5 of them (72%). The authors also performed an RNA-sequencing analysis of HLH-related genes and found that CD27 and STXBP2 (which are 2 HLH-related genes) were not upregulated in hyperactive CD8 T cells in EBV-HLH. In the HLH-94 regimen, these T cells are targeted by etoposide, but they are useful to fight against EBV. Nivolumab has the advantage of restoring the expression of HLH-associated degranulation and costimulatory genes in CD8 T cells. Nivolumab can be considered only for EBV-driven HLH, and further studies are needed to determine the efficacy and safety of nivolumab in EBV-related HLH compared with the HLH-94 regimen, DEP, ruxolitinib, and emapalumab. Moreover, since immune checkpoint inhibitors have been associated with HLH, choosing another therapeutic agent may be more prudent.
Alemtuzumab, a monoclonal antibody to the CD52 antigen, is another drug of interest for the treatment of adult HLH. CD52 is expressed on T cells, NK cells, and B cells, which are hyperactivated in HLH. In a retrospective study of 22 patients (17 pediatric and 5 adult patients) with refractory HLH treated with alemtuzumab, the rate of partial response was 64%, with no patient achieving complete response.45 Cytomegalovirus and adenovirus bloodstream infection were the most important side effects with alemtuzumab therapy. However, since alemtuzumab is also active on monocytes, cases of HLH secondary to alemtuzumab have been described, such as in patients with multiple sclerosis.65
The main characteristics and outcomes of studies with these new therapeutic regimens are summarized in Table 3. The studies included pediatric and adult patients; data from studies exclusively of adult patients are scarce. However, interesting trials are ongoing with these alternative and new therapeutics, especially trials with adult HLH patients: ruxolitinib (NCT03795909 [ruxolitinib combined or not with dexamethasone]; NCT04551131 [newly diagnosed HLH and relapsed/refractory HLH]; NCT03533790 [DEP-Ruxolitinib]; NCT04120090 [low-dose vs high-dose ruxolitinib in adult and pediatric patients with relapsed/refractory HLH]); anakinra (IL-1 blockade; NCT02780583 [patients with MAS in the context of systemic juvenile idiopathic arthritis]); alemtuzumab (NCT02472054 [familial HLH only], NCT02385110 [etoposide and dexamethasone combined or not with alemtuzumab/tocilizumab for adult HLH]); and emapalumab (NCT01818492 [patients <18 years old only], NCT03985423 [adult HLH]).
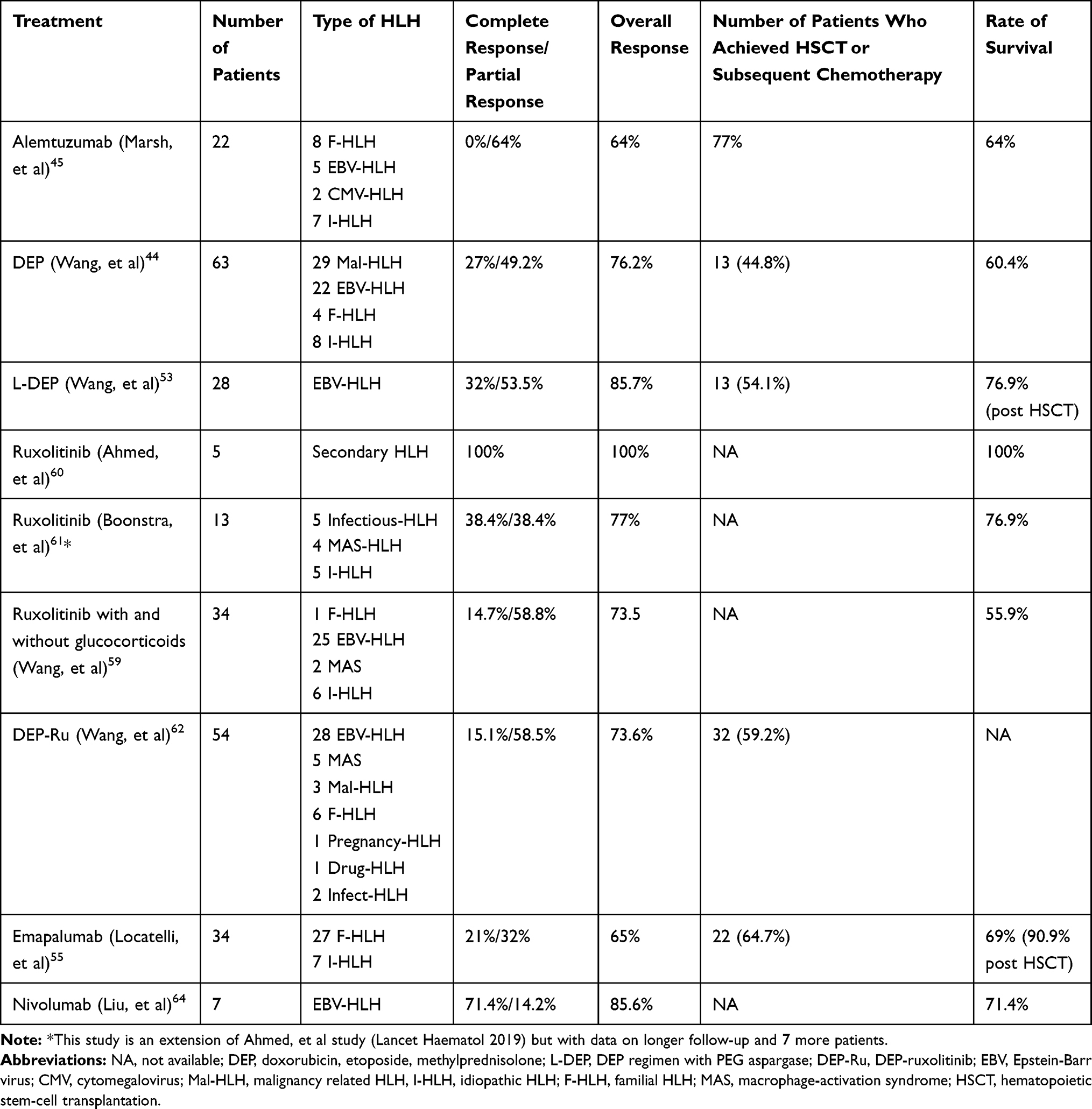 |
Table 3 Characteristics of Studies on Salvage Therapy for Refractory/Relapsed Adult HLH |
Other treatment strategies have been attempted with rituximab and infliximab but only in case reports or small case series.66,67 Nonetheless, these anecdotal reports highlight the therapeutic potential of cytokine-targeted therapies in HLH. Plasma exchange has been used as a salvage therapy for refractory HLH patients in intensive care units but much more as supportive care.68
Conclusion
Relapsed or refractory HLH in adult patients is associated with poor outcomes, and HSCT may be necessary in some cases, such as in EBV-related HLH, lymphoma-related HLH and even in late-onset primary HLH. HSCT with matched related donors and unrelated donors is associated with better outcome, but haploidentical donors can be used if matched donors are not available. Mortality with HSCT is mainly due to active HLH disease before HSCT and post HSCT complications, but studies in adult patients are lacking. The use of new drugs, such as empalumab, ruxolitinib, nivolumab, and alemtuzumab, will be interesting, as rates of partial response range from 14.2% to 100%, and the rate of complete response is variable according to the study performed and the medication used; the number of patients proceeding to HSCT ranged from 44.8% to 77%, with a survival rate of 55.9% to 100%. These studies are mainly composed of mixed-age patients (pediatric and adult patients), and studies including only adult patients are scarce. Studies are ongoing with these new therapies in treatment-naive primary/secondary HLH and in relapsed/refractory/HLH in adults. Although usually not relapsing, drug induced HLH-like cytokine storm has been described with immune checkpoint inhibitors (such as nivolumab) and alemtuzumab, so these drugs should be used with caution.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Humblet-Baron S, Franckaert D, Dooley J, et al. FN-γ and CD25 drive distinct pathologic features during hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. 2019;143(6):2215–2226.e7. doi:10.1016/j.jaci.2018.10.068
2. Janka GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematol Am Soc Hematol Educ Program. 2013;2013(1):605–611. doi:10.1182/asheducation-2013.1.605
3. Yildiz H, Van Den Neste E, Defour JP, Danse E, Yombi JC. Adult haemophagocytic lymphohistiocytosis: a review. QJM. 2020. doi:10.1093/qjmed/hcaa011
4. La Rosée P, Horne AC, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465–2477. doi:10.1182/blood.2018894618
5. Miao Y, Zhu HY, Qiao C, et al. Pathogenic gene mutations or variants identified by targeted gene sequencing in adults with hemophagocytic lymphohistiocytosis. Front Immunol. 2019;10:395. doi:10.3389/fimmu.2019.00395
6. Zhang K, Jordan MB, Marsh RA, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult onset familial HLH. Blood. 2011;118(22):5794–5798. doi:10.1182/blood-2011-07-370148
7. Chen X, Wang F, Zhang Y, et al. Genetic variant spectrum in 265 Chinese patients with hemophagocytic lymphohistiocytosis: molecular analyses of PRF1, UNC13D, STX11, STXBP2, SH2D1A, and XIAP. Clin Genet. 2018;94(2):200–212. doi:10.1111/cge.13363
8. Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503–1516. doi:10.1016/S0140-6736(13)61048-X
9. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20–26. doi:10.1016/j.coi.2017.08.004
10. Brisse E, Wouters CH, Matthys P. Advances in the pathogenesis of primary and secondary haemophagocytic lymphohistiocytosis: differences and similarities. Br J Haematol. 2016;174(2):203–217. doi:10.1111/bjh.14147
11. Zhang M, Behrens EM, Atkinson TP, Shakoory B, Grom AA, Cron RQ. Genetic defects in cytolysis in macrophage activation syndrome. Curr Rheumatol Rep. 2014;16(9):439–446. doi:10.1007/s11926-014-0439-2
12. Carvelli J, Piperoglou C, Farnarier C, et al. Functional and genetic testing in adults with HLH reveals an inflammatory profile rather than a cytotoxicity defect. Blood. 2020;136(5):542–552. doi:10.1182/blood.2019003664
13. Terrell CE, Jordan MB. Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8(+) T cells and dendritic cells. Blood. 2013;121(26):5184–5191. doi:10.1182/blood-2013-04-495309
14. Rood JE, Rao S, Paessler M, et al. ST2 contributes to T cell hyperactivation and fatal hemophagocytic lymphohistiocytosis in mice. Blood. 2016;127(4):426–435. doi:10.1182/blood-2015-07-659813
15. Mao H, Tu W, Qin G, et al. Influenza virus directly infects human natural killer cells and induces cell apoptosis. J Virol. 2009;83(18):9215–9222. doi:10.1128/JVI.00805-09
16. Jenkins MR, Rudd-Schmidt JA, Lopez JA, et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med. 2015;212(3):307–317. doi:10.1084/jem.20140964
17. Vastert SJ, van Wijk R, D’Urbano LE, et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology (Oxford). 2010;49(3):441–449. doi:10.1093/rheumatology/kep418
18. Kaufman KM, Linghu B, Szustakowski JD, et al. Whole exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheum. 2014;66(12):3486–3495. doi:10.1002/art.38793
19. Sieni E, Cetica V, Piccin A, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7(9):e44649. doi:10.1371/journal.pone.0044649
20. Cetica V, Sieni E, Pende D, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: report on 500 patients from the Italian registry. J Allergy Clin Immunol. 2016;137(1):188–196.e4. doi:10.1016/j.jaci.2015.06.048
21. Al-Samkari H, Berliner N. Hemophagocytic lymphohistiocytosis. Annu Rev Pathol Mech Dis. 2018;13(1):
22. Fazal F, Gupta N, Mittal A, Ray A. Haemophagocytic lymphohistiocytosis in human immunodeficiency virus: a systematic review of literature. Drug Discov Ther. 2020;14(5):226–231. doi:10.5582/ddt.2020.03069
23. Machaczka M, Vaktnäs J, Klimkowska M, Hägglund H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma. 2011;52(4):613–619. doi:10.3109/10428194.2010.551153
24. Ravelli A, Davì S, Minoia F, Martini A, Cron RQ. Macrophage Activation Syndrome. Hematol Oncol Clin North Am. 2015;29(5):927–941. doi:10.1016/j.hoc.2015.06.010
25. Sadaat M, Jang S. Hemophagocytic lymphohistiocytosis with immunotherapy: brief review and case report. J Immunother Cancer. 2018;6(1):49. doi:10.1186/s40425-018-0365-3
26. Laderian B, Koehn K, Holman C, Lyckholm L, Furqan M. Association of hemophagocytic lymphohistiocytosis and programmed death 1 checkpoint inhibitors. J Thorac Oncol. 2019;14(4):e77–e78. doi:10.1016/j.jtho.2018.11.035
27. Moon YW, Hajjar J, Hwu P, Naing A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J Immunother Cancer. 2015;3(1):51. doi:10.1186/s40425-015-0094-9
28. Put K, Brisse E, Avau A, et al. IDO1 deficiency does not affect disease in mouse models of systemic juvenile idiopathic arthritis and secondary hemophagocytic lymphohistiocytosis. PLoS One. 2016;11(2):e0150075. doi:10.1371/journal.pone.0150075
29. Hayden A, Park S, Giustini D, Lee AY, Chen LY. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: a systematic scoping review. Blood Rev. 2016;30(6):411–420. doi:10.1016/j.blre.2016.05.001
30. Karras A, Thervet E, Le Meur Y, Baudet-Bonneville V, Kessler M, Legendre C. Successful renal retransplantation after post-transplant lymphoproliferative disease. Am J Transplant. 2004;4(11):1904–1909. doi:10.1111/j.1600-6143.2004.00562.x
31. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613–2620. doi:10.1002/art.38690
32. Debaugnies F, Mahadeb B, Ferster A, et al. Performances of the H-score for diagnosis of hemophagocytic lymphohistiocytosis in adult and pediatric patients. Am J Clin Pathol. 2016;145(6):862–870. doi:10.1093/ajcp/aqw076
33. Chaturvedi V, Marsh RA, Lorenz AZ, et al. T cell activation profiles distinguish hemophagocytic lymphohistiocytosis and early sepsis. Blood. 2020:
34. Abdalgani M, Filipovich AH, Choo S, et al. Accuracy of flow cytometric perforin screening for detecting patients with FHL due to PRF1 mutations. Blood. 2015;126(15):1858–1860. doi:10.1182/blood-2015-06-648659
35. Rubin TS, Zhang K, Gifford C, et al. Perforin and CD107a testing is superior to NK cell function testing for screening patients for genetic HLH. Blood. 2017;129(22):2993–2999. doi:10.1182/blood-2016-12-753830
36. Merli P, Gentile L, Quagliarella F, et al. QuantiFERON-TB Gold can help clinicians in the diagnosis of haemophagocytic lymphohistiocytosis. Br J Haematol. 2020;191(2):e64–e67. doi:10.1111/bjh.17001
37. Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain K. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041–4052. doi:10.1182/blood-2011-03-278127
38. Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018;6(5):1508–1517. doi:10.1016/j.jaip.2018.05.031
39. Trottestam H, Horne A, Arico M, et al.; Histiocyte Society. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577–4584. doi:10.1182/blood-2011-06-356261
40. La Rosée P. Treatment of hemophagocytic lymphohistiocytosis in adults. Hematology Am Soc Hematol Educ Program. 2015;2015(1):190–196. doi:10.1182/asheducation-2015.1.190
41. Daver N, McClain K, Allen CE, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017;123(17):3229–3240. doi:10.1002/cncr.30826
42. Guangqiang M, Wang Y, Wang J, Wang Z. The DEP regimen is superior to the HLH-1994 regimen as first-line therapy for lymphoma-associated haemophagocytic lymphohistiocytosis. Leuk Lymphoma. 2020. doi:10.1080/10428194.2020.1849671
43. Delavigne K, Bérard E, Bertoli S, et al. Hemophagocytic syndrome in patients with acute myeloid leukemia undergoing intensive chemotherapy. Haematologica. 2014;99(3):474–480. doi:10.3324/haematol.2013.097394
44. Wang Y, Huang W, Hu L, et al. Multicenter study of combination DEP regimen as a salvage therapy for adult refractory hemophagocytic lymphohistiocytosis. Blood. 2015;126(19):2186–2192. doi:10.1182/blood-2015-05-644914
45. Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60(1):101–109. doi:10.1002/pbc.24188
46. Yuan L, Kan Y, Meeks JK, Ma D, Yang J. 18F-FDG PET/CT for identifying the potential causes and extent of secondary hemophagocytic lymphohistiocytosis. Diagn Interv Radiol. 2016;22(5):471–475. doi:10.5152/dir.2016.15226
47. Zheng Y, Hu G, Liu Y, et al. The role of 18F-FDG PET CT in the management of patients with secondary haemophagocytic lymphohistiocytosis. Clin Radiol. 2016;71(12):1248–1254. doi:10.1016/j.crad.2016.05.011
48. Henter JI, Samuelsson-Horne A, Aricò M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367–2373. doi:10.1182/blood-2002-01-0172
49. Horne A, Janka G, Maarten Egeler R, et al. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. 2005;129(5):622–630. doi:10.1111/j.1365-2141.2005.05501.x
50. Bergsten E, Horne A, Hed Myrberg I, et al. Stem cell transplantation for children with hemophagocytic lymphohistiocytosis: results from the HLH-2004 study. Blood Adv. 2020;4(15):3754–3766. doi:10.1182/bloodadvances.2020002101
51. Li Z, Wang Y, Wang J, Zhang J, Wang Z. Haploidentical hematopoietic stem cell transplantation for adult patients with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Leuk Lymphoma. 2018;59(1):77–84. doi:10.1080/10428194.2017.1330467
52. Oliansky DM, Czuczman M, Fisher RI, et al. The role of cytotoxic therapy with hematopoietic stem cell transplantation in the treatment of diffuse large B cell lymphoma: update of the 2001 evidence-based review. Biol Blood Marrow Transplant. 2011;17(1):20–47. doi:10.1016/j.bbmt.2010.07.008
53. Wang J, Wang Y, Wu L, Zhang J, Lai W, Wang Z. PEG-aspargase and DEP regimen combination therapy for refractory Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Hematol Oncol. 2016;9(1):84. doi:10.1186/s13045-016-0317-7
54. Zhao Y, Shi J, Li X, et al. Salvage therapy with dose-escalating ruxolitinib as bridge to allogeneic stem cell transplantation for refractory hemophagocytic lymphohistiocytosis. Bone Marrow Transplant. 2020;55(4):824–826. doi:10.1038/s41409-019-0577-2
55. Locatelli F, Jordan MB, Allen CE, et al. Emapalumab in Children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. 2020;382(19):1811–1822. doi:10.1056/NEJMoa1911326
56. Vallurupalli M, Berliner N. Emapalumab for the treatment of relapsed/refractory hemophagocytic lymphohistiocytosis. Blood. 2019;134(21):1783–1786. doi:10.1182/blood.2019002289
57. Meyer LK, Verbist KC, Albeituni S, et al. JAK/STAT pathway inhibition sensitizes CD8 T cells to dexamethasone-induced apoptosis in hyperinflammation. Blood. 2020;136(6):657–668. doi:10.1182/blood.2020006075
58. Sin JH, Zangardi ML. Ruxolitinib for secondary hemophagocytic lymphohistiocytosis: first case report. Hematol Oncol Stem Cell Ther. 2019;12(3):166–170. doi:10.1016/j.hemonc.2017.07.002
59. Wang J, Wang Y, Wu L, et al. Ruxolitinib for refractory/relapsed hemophagocytic lymphohistiocytosis. Haematologica. 2020;105(5):e210. doi:10.3324/haematol.2019.222471
60. Ahmed A, Merrill SA, Alsawah F, et al. Ruxolitinib in adult patients with secondary haemophagocytic lymphohistiocytosis: an open-label, single-centre, pilot trial. Lancet Haematol. 2019;6(12):e630–e637. doi:10.1016/S2352-3026(19)30156-5
61. Boonstra PS, Ahmed A, Merrill SA, Wilcox RA. Ruxolitinib in adult patients with secondary hemophagocytic lymphohistiocytosis. Am J Hematol. 2021;96(4). doi:10.1002/ajh.26091
62. Wang J, Wang Z. Multicenter study of ruxolitinib combined DEP regimen as a salvage therapy for refractory/relapsed hemophagocytic lymphohistiocytosis. Blood. 2019;134(Supplement_1):1042. doi:10.1182/blood-2019-130980
63. Kim ST, Cristescu R, Bass AJ, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018;24(9):1449–1458. doi:10.1038/s41591-018-0101-z
64. Liu P, Pan X, Chen C, et al. Nivolumab treatment of relapsed/refractory Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in adults. Blood. 2020;135(11):826–833. doi:10.1182/blood.2019003886
65. Saarela M, Senthil K, Jones J, et al. Hemophagocytic lymphohistiocytosis in 2 patients with multiple sclerosis treated with alemtuzumab. Neurology. 2018;90(18):849–851. doi:10.1212/WNL.0000000000005420
66. So MW, Koo BS, Kim YJ, Kim YG, Lee CK, Yoo B. Successful rituximab treatment of refractory hemophagocytic lymphohistiocytosis and autoimmune hemolytic anemia associated with systemic lupus erythematosus. Mod Rheumatol. 2014;24(5):855–857. doi:10.3109/14397595.2013.874740
67. Henzan T, Nagafuji K, Tsukamoto H, et al. Success with infliximab in treating refractory hemophagocytic lymphohistiocytosis. Am J Hematol. 2006;81(1):59–61. doi:10.1002/ajh.20462
68. Lorenz G, Schul L, Schraml F, et al. Adult macrophage activation syndrome-haemophagocytic lymphohistiocytosis: ‘of plasma exchange and immunosuppressive escalation strategies’ - a single centre reflection. Lupus. 2020;29(3):324–333. doi:10.1177/0961203320901594
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.