")
Back to Journals » The Application of Clinical Genetics » Volume 15
Clinical Importance of aCGH in Genetic Counselling of Children with Psychomotor Retardation
Authors Pasińska M , Łazarczyk E , Repczyńska A , Sobczyńska-Tomaszewska A, Zimowski J, Runge A, Haus O
Received 22 January 2022
Accepted for publication 13 April 2022
Published 14 May 2022 Volume 2022:15 Pages 27—38
DOI https://doi.org/10.2147/TACG.S357136
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Magdalena Pasińska,1 Ewelina Łazarczyk,1 Anna Repczyńska,1 Agnieszka Sobczyńska-Tomaszewska,2 Janusz Zimowski,3 Agata Runge,1 Olga Haus1
1Department of Clinical Genetics, Faculty of Medicine, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University, Bydgoszcz, Poland; 2Medical Center “Medgen”, Warszawa, Poland; 3Department of Genetics, Institute of Psychiatry and Neurology, Warszawa, Poland
Correspondence: Magdalena Pasińska, Department of Clinical Genetics, Faculty of Medicine, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University, ul. M. Skłodowskiej-Curie 9, Bydgoszcz, 85-094, Poland, Tel/Fax +48 52 585 35 68 ; +48 52 585 36 70, Email [email protected]; [email protected]
Introduction: The X and Y chromosomes are responsible for the determination and differentiation of the gonads, and their numerical and structural abnormalities may cause the abnormal development of secondary sex characteristics. The presence of abnormalities concerning X chromosome can also contribute to many genetically heterogeneous diseases associated with cognitive impairment and intellectual disability.
Purpose: This study shows the effect of aberrations of the maternal X chromosome on the abnormal development of the child.
Patients and Methods: Ten women aged 26 to 40 years were consulted in genetic counselling clinic and subsequently subjected to cytogenetic and molecular tests due to abnormal psychomotor development of their children, in whom structural aberrations of the X chromosome had been detected.
Results: Two women were diagnosed with changes in karyotype: 46,X,der(X)t(X;Y)(p22.3;q11.2) in one and 46,X,inv(X)(p21.2q13). Five women were diagnosed with microduplications in the short arm of the X chromosome; dupXp22.31 in one, and in four women dupXp22.33. The remaining three women were diagnosed with duplication in the long arm of the X chromosome; dupXq25 in one and dupXq26.3 in two women.
Conclusion: Genetic analysis of the X chromosome, based on cytogenetic and molecular methods of the highest available resolution, is extremely important in women with reproductive failure. These methods allow establishing accurately the breakpoints and rearrangements in chromosomes, and assessment of the copy number variation (CNV) can explain phenotypic variability with apparently similar aberrations. A more precise characterization of the alterations is necessary for the correct genetic diagnosis, as well as determination of the carrier status and genetic risk in family members.
Keywords: X chromosome, structural aberrations, genetic diagnostics, genetic counseling, intellectual disability
Introduction
Chromosomal abnormalities causing genomic imbalance are a major cause of congenital developmental defects and intellectual disability. The X and Y chromosomes are responsible for the determination and differentiation of the gonads, and their numerical and structural abnormalities may cause the abnormal development of secondary sex characteristics. Ovary dysfunction leads to the primary or secondary absence of menstrual period and infertility. The presence of abnormalities concerning X chromosome can also contribute to many genetically heterogeneous diseases associated with cognitive impairment and intellectual disability. The increased dosage of genes of any part of the X chromosome, except the pseudo-autosomal region, has been shown to lead to functional disomies of genes located within the aberration area, especially in boys. This increases the critical amount of protein affecting normal cognitive development.1,2
In standard clinical practice, conventional cytogenetic methods are still used to diagnose couples with recurrent miscarriages, stillbirths or births of children with psychomotor retardation. Due to their low resolution, these methods do not allow identification of submicroscopic lesions.3,4
The increasing popularity of cytogenetic molecular techniques such as array Comparative Genomic Hybridization (aCGH) improves the rates of detection of submicroscopic chromosomal abnormalities, especially in patients with intellectual disability. Although this method does not permit identification of balanced structural aberrations in potential carriers, it may increase the frequency of diagnosis of unbalanced subtelomeric rearrangements in sex chromosomes which, particularly in the mother, do not cause phenotypic manifestations.5
This study shows the effect of aberrations of the maternal X chromosome on the abnormal development of the child.
Material
Ten women aged 26 to 40 years were consulted in genetic counselling clinic and subsequently subjected to cytogenetic and molecular tests due to abnormal psychomotor development of their children, in whom structural aberrations of the X chromosome had been detected. In 7 cases, the tests were also carried out in the child’s father. All cases of both children and mothers were assessed by the same medical staff. The analysis of indications for genetic diagnostics in women and the methods used are shown in Table 1.
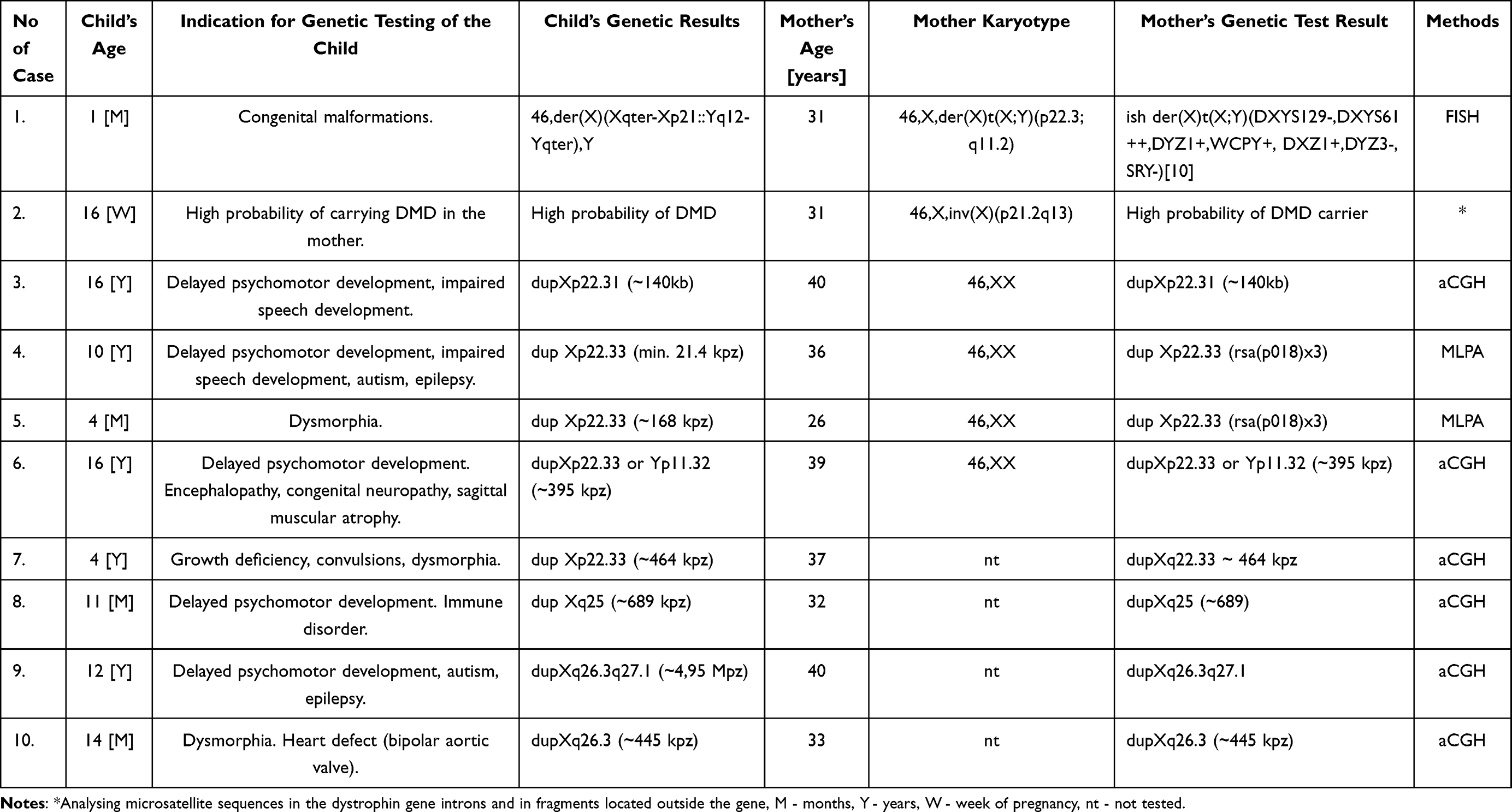 |
Table 1 Indications for Genetic Tests of Mother and Child |
For constitutional karyotype establishing and molecular analyses, we used peripheral blood lymphocytes from samples taken on heparin, whereas for molecular diagnostics – those from samples taken on EDTA; for karyotyping and molecular tests of fetuses, we used amniocytes from amniotic fluid.
DNA samples isolated from peripheral blood lymphocytes using the Roche MagNA Pure Compact device were the material for the dystrophin gene test.
Methods
The study used conventional cytogenetic methods to identify karyotypes. G-banded metaphases with a 550-band resolution from peripheral blood lymphocytes or amniocytes were prepared according to standard protocols.
Peripheral blood lymphocytes were cultured for 72 h with LF-7 or PHA mitogens, with the culture being conducted and terminated in standard conditions. Amniocytes, obtained by centrifuging amniotic fluid, were cultured for two weeks in AmnioMax from Gibco and Amnio Grow Plus from Cytogen complete media. Cytogenetic specimens were stained using the GTG technique, and further analysed using fluorescence in situ hybridisation (FISH) with molecular probes specific to the SRY gene, Y chromosome heterochromatin, short arm telomeres (XYpter), long arm telomeres (XYqter), centromeres (for the X and Y centromeres), Y chromosome euchromatin (painting probe) and Whole Chromosome Painting Probe from Cytocell and ToTelVysion MIX 3 from Vysis.
The FISH analysis was conducted according to the instructions from the manufacturers of the molecular probes used.
aCGH was conducted using a commercially available array (CytoSure, Constitutional v3 (8x60k), Oxford Gene Technology (OGT), Oxfordshire, UK), according to the manufacturer’s protocol. The CytoSure Interpret Software (OGT) was used for genomic copy-number analysis.
The search for extensive rearrangements of the SHOX (short stature homeobox-containing) gene (Gene/Locus OMIM no 312865) and adjacent regions (Xp22.33 region) was performed by MLPA using the MRC-Holland P018 kit.
All exons of the dystrophin gene and fragments of intron sequences adjacent to the tested exons were analysed using maternal DNA. Sanger sequencing was performed using the BigDye v1.1 reagents from Applied Biosystems. The search for deletions and duplications within the dystrophin gene was performed using the MLPA method.
Inheritance of the dystrophin gene within the family was investigated by analysing microsatellite sequences in the dystrophin gene introns and in fragments located outside the gene. Inheritance of the following sequences was analysed: DysIIa (promoter), Str45 (intron 45), Str50 (intron 50), StrHI (intron 62) and StrH3 (3’ UTR). The test was performed as follows: the selected gene segment was amplified, products were separated in a non-denaturing polyacrylamide gel against molecular weight markers, DNA was visualized by silver staining, and photographic documentation was performed. Results were collected in the Cyrillic program.
Case Description
Case 1
Patient aged 31; a set of congenital malformations diagnosed in a male child from normal, third pregnancy lasting 37 weeks. The child was born naturally, in serious general condition, with a birth weight of 2530 g. After birth, malformations of the spine and chest were identified, ie, butterfly vertebrae, funnel chest, scattered perispinal calcifications, also present in parenchymal organs, poorly developed nipples. Furthermore, dysmorphy (recessed nasal root, large tongue, small auricles with abnormal helices), oedemas and petechiae, slightly widened lateral ventricles of the brain with single calcifications, and enlarged liver were observed. The child had a hypotonia muscle tone and no breath. Based on these findings, Alagille syndrome was suspected. The child died at 65 days of age due to severe respiratory failure. In cytogenetic examination, the following abnormal karyotype was detected: 46,der(X)(Xqter-Xp21::Yq12-Yqter),Y.
The first two pregnancies of the mother resulted in the birth of healthy daughters. During the fourth and fifth pregnancy, invasive prenatal examination revealed normal karyotypes in both fetuses (female and male).
The mother has five healthy siblings. Her mother had two miscarriages in first trimester of gestation Figure 1.
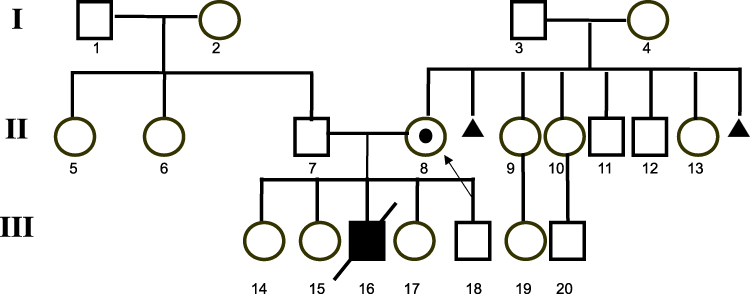 |
Figure 1 Pedigree of the patient in patient No 1 (III/8) with unbalanced insertion; 46,X,der(X)t(X;Y)(p22.3;q11.2). Notes: White symbol (square or circle) - a healthy person, A symbol (square or circle) with the black dot - carrier of the unbalanced insertion ins(X;Y), black symbol (square or circle) - a person with mental retardation (MR) and congenital malformations, oblique line (on a square or circle) - dead person, A little black triangle - early miscarriage, arrow - proband. |
Case 2
Patient aged 31, examined prenatally during first pregnancy due to family history: Duchenne/Becker muscular dystrophy (DMD/BMD) diagnosed based on clinical signs in the patient’s two brothers and her mother’s brother. No genetic tests were conducted previously. All three DMD patients died at approximately 16 years of age Figure 2.
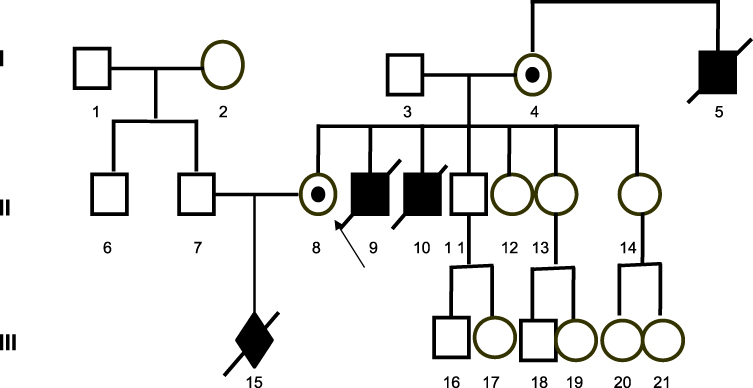 |
Figure 2 Pedigree of the patient in patient No 2 with (II/8) karyotype; 46,X,inv(X)(p21.2q13). Notes: White symbol (square or circle) - a healthy person, A symbol (square or circle) with the black dot - carrier of the unbalanced insertion ins(X;Y), black symbol (square) - a person with Duchenne muscular dystrophy (DMD), oblique line (on a square or circle) - dead person, black diamond - structural aberration in the fetus, arrow - proband. |
Based on the linkage analysis of the PCR-Dyslla, PCR-Str45, PCR-Str50, PCR-StrHI, PCR-STRH3 microsatellite sequences located within the dystrophin gene, the haplotypes of the patient’s healthy brother, mother and the pregnant patient herself were determined. The carrier status of Duchenne (DMD) muscular dystrophy was considered highly likely. Examination of the fetal constitutional karyotype using the amniotic fluid showed an abnormal male karyotype – 46,Y,inv(X)(p21.2q13) – pericentric inversion of the X chromosome, with the maternal karyotype being 46,X,inv(X)(p21.2q13).
Case 3
Patient aged 40, examined due to delayed psychomotor development in her son born naturally in the 41st week of her first pregnancy; birth weight 4100 g, Apgar score 10. The patient has also been diagnosed with mild intellectual disability, as well as difficulty expressing herself and interacting with other people. The patient’s brother is intellectually disabled and also has a child with psychomotor delay.
In a younger son, from the patient’s second relationship, psychomotor development was normal.
In the boy examined, at the age of 16 years, dysmorphic features were found: bitemporal narrowing; high forehead; large, protruding mandible; moderately delayed intellectual development; complex speech impairment complicating communication with other people. Since the age of 8, the boy has been studying at a special school. Psychological examination showed a progressive decline in intellectual development. Mental and perception processes were impaired. The boy knew letters but did not write or read on his own. He had serious problems predicting the consequences of his behaviour. He is easily influenced and persuaded by his peers.
Constitutional karyotype analysis performed in the boy revealed a normal male 46,XY karyotype with 6.7% of spontaneous chromosome fractures. A molecular test of the two most common mutations in the ARX gene – c.428_c.428dup(24bp) and c.304ins(GCG) in exon 2 – was also conducted, and no mutation was found. This was followed by a molecular karyotyping with aCGH in which a genomic modification was found – an interstitial duplication of the X chromosome in the Xp22.31 region, ~140 kb in size.
Case 4
Patient aged 36, examined due to delayed psychomotor development diagnosed in her son from first pregnancy. The boy was born naturally at 42 weeks of gestation; birth weight 3880 g, Apgar score 7. At 3 months of age, muscular hypertony was observed, and rehabilitation was introduced. The boy started walking at approximately 2 years of age, and a balance disorder was also observed by this time. In physical examination, slight dysmorphy was noted: triangular face with wide nose root, wide medial cleft, very thin upper lip, sunken chest and genu valgum. The boy’s gait was stiff. Speech development was disturbed and delayed. Epilepsy attacks occurred since the age of 6 years. Emotional disturbances and recurrent vomiting were also observed. The boy attended a special school. He had serious memory problems. A normal male 46,XY karyotype was diagnosed by a cytogenetic test. In contrast, a molecular test using the subtelomeric MLPA technique found genomic imbalance caused by a duplication in the pseudo-autosomal region PAR1 (located on the X and Y chromosomes). Subsequently, an MLPA analysis of the SHOX gene was conducted, and a heterozygous duplication of Xq22.33 of ~ 84.9 kbp in size was found in the upstream region of the SHOX gene, reaching and including exon 5.
Family history: the mother had two healthy siblings, and her fraternal nephew had psychomotor hyperactivity. Laboratory tests revealed an elevated level of 3-hydroxyvaleric acid.
Case 5
Patient aged 26, examined due to the presence of dysmorphic features in her son from the first pregnancy: hypoplastic scrotum and undescended testicles, borderline enlargement of the lateral ventricles of the brain, retrognathia, enlarged thymus, clubfoot, muscular hypotony tone and increased level of uric acid in blood serum. Boy was born by caesarean section; birth weight 3400 g, body length 60 cm, Apgar score 8. A normal male 46,XY karyotype was diagnosed by a cytogenetic test. In contrast, a molecular karyotyping with aCGH revealed genomic imbalance caused by a duplication in the pseudo-autosomal region. Duplication found covered a ~ 168 kbp region of the Xp22.33 band of the short arm of the X chromosome or Yp11.32 band of the short arm of the Y chromosome, and included the SHOX gene (OMIM: 322865).
Case 6
Patient aged 39 was examined due to the presence of encephalopathy, congenital neuropathy with loss of vision and sagittal muscle atrophy in her son.
The boy was born naturally at 26 weeks of gestation, following a preterm premature rupture of the membranes (PPROM), with a birth weight of 1010 g, in serious general condition (Apgar score 1). After birth, the boy was diagnosed with intrauterine hypoxia, congenital pneumonia and grade 4 haemorrhage into the fourth brain ventricle.
The boy started walking at the age of 2 years. Delayed speech development was observed. He spoke his first words after the age of 2. He underwent an ophthalmological surgery in the first month of life, and a strabismus surgery at the age of 7. He was blind in his left eye. In addition, he had left-sided hearing deficit. He was operated for inguinal hernia at the age of 3 and had a tonsillectomy at the age of 7. Up to 10 years of age, the boy had recurrent infections. Physical examination revealed asymmetry of the palpebral fissures, perilymph fistula, scoliosis in the Th–L segments of the spine, short halluxes of both feet, wide-set toes II and III. In addition, there were two residual supernumerary nipples, as well as stretch marks on the chest and lower legs. The boy has been attending a school for visually impaired children from the age of 6, and was diagnosed with severe intellectual disability. Supernumerary nipples were present also in other of family members – the grandfather and a maternal cousin. A molecular test using aCGH revealed genomic imbalance: a duplication in the pseudo-autosomal region, which covered a ~ 395 kbp fragment of the short arm of the X chromosome in the band Xp22.33 or the short arm of the Y chromosome in the band Yp11.32, and includes the SHOX gene (OMIM: 312865).
The boy’s younger sister is healthy.
Case 7
Woman aged 37, was examined due to the diagnosis of short stature and low body weight, mild dysmorphic features (hypertelorism, epicanthal fold) and decreased immunity in her son from the second pregnancy. There was a natural birth at week 40 of gestation; birth weight was 3020 g, and Apgar score 10. The child had twice shivering associated with fever.
A molecular test using aCGH found genomic imbalance caused by a duplication in the pseudo-autosomal region, which covers a ~464 kbp fragment of the X chromosome in the band Xp22.22.
The psychomotor development of the boy’s older brother is normal.
Case 8
Patient aged 32, was consulted and genetically examined at week 8 of her third pregnancy due to delayed psychomotor development, autism and dysmorphic features in her first son. The boy from her first pregnancy was born by caesarean section due to presentation of feet in the birth canal after water breaking; birth weight was 2980 g, Apgar score 9. The boy started walking at the age of 20 months, at the age of 11 years said single words. Due to frequent infections, he remained an outpatient of the Clinic for Immune System Disorders. A molecular test using a whole-genome microarray (Technology, GRCh37/hg19), with a mean resolution of 120 kbp found genomic imbalance caused by a duplication of the X chromosome arm, in the region Xq25 of ~ 689 kbp. The patient’s younger daughter was healthy. Family history: the patient’s sister, whose first child had multiple malformations, died of gallbladder cancer at the age of 45; the patient’s maternal cousin had ovarian cancer at the age of 43.
Case 9
Patient aged 40 was examined due to delayed psychomotor development in her son born naturally in the 42nd week of her fourth pregnancy, which was complicated by pregnancy-induced hypertension (PIH); birth weight 2700 g, Apgar score 9. Medical history: death of a son from her third pregnancy, born at 36 weeks of gestation with multiple defects, within 24 hours from birth. Two older children from the patient’s first relationship and a daughter from another relationship were healthy. The boy had dysmorphic features: bitemporal narrowing with wide occiput, dolichocephaly, right nasofrontal angle. A pigmented birthmark of 40 × 20 mm was visible above the right brow ridge. The boy had a short stature and low body weight. In addition, speech development was much delayed.
Head MRI revealed changes in the pituitary gland – ectopic posterior lobe. The boy attended a special school.
The boy’s father had a mental illness, as well as alcohol and tobacco addiction.
A cytogenetic test in the boy revealed a normal male 46,XY karyotype, while a molecular test using an oligonucleotide microarray found genomic imbalance caused by a duplication in the X chromosome long arm in the region Xq26.3q27.1 of ~ 4.95 Mbp, originating from the mother.
Case 10
Patient aged 33, underwent genetic diagnostics due to delayed psychomotor development and mild dysmorphy: long face with shortened frontal dimension, open mouth and heart defect (bicuspid aortic valve) in her son from the second pregnancy.
The boy was born at 40 weeks of gestation; birth weight was 3650 g, Apgar score 8. The boy started walking at the age of 13 months. At the age of 2, he was able to say single words. Rapid growth was observed in follow-up examinations. The boy’s older sister was healthy. Family history: the patient’s fraternal nephew has disturbed psychomotor development, and an aCGH test in him found a partial de novo monosomy of chromosome 13.
A molecular test using aCGH, conducted in both the patient and her son, found genomic imbalance caused by a duplication (dupXq26.3) of 445 kbp.
Summary of the Cytogenetic and Molecular Results
Cytogenetic evaluation of peripheral blood karyotype in patient 1 revealed a derived X chromosome with X;Y translocation, loss of telomeres of the short arms of the X and Y chromosomes, translocation of almost the entire long arm of the Y chromosome, with the whole heterochromatin and some part of euchromatin, onto the short arm of the X chromosome, and with absence of the SRY gene. The observed changes and the karyotype 46,X,der(X)t(X;Y)(p22.3;q11.2) were confirmed by the FISH technique, using probes specific to the telomeres of the short and long arms of the X and Y chromosomes, a painting probes specific to the heterochromatin and euchromatin of the Y chromosome, and a centromeric probe specific to the X and Y chromosomes. The use of a probe specific to the SRY gene showed absence of this gene, as shown in Figure 3.
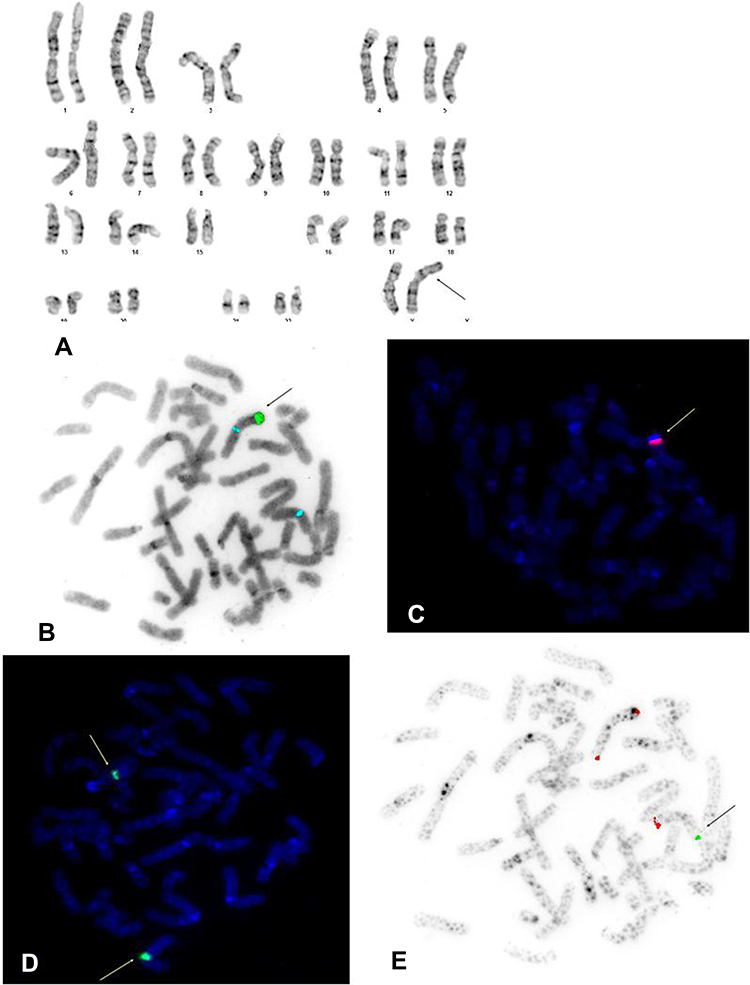 |
Figure 3 (A) Karyogram of patient No 1 in GTG-banding showing 46,X,der(X)t(X;Y)(p22.3;q11.2). The arrow indicates abnormal X chromosome. (B) FISH examination in patient No 1: ish der(X)t(X;Y)(DYZ1+,DXZ1+,SRY-). Probe specific for the SRY gene - red [lack], centromere probe for the centromere of the X chromosome - blue, probe specific for the heterochromatin of the Y chromosome - green. The arrow indicates the transferred heterochromatin from the Y on the X chromosome. (C) FISH examination in patient No 1: ish der(X)t(X;Y)(WCPY+), showing the absence of SRY probe on X chromosome. Painting probe for Y chromosome euchromatin - red. The arrow indicates the transferred heterochromatin from the Y chromosome on the X chromosome. (D) FISH examination in patient No 1: ish der(X)t(X;Y)(DXZ1+,DYZ3-), showing X centromere probes - green, for the Y centromere - red [lack]. The arrows indicate of the centromeres of X chromosome. (E) Probe specific for XYpter short arm telomeres - green, specific for XYqter long arm telomeres - red. The arrow indicates of the short arm of Y chromosome transferred on the X chromosome in patient No 1: ish der(X)t(X;Y)(DXYS129-,DXYS61++). |
Examination of the constitutional karyotype of patient 2 revealed an aberration of the X chromosome: a pericentric inversion, similar to the one found in her fetus, and the karyotype 46,X,inv(X)(p21.2q13).
In the third case, aCGH revealed a duplication in the region Xp22.31, and in four women (patients 4, 5, 6 and 7), MLPA and aCGH revealed duplications of Xp22.33 of various sizes. In patient 7, the alteration did not include any known gene. In three women and their sons, the continuous gene duplication involved the more distal regions of the long arm of the X chromosome. In the 8th case, dupXq25 of ~ 689 kbps was recognized, and in cases 9 and 10 there was found dupXq26.3, Table 1.
Discussion
Several types of translocations between homologous fragments of the Xp and Yq arms, resulting in the formation of a derivative X chromosome, have been described in the literature.1,6 One of them is the Xp;Yq translocation, which generates a derivative X chromosome with a partial Xp deletion and partial translocation of the long arm of the Y chromosome, such as that described in patient 1 with the karyotype 46,X,der(X)t(X;Y)(p22.3;q11.2).6,7
Most women with the karyotype 46,X,der(X)t(X;Y)(p22;q11) described in the literature had a short stature with a normal phenotype and usually retained normal function of gonads and fertility.6–8 Patients whose genomic imbalance includes the SRY gene have an increased risk of gonadoblastoma.6,8 The patient described as No 1 was of short stature (150 cm), with marked hirsutism, but did not report any fertility problems. Her two earlier and two later pregnancies led to the birth of healthy children, with a prenatally confirmed normal karyotype in two cases. A derivative X chromosome is most often found in male offspring with the phenotypic abnormalities due to partial Xp nullisomy always present. These abnormalities include short stature and complex clinical phenotypes resulting from the lack of Xp gene cluster: developmental delay and intellectual disability, dyschondrosteosis (associated with SHOX), chondrodysplasia punctata (associated with ASS), hypogonadotropic hypogonadism, anosmia (associated with KAL1) and ocular albinism (associated with OA1).7,10 Chen et al presented a fetus with female phenotype, and the development of the long limb bones (humerus and femur) delayed by approximately 2 weeks compared with other biometric parameters, observed at week 17 of pregnancy in a US examination. aCGH, GTG and FISH identified an abnormal karyotype: 46,X,der(X)t(X;Y)(p22.31;q11.22.1) generated de novo.4,7 The same authors also described two women with a derivative X chromosome, with the additional material originating from the long arms of the Y chromosome. One of these women was examined due to giving birth to a son with multiple defects, while the other was examined due to secondary infertility. Both women had given birth to healthy children. One of them became pregnant naturally, and prenatal diagnostics of the fetus were conducted; the other became pregnant using assisted reproduction technology (ART), with embryos used following prenatal pre-implantation diagnostic (PGD) procedures.4,7
The identification of inv(X) and the karyotype 46,X,inv(X)(p21.2q13) in the fetus and then in the mother (in case 2) explained the familial occurrence of progressive muscle dystrophy, and the clinical diagnosis of DMD. One breakpoint in the study participants was Xp21.2, which houses the DMD gene which mutations cause DMD. Saito-Ohara et al described a boy diagnosed with DMD due to the presence of inv(X) and the karyotype 46,Y,inv(X)(p21.2q22.2), accompanied by a severe disturbance of the psychomotor development, athetosis, nystagmus and severe congenital hypotonia. Based on a cytogenetic analysis, the authors hypothesized that the neurological symptoms accompanying DMD were associated with a break in Xq22.11,12 In patient 2, the second breakpoint in the identified aberration was located at Xq13. Based on the information provided by the family, the mental development of patients with DMD was normal. As it was impossible to perform aCGH in the patient and the fetus, the possible genomic imbalance could not be accurately located.12
There are many reports related to the loss of adjacent genes in the pseudo-autosomal region Xp22.33, especially involving the SHOX gene, but no cases involving pathogenic duplication of this region have been described. The SHOX gene is active on both copies of the X chromosome in females and on the X and Y chromosomes in males. Spranger et al reported a mother and her 5-year-old son with a short stature associated with the SHOX gene and Madelung’s deformity, in whom terminal deletion of the short arm of the chromosome including SHOX gene was detected. The nullisomy of this region in the boy also caused intellectual disability, presumably related to the MRR49 gene, as well as achondroplasia punctata related to the ARSE gene.8,12 Bleyl et al described a mother and her son with abnormal structure of the anterior chamber of the eye and skeletal defects.14
The homeodomain protein encoded by the SHOX gene acts as a transcriptional activator which controls the expression of genes responsible for growth. Loss of a copy of the gene or mutation in one of its alleles results in shortened fourth metacarpal bones, abnormalities of cranial bones, high palate, short and wide neck, as well as scoliosis. Mesomelia, in which the middle section of the limb is shortened in relation to the proximal and distal sections, may first become visible in school-age children and increases with age. Madelung’s deformity (incorrect positioning of the radius, ulna and carpal bones) usually develops until late childhood and is more frequent and pronounced in females. The growth phenotype caused by the SHOX deficiency/excess, in the absence of mesomelia and Madelung’s deformity, is highly variable, even within the same family. In the cases reported in the literature, delayed speech development and behavioural problems also occurred in women.13–15 The most common aberrations of the SHOX gene are deletions of different sizes, including SHOX and the regulatory region. Loss of both SHOX alleles causes a complete deficiency of the SHOX protein and the extreme osteodysplasia phenotype described as Langer syndrome.13–15
There are few reports in the literature regarding the psychomotor development of patients with the Xq22.33 duplication or the influence of the carrier status for this alteration on the parent phenotype. In our study, we presented patients with an inverted set of additional gene copies in this region. Ogata et al, in their studies in patients with gonadal dysgenesis in association with data on the growth of people with aneuploidy or polysomy of the sex chromosomes, suggested that tall stature can be caused by an additional copy of the SHOX gene with a shortage of oestrogens.8,16 In our study group, one boy was found to have a short stature and low body weight (case 7), while the others with dupXp22.33 showed a rapid growth rate and intellectual disability, which was also found in patient 3. Three (patients 3, 4 and 6) out of five women with identified duplications of Xp22.31 or Xp22.33 reported delayed speech development and learning difficulties in their childhood.
A continuous cluster of dupXq25 of ~ 689 kbp was diagnosed in patient 8 and her son. There are many reports describing male patients with duplications of this region.17–19 In all cases, authors observed neurodevelopmental disorders characterized by delayed speech development and intellectual disability in association with behavioural problems and dysmorphic facial features. In addition, variations included thin corpus callosum in brain imaging and sleep disorders. Female carriers may have milder manifestations. Leroy et al noticed that mothers of the boys described in their study had moderate learning difficulties, and one had epileptic seizures which did not occur in any of the boys. The size of duplications in the patients was 465–662 kbp and was similar to those observed in our study in which the women did not manifest any worrying clinical signs.
Another literature report concerned one patient with triplicate Xq25 and more pronounced manifestations of the dupXq25 syndrome.17–19 Regardless of the extent of duplication, the critical region in all patients described included the STAG2 gene and at least a fragment of the adjacent XIAP gene. Leroy et al noticed that the STAG2 protein is a subunit of the cohesin complex, which is a protein ring surrounding chromatin, regulating the separation of sister chromatids into the two daughter cells during cell division. The increased copy number of the STAG2 gene and dysregulation of its downstream target genes may be responsible for the specific clinical symptoms of this syndrome.19
The dupXq26.3 syndrome was diagnosed in two women and their sons. In patient 9, the extent of imbalance covers multiple genes, including at least 8 dosage-sensitive genes: SLC9A6 (OMIM:300231), FHL1 (OMIM: 300163), HTATSF1 (OMIM 300346), VGLL1 (OMIM: 300583), CD40LG (OMIM: 300386), ZIC3 (OMIM: 300265), ATP11C (OMIM: 300516) and SOX3 (OMIM: 313430), and is of approximately 4.95 Mbp. The region of duplication includes: critical region of the Xq26.3 duplication cluster (OMIM: 300942) and partially the critical region of the Xq27.3q28 duplication (OMIM: 300869).20 In patient 10, the duplication included the following genes: CD40 (OMIM: 300386), ARHGEF6 (OMIM: 300267), RBMX (OMIM: 300199), GPR101 (OMIM: 300393) – with the size of the imbalance of 445 kbp.
The Xq26.3 duplications have been described in patients with endocrine disorders and pituitary adenomas, which may lead to excessive secretion of the growth hormone as well as pituitary hypertrophy and/or co-occurrence of a macroadenoma. In such cases, excessive growth is observed, usually seen already in the first year of life.20,21 The Xq27.3q28
duplication syndrome is characterized by a neurodevelopmental disorder: mild to moderate mental disability, discrete facial dysmorphy, short stature and primary testicular failure leading to hypergonadotropic hypogonadism (absent or delayed puberty, thin body hair, abdominal obesity and small testicles). In cases described in the literature, manifestations have most often been identified after the age of 2.20,21 Moreover, patients have delayed speech development; behavioural problems, including: aggression, impulsivity, attention deficit; as well as hyperkinetic and autistic features, such as arm flapping, limited interests, and repetitive behaviour, social communication and interaction disorders.20,21 Patient 10 had a short stature (153 cm), and learning difficulties. Her son, besides his psychomotor development delay, has been diagnosed with short stature and low body weight. Therefore, it may be concluded that the phenotypic features occurring in both of them were a result of the overlapping of two syndromes of double-dose genes.
Conclusion
Genetic analysis of the X chromosome, based on cytogenetic and molecular methods of the highest available resolution, is extremely important in women with reproductive failure. These methods allow establishing accurately the breakpoints and rearrangements in chromosomes, and assessment of the copy number variation (CNV) can explain phenotypic variability with apparently similar aberrations.
A more precise characterization of the alterations is necessary for the correct genetic diagnosis, as well as determination of the carrier status and genetic risk in family members. Genetic counseling for women carriers of changes on the X chromosome must contain clear and complete information about the increased risk of having a child with a physical or intellectual disability. It can benefit families greatly by providing information that can influence reproductive decisions and even clinical management.22,23 It allows couples with a known carrier status to make informed reproduction choices, including PGD or prenatal diagnostics, acceptance of genetic risk and preparation for the possibility of having a child with a specific disease. Other options may include the use of a gamete banks, adoption or voluntary childlessness.
Ethics Approval and Consent to Participate
The study was performed in accordance with the Declaration of Helsinki and accepted standards of ethics. The consent of the relevant bioethics committee was obtained: Bioethics Committee of Nicolaus Copernicus University, Toruń, Poland. NCU Committee of Bioethics KB 61/2021.
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
All patients signed written informed consent to take part in the study. For patients with intellectual disabilities, written consent for part in the study was signed on the study by the parents/legal guardians.
Consent to Publish
All patients signed written informed consent for the publication of their clinical data. For patients with intellectual disabilities, written consent for publication was obtained by the parents/legal guardians.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Disclosure
The authors declare that they have no competing interests.
References
1. Poriswanish N, Neumani R, Wetton JH, et al. Recombination hotspots in an extended human pseudoautosomal domain predicted from double-strand break maps and characterized by sperm-based crossover analysis. PLoS Genet. 2018;14(10):1–20. doi:10.1371/journal.pgen.1007680
2. Tarpey PS, Smith R, Pleasance E, et al. A systematic, large-scale resequencing screen of X chromosome coding exons in mental retardation. Nat Genet. 2009;41(5):535–543. doi:10.1038/ng.367
3. Vandewalle J, Van Esch H, Govaerts K, et al. Dosage-dependent severity of the phenotype in patients with mental retardation due to a recurrent copy-number gain at Xq28 mediated by an unusual recombination. Am J Hum Genet. 2009;85(6):809–822. doi:10.1016/j.ajhg.2009.10.019
4. Chen CP, Su YN, Chern SR, et al. Prenatal diagnosis and array comparative genomic hybridization characterization of a de novo X;Y translocation. Taiwan J Obstet Gynecol. 2012;51(3):485–488. doi:10.1016/j.tjog.2012.07.037
5. Pasińska M, Łazarczyk E, Jułga K, Bartnik-Głaska M, Nowakowska B, Haus O. Multiple occurrence of psychomotor retardation and recurrent miscarriages in a family with a submicroscopic reciprocal translocation t(7;17)(p22;p13.2). BMC Med Genom. 2018;11(69):1–7. doi:10.1186/s12920-018-0384-4
6. Cheng DH, Gong F, Tan K, et al. Karyotype determination and reproductive guidance for short stature women with a hidden Y chromosome fragment. Reprod Biomed Online. 2013;27(1):89–95. doi:10.1016/j.rbmo.2013.03.015
7. Chen CP, Chen CY, Chern SR, et al. Molecular cytogenetic characterization of Xp22.32→pter deletion and Xq26.3→qter duplication in a male fetus associated with 46,Y,rec(X)dup(Xq) inv(X)(p22.3q26.3), a hypoplastic left heart, short stature, and maternal X chromosome pericentric inversion. Taiwan J Obstet Gynecol. 2016;55(5):705–711. doi:10.1016/j.tjog.2016.05.009
8. Ogata T, Kosho T, Wakui K, Fukoshima Y, Yoshimoto M, Miharu N. Short stature homeobox-containing gene duplication on the der(X) chromosome in a female with 45,X/46,X,der(X), gonadal dysgenesis, and tall stature. J Clin Endocrinol Metab. 2000;85(8):2927–2930. doi:10.1210/jcem.85.8.6745
9. Zhang L, Ren M, Song G, et al. Prenatal diagnosis of sex chromosomal inversion, translocation and deletion. Mol Med Rep. 2018;17(2):2811–2816. doi:10.3892/mmr.2017.8198
10. Hori T, Kawakita N, Minowada S, et al. Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocr Metab. 2004;89:1079–1088. doi:10.1210/jc.2003-030476
11. Saito Ohara F, Fukuda Y, Ito M, et al. The Xq22 inversion breakpoint interrupted a novel ras-like GTPase gene in a patient with Duchenne muscular dystrophy and profound mental retardation. Am J Hum Genet. 2002;71(3):637–645. doi:10.1086/342208
12. Tran TH, Zhang Z, Yagi M, et al. Molecular characterization of an X(p21.2;q28) chromosomal inversion in a Duchenne muscular dystrophy patient with mental retardation reveals a novel long non-coding gene on Xq28. J Hum Genet. 2013;58(1):33–39. doi:10.1038/jhg.2012.131
13. Spranger S, Schiller S, Jauch A, et al. Leri-Weill syndrome as part of a contiguous gene syndrome at Xp22.3. Am J Med Genet. 1999;83:367–371. doi:10.1002/(SICI)1096-8628(19990423)83:5<367::AID-AJMG5>3.0.CO;2-K
14. Bleyl SB, Byrne JLB, South T, et al. Brachymesomelic dysplasia with Peters anomaly of the eye results from disruptions of the X chromosome near the SHOX and SOX3 genes. Am J Med Genet. 2007;143A:2785–2795. doi:10.1002/ajmg.a.32036
15. Gürsoy S, Hazan F, Aykut A, et al. Detection of SHOX gene variations in patients with skeletal abnormalities with or without short stature. J Clin Res Pediatr Endocrinol. 2020;12(4):358–365. doi:10.4274/jcrpe.galenos.2020.2019.0001
16. Savarese E, Di Felice B, Miconi F, et al. An association of PTPN11 and SHOX mutations in a male presenting with syndromic growth failure. Front Endocrinol (Lausanne). 2018;9:557–560. doi:10.3389/fendo.2018.00557
17. Cabezas D, Slaugh R, Abidi F, et al. A new X linked mental retardation (XLMR) syndrome with short stature, small testes, muscle wasting, and tremor localises to Xq24-q25. J Med Genet. 2000;37(9):663–668. doi:10.1136/jmg.37.9.663
18. Kumar R, Corbett MA, Van Bon BWM, et al. Increased STAG2 dosage defines a novel cohesinopathy with intellectual disability and behavioral problems. 2015. Hum Molec Genet. 2015;24:7171–7181. doi:10.1093/hmg/ddv414
19. Leroy C, Jacquemont ML, Doray B, et al. Xq25 duplication: the crucial role of the STAG2 gene in this novel human cohesinopathy. 2016. Clin Genet. 2016;89:68–73. doi:10.1111/cge.12567
20. Stagi S, Lapi E, Pantaleo M, et al. A SOX3 (Xq26.3-27.3) duplication in a boy with growth hormone deficiency, ocular dyspraxia, and intellectual disability: a long-term follow-up and literature review. Hormones. 2014;13(4):552–560. doi:10.14310/horm.2002.1523
21. Trivellin G, Sharwood E, Hijazi H, et al. Xq26.3 duplication in a boy with motor delay and low muscle tone refines the X-Linked acrogigantism genetic locus. J Endocr Soc. 2018;2(10):1100–1108.
22. Griesi-Oliveira K, Laurato Sertié AL. Autism spectrum disorders: an updated guide for genetic counseling. Einstein. 2017;15(2):233–238. doi:10.1590/s1679-45082017rb4020
23. Barrosa F, Carvalho F, Barros A, Dória S. Premature ovarian insufficiency: clinical orientations for genetic testing and genetic counseling. Porto Biomed J. 2020;5(3):1–5. doi:10.1097/j.pbj.0000000000000059
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.