Back to Journals » Cancer Management and Research » Volume 11
Clinical genetic features and related survival implications in patients with surgically resected large-cell lung cancer
Authors Wang F, Lu JB, Wu XY, Feng YF, Shao Q, An X ![]() , Wang HY
, Wang HY ![]()
Received 3 January 2019
Accepted for publication 10 April 2019
Published 14 June 2019 Volume 2019:11 Pages 5489—5499
DOI https://doi.org/10.2147/CMAR.S200263
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Fang Wang,1,2 Jia-Bin Lu,3 Xiao-Yan Wu,2 Yan-Fen Feng,1,3 Qiong Shao,2 Xin An,4 Hai-Yun Wang1,2
1State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center, Guangzhou 510060, People’s Republic of China; 2Department of Molecular Diagnostics, Sun Yat-sen University Cancer Center, Guangzhou 510060, People’s Republic of China; 3Department of Pathology, Sun Yat-sen University Cancer Center, Guangzhou 510060, People’s Republic of China; 4Department of Medical Oncology, Sun Yat-sen University Cancer Center, Guangzhou 510060, People’s Republic of China
Background: Large-cell lung carcinomas (LCLCs) were reclassified by the World Health Organization 2015 criteria. and remain fairly unknown at the molecular level and targeted-therapeutic options.
Methods: Data of 184 lung cancer patients were retrieved from clinical records, of which 54 were found to be pathologically diagnosed as LCLC. The genetic alterations EGFR/KRAS/BRAF mutations, MET copy number, and exon 14 mutation, ALK and ROS1 rearrangements, and PDL1 expression were investigated using clinical technologies. The relationship between clinicopathologic and genetic features was analyzed, and the Kaplan–Meier method with log-rank test was used for analyzing patient survival.
Results: Major events, including EGFR, KRAS, and BRAF mutations and MET copy-number gain, were found in 5.6%, 16.7%, 1.9%, and 18.5% in LCLC, respectively. No ALK or ROS1 translocation was detected. PDL1 expression in tumor cells and in tumor-infiltrating lymphocytes was observed in 24 (44.4%) and 16 (29.6%) patients. Kaplan–Meier analysis showed that patients with a KRAS mutation had ower 5-year overall survival than those with wild-type KRAS (25.4% vs 47.8%, P=0.028) and that patients with negative PDL1 stained in tumor cells but positive for tumor-infiltrating lymphocytes had significantly favorable overall survival compared to those with solitary and positive PDL1 stained in tumor cells (62.5% vs 20.6%, P=0.044).
Conclusion: KRAS mutations and PDL1 expression can predict patient survival and be potential target options in LCLC.
Keywords: large-cell lung cancer, driver mutations, PD-L1, KRAS
Introduction
Large-cell lung cancer (LCLC) is the third-most common subtype of non-small-cell lung carcinoma (NSCLC) after adenocarcinoma and squamous-cell carcinoma, representing 2%–3% of NSCLCs.1 LCLCs comprise many different subtypes, lack morphological features, and are poorly differentiated. In order to achieve accurate distinction of the histological subtypes of LCLC, cases were regrouped using the 2015 World Health Organization (WHO) classification of lung tumors, characterized by a lacking of histological features and immunomarkers for neuroendocrine, squamous, or glandular differentiation.2 Eventually, there has been a decrease in the number of confirmed LCLC cases. In addition, in comparison to other NSCLC subgroups, LCLC showed an inferior survival, independently from the type of chemotherapy.3
The combination of different therapeutic modalities in the treatment of lung cancer has undergone tremendous progress.4 Tyrosine kinase–inhibitor therapies targeting EGFR mutations and ALK gene rearrangements have strikingly improved patients’ quality of life and survival. The progression of NSCLCs may be primarily driven by the MET pathway, which mainly includes a MET exon 14–skipping mutation and de novo MET amplification with a reported prevalence of 1%–5%.5 More recent studes have focused on MET mutations and amplifications for favorable clinical responses to METinhibitor therapy.6,7 In addition, antibodies targeting PD1 have shown unprecedented durable clinical responses in lung cancer.8,9 Extensive efforts have been made to identify predictive markers for anti-PD1 therapies, such as expression of PDL1 by tumor cells,8,9 tumor mutational burden,10 and DNA mismatch–repair deficiency.11 Moreover, PD1 antibodies were approved as first-line therapy in NSCLC patients with elevated PDL1 expression on tumor-cell surfaces.12 LCLC remains unclear with regard to genetic alterations, and there have been few reports based on PDL1 expression and its prognostic relevance.4,5
In this study, we reclassified LCLCs based on the 2015 WHO classification of tumors, and investigated the overall rate of targetable alterations in LCLC using routine clinical laboratory technologies to evaluate probable therapeutic options and their potential prognostic value.
Methods
Study patients
Cases who had undergone surgical resection between January 2005 and April 2017 were retrieved from the pathological and diagnostic database of Sun Yat-sen University Cancer Center using the keywords “lung” and “large cell carcinoma”. Data on clinical information, sex, age, smoking status, and TNM stage (eighth Union for International Cancer Control TNM staging system for NSCLC) were recorded. Smoking status was categorized as ever or never (<100 lifetime cigarettes). Clinical follow-up information was obtained from the medical records of inpatient or outpatient visits, as well as telephone interviews until September 10, 2018. Overall survival (OS) was calculated from the time of primary surgical resection to patient death from any cause or last contact. The study was approved by the Research Ethics Committee of the Sun Yat-sen University Cancer Center (B2016-069-01).
Immunohistochemistry
Immunohistochemistry (IHC) experiments in each case were conducted using the BenchMark XT automated immunostainer (Ventana, Tucson, AZ, USA) with antibody chromogranin A (CgA; clone LK2H10; Ventana), Syn (clone SP11, 1:50; Geneentech), CD56 (clone 123C3D5; Ventana), TTF1 (clone SP141; Ventana), p63 (clone 4A4 recognizing all p63 gene isoforms, 1:200; Geneentech Company Limited), napsin A (polyclonal; Ventana), p40 (rabbit polyclonal, 1:100; Maixin), and CK5/6 (clone MX040; Maixin). The determination of PDL1 expression was performed using the rabbit monoclonal anti-PDL1 antibody (E1L3N, dilution 1:200; Cell Signaling Technology, Danvers, MA, USA). Negative and positive controls were included in each staining batch. For each marker, the percentage and intensity of positive cells were recorded. PDL1 expression in tumor cells and tumor-infiltrating lymphocytes (TILs) was positive when moderate–strong membrane staining was observed in ≥5% of the tumor cells and ≥1% of TILs, respectively.13 A tumor was considered positive for other diagnostic markers when moderate–strong staining was observed in ≥10% of the tumor cells. IHC analysis was independently performed by two pathologists (JBL and YFF). When disagreement arose, a third pathologist (FW) reconfirmed.
Identification of LCLC
All original H&E and IHC staining slides were reviewed by two experienced pathologists (JBL and YFF), and cases with neuroendocrine morphological features were evaluated for IHC staining with the neuroendocrine markers CD56, CgA, and Syn. Cases without neuroendocrine features were subsequently analyzed for IHC staining with TTF1, napsin A, p40, and CK5/6 to identify glandular or squamous differentiation. In all cases with negative immunomarkers, alcian blue–periodic acid Schiff staining was performed to rule out solid adenocarcinoma with mucin production. Marker-null NSCLC cases reclassified as LCLC according to the 2015 WHO guidelines were further detected for genetic variation.2 Representative images of each diagnostic category are provided in Figure S1.
DNA extraction and genetic variations
AS QIAamp DNA FFPE tissue kit (Qiagen, Hilden, Germany) was used to extract DNA from paraffin-embedded tissue samples, and the presence of tumor cells (>70%) was obtained by trimming the normal and necrotic tissues. Genetic analyses of EGFR, KRAS, and BRAF were performed using the OncoCarta Panel14 (version 1.0; Sequenom, San Diego, CA, USA). Mutation data were analyzed using MassArray Typer software version 4.0 (Sequenom). MET skipping mutation was detected by direct sequencing6 using a PTC-200PCR (Bio-Rad, Hercules, CA, USA) with the forward primer 5’-CTTTGTACGTCTCATGTTAT-3’ and reverse primer 5’-CTCCTAGCGACCTAAC-3’. PCR products were purified and labeled using a BigDye Terminator 3.1 cycle-sequencing kit (Applied Biosystems, Foster City, CA), followed by sequencing in an ABI 3500XL Genetic Analyzer (Applied Biosystems).15
Fluorescence in situ hybridization (FISH)
MET gene copy number per cell was investigated with fluorescence in situ hybridization (FISH) using an MET/chromosome 7 centromere probe (Kreatech Diagnostics, Amsterdam, Netherlands). ALK and ROS1 genes were examined separately with a LSI ALK dual-color, break-apart rearrangement probe and LSI ROS1 dual-color, break-apart rearrangement probe (Vysis; Abbott Laboratories, Chicago, IL, USA). FISH analysis was performed independently by two pathologists (XA and QS). Rearrangement-positive cells were defined as described previously.16,17 Copy number or apart signals per cell were counted in 100 nonoverlapping tumor-cell nuclei. FISH signals were assessed under microscopy (BX51 TRF; Olympus, Tokyo, Japan) equipped with a triple-pass filter (DAPI/green/orange; Vysis). Tumors with five or more MET signals per cell were classified as FISH+ according to the Capuzzo scoring system, including MET polysomy with MET signals five orfewer and fewer than ten and MET amplification characterized by tumor cells with MET:CEP7 ratio greater than or equal to two and ten signals per cell in >10% tumor cells or tight gene clusters (Figure S2).6,18,19
 | Figure S2 Fluorescent in situ hybridization for MET gene (orange) and centromere 7 (green). Notes: Negative (A); MET polysomy (B); MET amplification (C); tight gene clusters (D). Magnification 1,000×. |
Statistical analysis
Differences in distributions of LCLC patients’ characteristics between groups were evaluated by χ2 or Fisher’s test. Cases with second primary cancer were excluded from survival analysis. The Kaplan–Meier method was used to estimate 5-year OS with 95% CIs of LCLC patients in different groups, and two-sided log-rank tests were applied to determine statistical significance. Each genetic and clinical feature is the estimated regression coefficient of a prognostic factor in multivariate Cox regression analysis. All statistical analyses were performed using SPSS 23.0, and P<0.05 was considered statistically significant.
Results
Patient characteristics
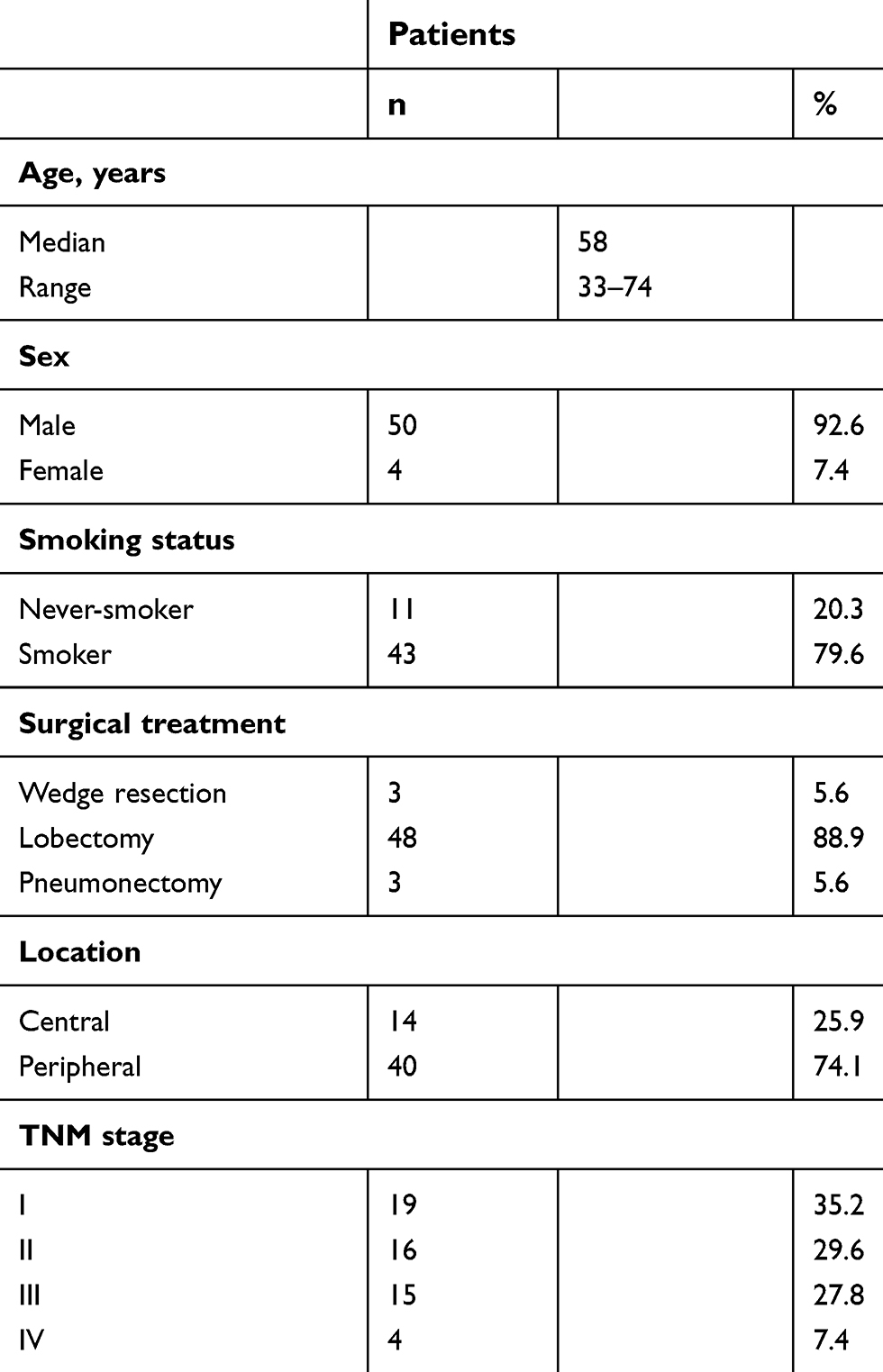
The records of 184 lung cancer patients were retrieved, and based on the study requirements (Figure 1) 54 patients eventually diagnosed with LCLC were found eligible. The clinicopathological characteristics of these LCLC patients are summarized in Table 1. The median age was 58 years (range 33–74 years). The majority of patients were male (92.6%, 50 of 54) and smokers (79.6%, 43 of 54). A total of 48 patients had received lobectomy, three pneumonectomy, and three wedge resection. Nineteen patients (35.2%) were diagnosed as stage I, 16 (29.6%) as stage II, 15 (27.8%) as stage III, and four (7.4%) as stage IV. Fourteen patients received platinum-based adjuvant chemotherapy, two patients radiotherapy, one radiochemotherapy, and three neoadjuvant chemotherapy (one received radiochemotherapy after operation) during the course of the surgical treatment. Additionally, one patient received gefitinib as subsequent treatment, due to a recurrence after resection. The median follow-up was 21.8 months (range 1–147.7 months), and 25 patients (46.3%) died of tumors.
 | Table 1. Baseline characteristics of 54 LCLC patients |
 | Figure 1 Flowchart illustrating LCLC patient enrollment. Abbreviations: SCC, squamous-cell carcinoma; LCNEC, large-cell neuroendocrine carcinoma; AD, adenocarcinoma; LCLC, large-cell lung carcinoma. |
Molecular profiling
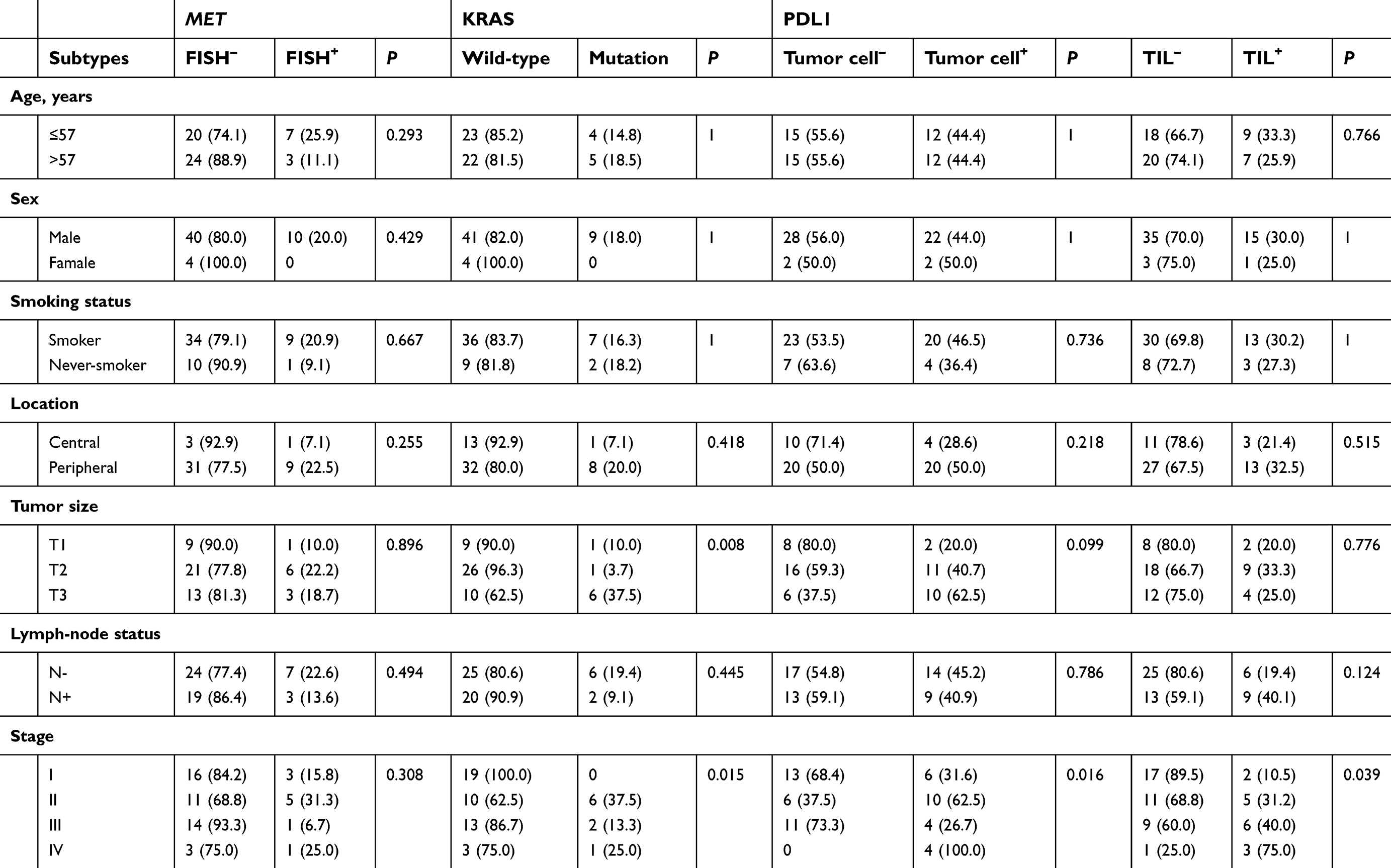
Nine patients (16.7%, nine of 54) harbored KRAS mutations in codon 12 (three G12V, three G12D, two G12C, and one G12A), which were significantly associated with larger tumors (P=0.008, Table 2) and advanced stage (P=0.019, Table 2). Three female patients had an EGFR mutation (two L858R and one E746-A750del), one a BRAFD594N mutation, and there were no cases of ALK, ROS1, or RET rearrangement or MET exon 14-skipping mutations. Ten cases were classified as MET FISH+, with two distinct patterns: amplification (n=5) and polysomy (n=5). In addition, two cases were identified with MET polysomy (signals 5.7 and 6.2) coexisting with KRAS mutation. However, the MET FISH+ had no association with clinicopathological features. PDL1 expression in tumor cells was positive in 24 patients (44.4%, 24 of 54), of which were high expression, with ≥50% of tumor cells with moderate–strong staining (27.8%, 15 of 54). PDL1 expression in TILs was positive in 16 patients (TIL+, 29.6%). The representative images of PDL1 expression are shown in Figure 2. Positive PDL1 expression in both tumor cells and TILs occurred more frequently in patients with stage IV (P=0.016 and 0.039, respectively; Table 2), but showed no significant relationship with other clinicopathological features.
 | Table 2. Correlation of clinical characteristcs of 54 LCLC patients with MET FISH status, KRAS mutations, and PDL1 expression |
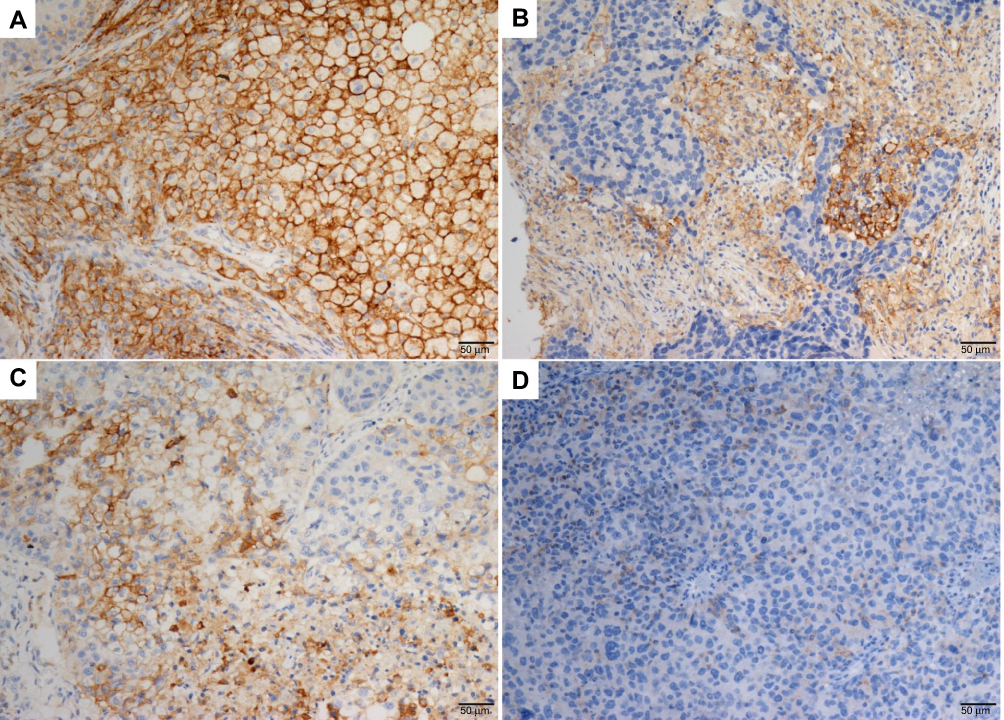 | Figure 2 Representative IHC staining of for PDL1 expression shown in LCLC tissue. Notes: Membranous positive staining of PDL1 in tumor cells (A) and TILs (B). Membranous positive and negative staining of PDL1 in tumor cells (C) and TILs (D). Original magnification ×20. Abbreviations: IHC, immunohistochemistry; LCLC, large-cell lung carcinoma; TILs, tumor-infiltrating lymphocytes. |
LCLC patient survival
Patients with a KRAS mutation had lower OS than those with wild-type KRAS (25.4% vs 47.8%, P=0.028; Figure 3A). There was no significant difference in OS between patients based on MET FISH status (46.7% vs 36.0%, P=0.635; Figure 3B). Positive PDL1 status in tumor cells trended marginally toward poorer OS (56.2% vs 34.0%, P=0.085; Figure 3C). PDL1 expression in TILs showed no significance on 5-year OS (53.0% vs 40.6%, P=0.314; Figure 3D). Further, patients with negative PDL1 staining in tumor cells, but positive expression in TILs (n=6) showed significantly favorable OS compared to those with PDL1-positive tumor cells but negative TILs (n=14, 62.5% vs 20.6%, P=0.044; Figure 3E). There were no significant differences in OS between patients with positive PDL1 in both compartments (n=10) those with both negative PDL1 (n=24, 30% vs 25%; P=0.720). When clinical and genetic variables were considered as prognostic factors, univariate analysis revealed that only KRAS mutation was an independent risk factor for poor OS (HR 2.72, 95% CI 1.07–6.91, P=0.035; Table 3).
 | Table 3. Univariate associations of clinicopathological characteristics, and status of EGFR, KRAS and PDL1 expression with OS in the 54 LCLC patients |
 | Figure 3 OS analysis in LCLC patients with alterations in different genes. Notes: (A, B) Kaplan–Meier model of OS according to KRAS and MET status, respectively. (C, D) Kaplan–Meier model for OS in patients with different PDL1 expression in tumor cells and TILs, respectively. (E) Kaplan–Meier model of OS among patients with different PDL1 expression in both tumor cells and TILs. |
Discussion
Diagnostic terminology on LCLC has been inconsistently applied in clinics, based only on morphology and insufficient IHC markers. This study revealed that KRAS mutations and PDL1 expression were related to patient survival, indicating that KRAS and PDL1 might be therapeutic targets for LCLC patients.
The oncological efficacy of genes in LCLC is generally limited; therefore, LCLC was incorporated in the adenocarcinoma category for treatment and molecular testing in current recommendations in the National Comprehensive Cancer Network guidelines for NSCLC (version 3.2018). EGFR mutations and ALK rearrangement are the key predictive molecular variables for targeted therapy in lung cancer, but their occurrence in LCLC is comparatively rare. One explanation could be that the majority of patients with LCLC are male and smokers. Several studies have reported the mutational landscape of LCLC in comparison with adenocarcinoma or squamous-cell carcinoma. Rekhtman et al found five KRAS codon 12 missense mutations and one BRAFV600E mutation in a set of 20 marker-null large-cell carcinomas (immunomarkers negative for TTF1 and p40).20 Karlsson et al found one each of BRAFQ456K and METT1010I in a set of 12 large-cell carcinomas (immunomarkers negative for TTF1, p40, napsin A, and CK5).21 Driver et al found two BRAFD594N and one KRASG12C in two regrouped large-cell carcinomas using next-generation sequencing covering 50 genes.22 Karlsson et al found only one each of BRAFQ546K and PTENF257L in a set of ten reclassified large-cell carcinomas through gene expression–profiling analysis.23 In the present study, KRAS mutations (16.7%) were identified as showing significant correlation with worsened prognosis, which was consistent with previous studies. KRAS activation is the most common oncogenic alteration in LCLC, but remains controversial as a prognostic marker.24 However, most studies have shown that KRAS mutation was a negative prognostic marker in NSCLC.25,26
MET gene amplification has been identified as one of the mechanisms of acquired resistance to anti-EGFR therapy in patients with activating EGFR mutations.27 Nevertheless, de novo MET amplification is rare, ranging 1%–5% depending on the assay and positivity cut point used, and has been associated with poor outcomes in NSCLC.28 Only this high-level amplification category and MET exon 14 alterations are likely to be predictive of response to MET inhibition, such as with crizotinib.29 In our study, MET FISH+ was determined in ten patients (11.1%), including five cases (9.3%) with true gene amplification, using the Cappuzzo scoring system. Although no MET exon 14 mutation was identified in this study, MET amplification as a relatively high-frequency alteration was found in LCLC compared with other NSCLC, suggesting these patients may benefit from MET inhibitors.
Various issues and clinical trials of PD1 blockade have been reported regarding tumor-cell PDL1 expression as a putative biomarker of therapeutic response. Also, TILs indicating a “T cell–inflamed phenotype” have been assessed for their association with anti-PD(L)1 therapy.30 However, most research has concerned PDL1 expression in adenocarcinoma, squamous-cell carcinoma, small-cell carcinoma, or large-cell neuroendocrine carcinoma, with little on large-cell carcinoma. We used the E1L3N antibody and observed 44.4% and 27.8% positive PDL1 expression on tumor cells and TILs, respectively. Although IHC is a good method for assessment of PDL1 expression, different staining assays may cause differences in PDL1 expression.31,32 The E1L3N assay has been reported to be less sensitive, especially for lower-intensity staining levels, than the SP263 assay. To our knowledge, this is the first integrated report about PDL1 expression of LCLC. Elevated PDL1 in TILs indicated that the PD1–PDL1 pathway might be activated in patients with negative PDL1 expression in tumor cells, and this subgroup with PDL1 expression in solitary TILs obtained an optimal outcome. This found was similar with large-cell neuroendocrine carcinoma.33 In addition, combining PDL1 expression in tumor cells and TILs would help greatly in classifying cancers and selecting patients for anti-PD(L)1 monotherapy or combination therapies.34 Herbst et al found that PDL1 expression on TILs could predict favorable responses to atezolizumab better than PDL1 expression on tumor cells.13
The present study covered a relatively large number of cases with LCLC. However, there are also some limitations that may be of concern. Firstly, it could not entirely uncover the mutational landscape of LCLC using routine clinical technology. Next-generation sequencing technology should be performed to obtain comprehensive knowledge about tumor mutational burden and the tumor microenvironment in further investigations. Secondly, most samples of the advanced cases obtained from small biopsies were excluded, since LCLC can be diagnosed onlyin a surgically resected tumor according to the WHO 2015 criteria. As such, the survival analysis may be biased to a large extent.
Conclusion
We detected main driver-gene alterations and PDL1 status in the 54 patients with marker-null LCLC according to the 2015 WHO classification. KRAS mutation was found to be associated with higher stage of disease and poorer OS, and on the other hand negative PDL1 expression in tumor cells but positive expression in TILs were related with a favorable outcome. Improved genetic understanding can help LCLC patients evaluate probable targeted and immunotherapy regimens.
Ethics approval and consent to participate
The ethics committee of Sun Yat-sen University Cancer Center approved our research (B2016-069-01). Written informed consent was obtained from all individual participants included in the study. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Acknowledgments
This work was supported by grants from the Guangdong Natural Science Foundation (2017A030310192) and Fundamental Research Funds for the Central Universities (17ykpy84).
Author contributions
Concept and design: FW, HYW. Drafting the manuscript: FW, HYW. Acquisition of data: JBL, XA. Analysis and interpretation of data: HYW, YFF, FW. Performing the experiment: JBL, XYW. Final approval of manuscript: all authors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2013. Bethesda (MD): National Cancer Institute.; 2015.
2. Travis WD, Marx A, Nicholson AG. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. Lyon: IARC Press; 2015.
3. Righi L, Vavala T, Rapa I, et al. Impact of non-small-cell lung cancer-not otherwise specified immunophenotyping on treatment outcome. J Thorac Oncol. 2014;9(10):1540–1546. doi:10.1097/JTO.0000000000000271
4. Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389(10066):299–311. doi:10.1016/S0140-6736(16)30958-8
5. Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in lung cancer: will expectations finally be MET? J Thorac Oncol. 2017;12(1):15–26. doi:10.1016/j.jtho.2016.10.014
6. Tong JH, Yeung SF, Chan AW, et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin Cancer Res. 2016;22(12):3048–3056. doi:10.1158/1078-0432.CCR-15-2061
7. Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–859. doi:10.1158/2159-8290.CD-15-0285
8. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi:10.1056/NEJMoa1507643
9. Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi:10.1056/NEJMoa1606774
10. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. doi:10.1126/science.aaa1348
11. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi:10.1126/science.aan6733
12. Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–1550. doi:10.1016/S0140-6736(15)01281-7
13. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi:10.1038/nature14011
14. Zhang ZC, Fu S, Wang F, Wang HY, Zeng YX, Shao JY. Oncogene mutational profile in nasopharyngeal carcinoma. Onco Targets Ther. 2014;7:457–467. doi:10.2147/OTT.S58791
15. Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–5257.
16. Davies KD, Le AT, Theodoro MF, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. 2012;18(17):4570–4579. doi:10.1158/1078-0432.CCR-12-0550
17. Kim MH, Shim HS, Kang DR, et al. Clinical and prognostic implications of ALK and ROS1 rearrangements in never-smokers with surgically resected lung adenocarcinoma. Lung Cancer. 2014;83(3):389–395. doi:10.1016/j.lungcan.2014.01.003
18. Go H, Jeon YK, Park HJ, Sung SW, Seo JW, Chung DH. High MET gene copy number leads to shorter survival in patients with non-small cell lung cancer. J Thorac Oncol. 2010;5(3):305–313. doi:10.1097/JTO.0b013e3181ce3d1d
19. Tanaka A, Sueoka-Aragane N, Nakamura T, et al. Co-existence of positive MET FISH status with EGFR mutations signifies poor prognosis in lung adenocarcinoma patients. Lung Cancer. 2012;75(1):89–94. doi:10.1016/j.lungcan.2011.06.004
20. Rekhtman N, Tafe LJ, Chaft JE, et al. Distinct profile of driver mutations and clinical features in immunomarker-defined subsets of pulmonary large-cell carcinoma. Mod Pathol. 2013;26(4):511–522. doi:10.1038/modpathol.2012.195
21. Karlsson A, Brunnstrom H, Lindquist KE, et al. Mutational and gene fusion analyses of primary large cell and large cell neuroendocrine lung cancer. Oncotarget. 2015;6(26):22028–22037. doi:10.18632/oncotarget.4314
22. Driver BR, Portier BP, Mody DR, et al. Next-generation sequencing of a cohort of pulmonary large cell carcinomas reclassified by World Health Organization 2015 criteria. Arch Pathol Lab Med. 2016;140(4):312–317. doi:10.5858/arpa.2015-0361-OA
23. Karlsson A, Brunnstrom H, Micke P, et al. Gene expression profiling of large cell lung cancer links transcriptional phenotypes to the new histological WHO 2015 classification. J Thorac Oncol. 2017;12(8):1257–1267. doi:10.1016/j.jtho.2017.05.008
24. Zer A, Ding K, Lee SM, et al. Pooled analysis of the prognostic and predictive value of KRAS mutation status and mutation subtype in patients with non-small cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitors. J Thorac Oncol. 2016;11(3):312–323. doi:10.1016/j.jtho.2015.11.010
25. Izar B, Zhou H, Heist RS, et al. The prognostic impact of KRAS, its codon and amino acid specific mutations, on survival in resected stage I lung adenocarcinoma. J Thorac Oncol. 2014;9(9):1363–1369. doi:10.1097/JTO.0000000000000266
26. Wood K, Hensing T, Malik R, Salgia R. Prognostic and predictive value in KRAS in non-small-cell lung cancer: a review. JAMA oncol. 2016;2(6):805–812. doi:10.1001/jamaoncol.2016.0405
27. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi:10.1126/science.1141478
28. Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27(10):1667–1674. doi:10.1200/JCO.2008.19.1635
29. Ou SH, Kwak EL, Siwak-Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6(5):942–946. doi:10.1097/JTO.0b013e31821528d3
30. Taube JM. Unleashing the immune system: PD-1 and PD-Ls in the pre-treatment tumor microenvironment and correlation with response to PD-1/PD-L1 blockade. Oncoimmunology. 2014;3(11):e963413. doi:10.4161/21624011.2014.963413
31. Hutarew G. PD-L1 testing, fit for routine evaluation? From a pathologist‘s point of view. Memo. 2016;9(4):201–206. doi:10.1007/s12254-016-0292-2
32. Rimm DL, Han G, Taube JM, et al. A prospective, multi-institutional, pathologist-based assessment of 4 immunohistochemistry assays for PD-L1 expression in non-small cell lung cancer. JAMA oncol. 2017;3(8):1051–1058. doi:10.1001/jamaoncol.2017.0013
33. Eichhorn F, Harms A, Warth A, Muley T, Winter H, Eichhorn ME. PD-L1 expression in large cell neuroendocrine carcinoma of the lung. Lung Cancer. 2018;118:76–82. doi:10.1016/j.lungcan.2018.02.003
34. Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on t-cell infiltration and PD-L1. Cancer Res. 2015;75(11):2139–2145. doi:10.1158/0008-5472.CAN-15-0255
Supplementary material
 | Figure S1 Representative images of each diagnostic category for LCLC. Notes: According to the 2015 World Health Organization classification of tumors, the morphological features of LCLC were considered through IHC: (A) H&E staining of LCLC; (B) CK staining positive observed in tumors characterized by epithelial origin; (C–E) negative IHC staining with neuroendocrine markers, CD56, CgA, and Syn; (F–J) negative IHC staining with TTF1, napsin A, p40, CK5/6, and p63 to identify glandular or squamous differentiation.Abbreviations: LCLC, large-cell lung carcinoma; IHC, immunohistochemistry. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.