Back to Journals » Clinical Epidemiology » Volume 7
Clinical features, epidemiology, and therapy of lymphangioleiomyomatosis
Authors Taveira-DaSilva AM, Moss J
Received 12 July 2014
Accepted for publication 10 September 2014
Published 7 April 2015 Volume 2015:7 Pages 249—257
DOI https://doi.org/10.2147/CLEP.S50780
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Henrik Toft Sørensen
Angelo M Taveira-DaSilva, Joel Moss
Cardiovascular and Pulmonary Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA
Abstract: Lymphangioleiomyomatosis (LAM) is a multisystem disease of women, characterized by proliferation of abnormal smooth muscle-like LAM cells, leading to the formation of lung cysts, fluid-filled cystic structures in the axial lymphatics (eg, lymphangioleiomyomas), and renal angiomyolipomas. LAM is caused by mutations of the TSC1 or TSC2 genes, which encode, respectively, hamartin and tuberin, two proteins with a major role in control of the mammalian target of rapamycin (mTOR) signaling pathway. LAM occurs sporadically or in association with tuberous sclerosis complex, an autosomal-dominant syndrome characterized by widespread hamartomatous lesions. LAM may present with progressive dyspnea, recurrent pneumothorax, or chylothorax. Pulmonary function tests show reduced flow rates (forced expiratory volume in the first second) and diffusion capacity. Exercise testing may reveal gas exchange abnormalities, ventilatory limitation, and hypoxemia. The severity and progression of disease may be assessed by lung histology scores, quantification of computed tomography, pulmonary function testing, 6-minute walk tests, cardiopulmonary exercise testing, and measurement of serum vascular endothelial growth factor D levels. Sirolimus and everolimus, two mTOR inhibitors, are effective in stabilizing lung function and reducing the size of chylous effusions, lymphangioleiomyomas, and angiomyolipomas. However, inhibition of mTOR complex 1 increases autophagy, possibly enhancing LAM cell survival. Inhibition of autophagy with hydroxychloroquine, in combination with sirolimus, has been proposed as a possible treatment for LAM. Deficiency of tuberin results in increased RhoA GTPase activity and cell survival, an effect that is mediated through mTOR complex 2 signaling. Because sirolimus and everolimus only affect the activity of mTOR complex 1, therapies targeting RhoA GTPases with simvastatin, which inhibits Rho GTPases and promotes apoptosis, are being investigated. As in the case of cancer, LAM may be best treated with multiple drugs targeting signaling pathways considered important in the pathogenesis of disease.
Keywords: lymphangioleiomyomatosis, tuberous sclerosis, TSC1 and TSC2 mutations, mammalian target of rapamycin signaling pathway
Introduction
Lymphangioleiomyomatosis (LAM), a multisystem disorder affecting predominantly women, is characterized by cystic lung destruction and extrapulmonary disease consisting of angiomyolipomas (AMLs), lymphatic tumors, eg, lymphangioleiomyomas, and chylous effusions.1–6 The pathologic features of LAM result from the proliferation of neoplastic cells (LAM cells), which have characteristics of both smooth muscle cells and melanocytes.2,4–6 LAM occurs with increased frequency in patients with tuberous sclerosis complex (TSC), an autosomal-dominant disorder caused by mutations in the TSC1 or TSC2 genes, and characterized by mental retardation, autism, seizures, and hamartomatous lesions in the brain, heart, skin, kidney, eyes, lungs, and liver.7–10 A noninherited form of LAM called sporadic LAM is caused by somatic mutations of the TSC2 gene8–10 and is estimated to have a prevalence of approximately 3.3–7.7 per 1,000,000 women.11
Epidemiology
LAM was once considered to be a fatal disease of women of childbearing age for which there was no effective treatment except for lung transplantation.12–14 Thanks to intensive study in the past 2 decades,1,3,15 LAM is now considered to be a chronic disease that can affect both pre- and postmenopausal women, with a median transplant-free survival of approximately 29 years from the onset of symptoms and a 10-year transplant-free survival of 86%.1,15,16
TSC is a multisystem, autosomal-dominant disorder that occurs in 1 in 12,000–14,000 children aged <10 years or 1 in 6,000 live births.7 TSC is characterized by facial angiofibroma, periungual fibromas, Shagreen patches, cortical tubers, cardiac rhabdomyomas, giant cell astrocytomas, mental retardation, and seizures, in addition to clinical features found in sporadic LAM.7 The prevalence of LAM in TSC was once thought to be 1%–4%,17–20 but subsequent studies showed that the occurrence of cystic lung disease in women with TSC ranges from 26% to 38%.21–23 In a recent study, it was estimated that as many as 80% of women with TSC will develop lung cysts.24 Lung cysts occur in only about 13% of men with TSC,25 who show much less clinically significant disease than women.
Pathology
Histologically, lung lesions consist of LAM cells in the walls of cysts and along blood vessels, lymphatics, and bronchioles, leading to narrowing of the airways, thickening of the vascular walls, lymphatic disruption, and venous occlusion.5,6 Noncystic lesions consist of nodular infiltrates of LAM cells. The center of the nodules contains a preponderance of small, spindle-shaped cells, whereas epithelioid cells with large cytoplasm predominate in the periphery.5,6 Both types of cells react with antibodies against smooth muscle antigens, eg, α-actin, vimentin, and desmin. Epithelioid cells react with human melanin black antibody (HMB-45), a monoclonal antibody that recognizes gp100, a premelanosomal protein encoded by the Pmel17 gene.5,6 The spindle-shaped cells react with antiproliferation cell nuclear antigen antibodies, indicating a proliferative phenotype.5,6
AMLs are characterized histologically by proliferation of smooth muscle-like LAM cells, poorly differentiated blood vessels, and adipocytes.4–6 The blood supply of AML frequently originates from the renal arteries or aberrant blood vessels.4–6,26 Tumors may vary in size from a few millimeters to >20 cm in diameter.26
Lymphangioleiomyomas consist of chyle-filled lymphatic masses of varying sizes that are most frequently localized to the retroperitoneum, pelvis, and posterior mediastinum.4–6,26 Proliferation of epithelioid cells in the walls of lymphatic vessels leads to obstruction of chyle flow and formation of chyle-filled masses.4–6,26
Clinical presentation
Patients often present with a history of recurrent pneumothoraces or progressive dyspnea (Table 1).1–3 Less commonly, the first sign of LAM is a chylous effusion, hemoptysis, abdominal hemorrhage originating from an AML, or the discovery of abdominal or pelvic tumor masses.1–6 Pneumothoraces occur in about 50%–60% of patients and tend to recur.1–3 Patients with larger cysts are more likely to present with pneumothoraces.27 Another frequent mode of presentation is dyspnea on exertion, which worsens with time. These patients show more advanced disease than those presenting with pneumothorax, perhaps because the occurrence of a pneumothorax prompts lung imaging studies that uncover the presence of lung cysts and lead to diagnosis.15
 | Table 1 Clinical features of LAM |
Other modes of presentation include chylothorax, chylous ascites, hemoptysis, chyluria, chyloptysis, abdominal lymphangioleiomyomas suggesting malignancy, and abdominal hemorrhage caused by renal AMLs (Table 1).1,4,26
Lymphatic involvement in LAM occurs in the posterior mediastinal, retroperitoneal, and pelvic areas and consists of lymphangioleiomyomas, lymphadenopathy, pleural effusions, and ascites.4,26,28
In patients with TSC, typical skin lesions include Shagreen patches and facial and periungal angiofibromas (Figure 1); neurologic involvement includes tubers and giant cell astrocytomas.7 The prevalence of extrapulmonary LAM in TSC-LAM patients is different from that in patients with sporadic LAM. In a study comparing computed tomography (CT) scan features of TSC-LAM and sporadic LAM patients, AMLs were found in approximately 93% of patients with TSC and LAM, and 32% of those with sporadic LAM (Table 1).1,4,28 Lymphangioleiomyomas and chylous effusions were more common in sporadic LAM.28 The number of sclerotic bone lesions was greater in patients with TSC-LAM than in those with sporadic LAM.28 Lung disease in TSC may consist of only a few cysts scattered throughout the lungs. These patients may have no clinical symptoms and are usually diagnosed by CT scans. However, those patients with TSC who initially present with pulmonary symptoms may have a clinical course similar to patients with sporadic LAM.24
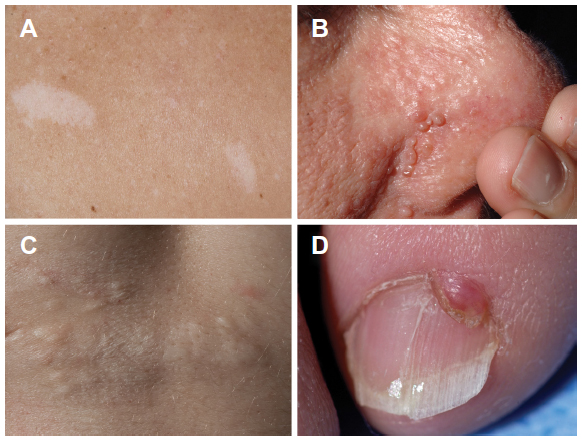 | Figure 1 Skin manifestations of tuberous sclerosis complex in adult women |
Pathogenesis
LAM in the presence of TSC is caused by proliferation of neoplastic LAM cells that have mutations or deletions in one of the tuberous sclerosis genes, TSC1 localized on chromosome 9q34 or, more frequently, TSC2, which is localized on chromosome 16p13.8–10,29 Only TSC2 abnormalities have been found to be associated with sporadic LAM.8–10,29 Loss of heterozygosity of TSC2 has been demonstrated in LAM cells isolated from lung, AMLs, blood, chyle, and urine from patients with sporadic LAM and TSC-LAM.8–10,30,31 LAM cell clusters, consisting of LAM cell aggregates covered by lymphatic endothelial cells, have been identified in chylous fluid.32 Although LAM cells exhibit low-grade features of malignancy, they have metastatic properties. Indeed, LAM cells have been detected in donor lungs of patients who had lung transplantation, suggesting migration to the lungs of cells from other sites, such as the kidney, lymphatic system, or uterus.33–35 Identical TSC2 mutations have been found in the lungs and kidneys of the same patient with sporadic LAM.9
Dysregulation of the mammalian target of rapamycin (mTOR) signaling pathway is the cause of abnormal LAM cell proliferation. TSC1 and TSC2 genes encode, respectively, hamartin and tuberin (Figure 2),36–39 two proteins that regulate the intracellular serine/threonine kinase mTOR signaling pathway. mTOR regulates cell size, proliferation, and survival by integrating signals from growth factors, energy, and stress.36,37 Tuberin is a GTPase-activating protein for the guanine nucleotide-binding protein Rheb (Ras homologue enriched in brain), which promotes the formation of inactive Rheb-GDP from active Rheb-GTP.36 Inhibition or absence of tuberin, as occurs with TSC2 gene mutations, results in accumulation of active Rheb-GTP, stimulation of mTOR, phosphorylation of S6 kinase and eukaryotic initiation factor 4E-binding protein, and increased translation, cell size, and proliferation.37
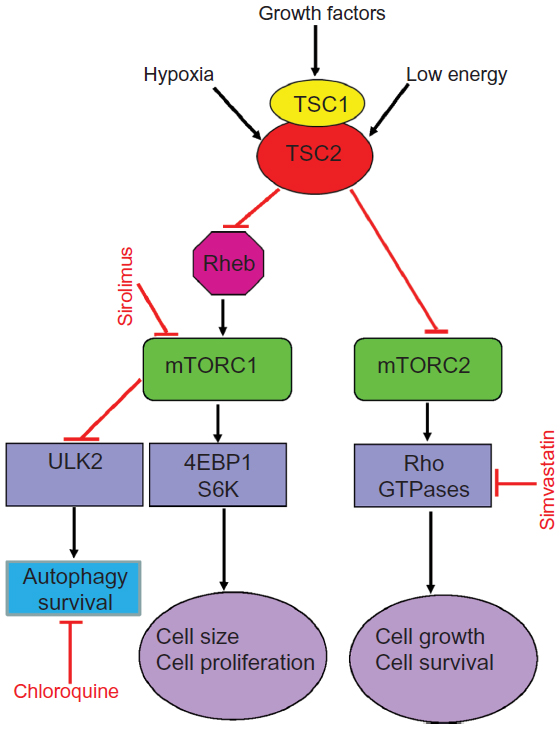 | Figure 2 Simplified scheme of the mTOR signaling pathways showing potential sites of action of some agents tested or in use in LAM. |
Since LAM occurs almost exclusively in women, a role of estrogens in the pathogenesis of LAM was suspected.2 Further, estrogen and progesterone receptors were seen in lung nodules and AML,40–42 predominance of LAM was observed in premenopausal women,1 and worsening of pulmonary symptoms was observed during pregnancy or following the administration of exogenous estrogens.43,44 Moreover, the rate of decline in lung function tends to be greater in premenopausal women than in postmenopausal women.45 Furthermore, in vitro and animal data have shown that estrogens promote the proliferation of TSC-null rat ELT3 leiomyoma-derived cells and human AML TSC2–/– cells in vitro and the growth of subcutaneous tumors in animal models of TSC.46–49 However, there is yet no definite proof that antiestrogen therapies are effective in the treatment of LAM.45
The mechanisms by which LAM cell proliferation causes formation of lung cysts and destruction of the lung parenchyma are under investigation.29,50 Compression of the airways by LAM cells, leading to distention of the terminal airspaces upstream from the occluded airway, has been proposed as a cause of cyst formation.2,5,6 Another potential cause of cystic lesions is degradation of lung elastic fibers caused by proteinases.29,50 This degradation may result from an increase in matrix metalloproteinase (MMP) activity in the lungs, where they have an important role in lung remodeling and lymphangiogenesis.50–52 LAM nodules contain MMP2, MMP9, MMP1, MMP activators (MT1-MMP), and MMP inhibitors (TIMPs).51–53 A reduction in levels of an MMP inhibitor (TIMP-3) was reported in LAM lesions,53,54 and serum levels of MMP-9 were reported to be elevated in patients with LAM.55 An imbalance between MMP and MMP inhibitors may be a contributing factor to destruction of the lung parenchyma.56
Disorganized lymphangiogenesis has an important role in the pathogenesis of LAM.32,50,57–60 Lymphatic spaces are present in LAM lung nodules that have, in addition, immunoreactivity toward vascular endothelial growth factor (VEGF)-C and VEGF-D.32,50,57–60 Vascular endothelial growth factor receptor (VEGFR)-3 and other markers of lymphatic endothelial cells are also noted in the channels found in the lung nodules.50,57–60
Physiologic features
According to the National Heart, Lung, and Blood Institute Lymphangioleiomyomatosis Registry, airflow obstruction was seen initially in approximately 61% of patients with sporadic LAM.1,45 Normal spirometry was present in about 31% of patients with sporadic LAM, whereas normal lung function was observed in 53% of the patients with TSC-LAM (Table 2).1 Air trapping may be associated with severe airflow obstruction. Reduced lung diffusing capacity (DLCO) was reported in about 57% of patients with sporadic LAM,1 with some patients having reduced DLCO and normal flow rates.45
 | Table 2 Pulmonary function abnormalities in LAM |
Gas exchange abnormalities, especially during exercise, are frequently observed in LAM (Table 3).61,62 These abnormalities are characterized by reduced exercise capacity, decreased peak oxygen uptake (VO2 max), reduced breathing reserve, increased ventilatory equivalent for carbon dioxide (VE/VCO2), and hypoxemia (Table 3).61,62
 | Table 3 Cardiopulmonary exercise abnormalities in LAM |
Airflow limitation in LAM had been attributed to alveolar destruction with consequent loss of elastic recoil.63 Studies of lung mechanics showed, however, that airway resistance was increased and lung elastic recoil was not significantly reduced.64 The primary causes of dyspnea and exercise limitation are reduced breathing reserve, dynamic hyperinflation, and an exaggerated ventilatory response to exercise because of limitation in oxygen transfer due to loss of alveolar capillary surface area.61,62,65 Exercise-induced pulmonary hypertension seen in patients with severe disease may also be a factor in reducing oxygen transfer.66
Radiologic findings
Chest radiographic findings may be normal or show an interstitial pattern or cystic changes. CT shows well-defined, round, thin-walled cysts scattered throughout the lungs, which may vary in size from a few millimeters to 2 cm (Figure 3).67–69 In patients with lymphatic involvement, pleural effusions and lung infiltrates due to chyle in the interstitium may be present. There is correlation between the extent of the cystic destruction, pulmonary function, and exercise performance.62,67–72
 | Figure 3 Computed tomography scans showing pulmonary and extrapulmonary images of patients with LAM. |
AMLs, lymphadenopathy, lymphangioleiomyomas, and ascites may be visualized by ultrasonography, CT scans, or magnetic resonance imaging.26,28 AMLs occur predominantly in the kidney and liver, appearing as tumor masses comprising fatty areas, mixed with kidney parenchyma (Figure 3).26,28 Atypical AMLs lack fatty tissue, comprising predominantly epithelioid LAM cells that may suggest renal cell carcinoma.4–6,73
Lymphangioleiomyomas appear as well-circumscribed masses of variable dimensions, comprising an outer wall and a fluid-rich region central region (Figure 3).26,28,74–76 The size of lymphangioleiomyomas tends to be greater in the evening than in the morning, which may help differentiating them from malignant tumors.75,76
Diagnosis
The diagnosis of LAM should be strongly suspected in any woman who presents with progressive dyspnea, recurrent pneumothoraces, or chylous pleural effusions.1,4,6 The diagnosis of definite LAM may be made in the presence of a characteristic CT scan and a transbronchial, thoracoscopic, or open lung biopsy29,77–79 showing the characteristic histological features of LAM and reactivity with monoclonal antibody HMB-45.4–6 Alternatively, the presence of a characteristic lung CT and AML, chylous effusions, lymphangioleiomyoma, or TSC are sufficient to establish the diagnosis.79 The presence of a characteristic CT scan in the absence of extrapulmonary findings is not diagnostic of LAM.79
An important biomarker of value in diagnosing LAM is VEGF-D, a lymphangiogenic growth factor that is increased in the serum of patients with LAM, especially those with lymphatic involvement.58–60 In the appropriate clinical and radiologic setting, a VEGF-D serum level ≥800 pg/mL is rarely found in other cystic lung diseases.60 VEGF-D may be of value in grading the severity of disease and monitoring therapeutic response to mTOR inhibitors.60
The differential diagnosis of LAM includes emphysema, sarcoidosis, Langerhans cell histiocytosis, Birt–Hogg–Dubé syndrome, follicular bronchiolitis, light chain disease, Sjögren’s syndrome, allergic alveolitis, bronchiectasis, Mounier–Kuhn syndrome, pneumoconiosis, and chronic lung infectious processes. In general, most of these conditions can be easily excluded on the basis of clinical, histopathological, and radiologic findings.2,79
Prognosis
Clinical, pathologic, and physiologic data may assist in estimating the prognosis of newly diagnosed patients with LAM. Premenopausal patients may have a faster decline in lung function than older, postmenopausal women.45,80 Patients who give a history of pneumothorax as the first sign of LAM present with better lung function than those experiencing dyspnea on exertion.15,79 Indeed, patients presenting with dyspnea have lower survival rates than patients whose initial symptom is related to a pneumothorax.81 Moreover, in patients with forced expiratory volume in the first second (FEV1) and DLCO >40% predicted, the rate of functional decline in patients presenting with dyspnea on exertion was greater than in those presenting with pneumothorax.81
In patients who have had a lung biopsy, a predominance of cystic lesions instead of cellular infiltrates is associated with worse prognosis.82,83 The presence of hemosiderin-laden macrophages in the biopsy tissue is also associated with more severe disease.82 Patients with more cystic disease tend to have lower FEV1 and DLCO, lower VO2 max, greater exercise-induced hypoxemia, and lower survival than patients with predominantly nodular lesions.82,83 Findings on CT scans also correlate with lung function tests, gas exchange, and exercise performance,62,67–69 especially if the percentage of lung volume affected by cysts and texture changes in the vicinity of cysts are quantified.71,72
Overall, pulmonary function testing is the most practical method of assessing the severity of lung disease in LAM.1,45 In patients with near normal flow rates and a reduced diffusion capacity, the severity of lung disease is best graded by a 6-minute walk test or cardiopulmonary exercise testing.61,62,70 Estimation of rates of FEV1 and DLCO declining over time may help in defining whether lung disease is rapidly or slowly progressing.45,80 A positive response to bronchodilators, which occurs in 25%–30% of LAM patients, may be a predictor of disease severity and rate of progression.83,84
VEGF-D levels show a correlation with DLCO and CT scan as measures of severity of lung disease.58,59 Analysis of data from the Multicenter International Lymphangioleiomyomatosis Efficacy and Safety of Sirolimus (MILES) trial showed that serum levels of VEGF-D correlated positively with use of oxygen, bronchodilator response, lower quality of life, decreased ability to perform daily living activities, lower diffusion capacity, and more severe airflow obstruction.60
Treatment
Not long ago, LAM was defined as a fatal disease of young women, for which the only treatment option was lung transplantation. The finding that LAM is caused by mutations of the TSC1 or TSC2 genes, which encode hamartin and tuberin, two proteins with a major role in control of the mTOR signaling pathway, led to therapies targeting mTOR (Figure 2).8–10,36 mTOR is the catalytic subunit of two distinct complexes named mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (Figure 2).85 Sirolimus and everolimus, two mTORC1 inhibitors, have been shown to be effective in stabilizing lung function and reducing the size of chylous effusions, lymphangioleiomyomas, and AMLs.86–88 Treatment with sirolimus or everolimus is now recognized as the standard therapy for patients with declining lung function, symptomatic chylous effusions, lymphangioleiomyomas, or large AMLs.86–88 However, discontinuation of mTOR inhibitor therapy results in further decline in lung function or return of AMLs to pretherapy tumor size, suggesting that treatment has to be continued.86,88 Inhibition of mTOR results in increased autophagy and possibly enhanced LAM cell survival, reducing the beneficial effects of mTOR inhibitors (Figure 2).50,89 Inhibition of autophagy with chloroquine in combination with mTOR inhibition is now being examined as a possible treatment for LAM.89 Deficiency of tuberin due to TSC2 mutations results in increased RhoA GTPase activity and increased cell survival,90 an effect mediated through mTORC2 signaling (Figure 2).90 Because sirolimus and everolimus only inhibit the activity of mTORC1, it has been suggested that therapies targeting RhoA GTPases should be considered (Figure 2).90,91 Statins, namely simvastatin, inhibit Rho GTPases and promote apoptosis.56,92 In combination with sirolimus, simvastatin was reported to prevent lung destruction in a mouse model of LAM.56 A Phase I clinical trial testing the effect of simvastatin combined with either sirolimus or everolimus in patients with LAM is underway.
Other treatments that are under investigation are estrogen receptor blockers and aromatase inhibitors.50,57,91 Because of the proposed role of lymphangiogenesis in the pathogenesis of LAM,57 and evidence that increased levels of VEGF-D in the serum of LAM patients correlate with disease severity and clinical course, blockade of VEGFRs or anti-VEGF therapies are also being considered.50,57 In an animal model of LAM, combination of VEGFR1, VEGFR2, and VEGFR3 inhibitor and sirolimus decreased tumor volume and increased survival more effectively than sirolimus alone.93
LAM is considered to be a low-grade malignancy and, as in the case of cancer, treatment with drugs targeting signaling pathways considered important in the pathogenesis of the disease may eventually turn out to be effective. However, most ongoing clinical studies are in a preliminary stage, and there is yet no evidence that they will prove to be effective.
Acknowledgment
The investigators were supported by the Intramural Research Program, National Institutes of Health, National Heart, Lung, and Blood Institute.
Disclosure
The authors report no conflicts of interest in this work.
References
Ryu JH, Moss J, Beck GJ, et al. The NHLBI Lymphangioleiomyomatosis Registry. Characteristics of 230 patients at enrollment. Am J Respir Crit Care Med. 2006;173(1):105–111. | |
McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest. 2008;133(2):507–516. | |
Urban T, Lazor R, Lacronique J, et al. Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM“O”P). Medicine. 1999;78(5):321–327. | |
Matsui K, Tatsuguchi A, Valencia J, et al. Extrapulmonary lymphangioleiomyomatosis (LAM): clinicopathologic features in 22 cases. Hum Pathol. 2000;31(10):12421248. | |
Abbott GF, Rosado-de-Christenson ML, Frazier AA, Franks TJ, Pugatch RD, Galvin JR. From the archives of the AFIP: lymphangioleiomyomatosis: radiologic-pathologic correlation. Radiographics. 2005;25(3):803–828. | |
Ferrans VJ, Yu ZX, Nelson WK, et al. Lymphangioleiomyomatosis (LAM). A review of clinical and morphological features. J Nippon Med Sch. 2000;67(5):311–329. | |
Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–668. | |
Smolarek TA, Wessner LL, McCormack FX, Mylet JC, Menon AG, Henske EP. Evidence that lymphangiomyomatosis is caused by TSC2 mutations, chromosome 16p13 loss of heterozygosity in angiomyolipomas and lymph nodes from women with lymphangiomyomatosis. Am J Hum Genet. 1998;62(4):810–815. | |
Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2000;97(11):6085–6090. | |
Yu J, Astrinidis A, Henske EP. Chromosome 16 loss of heterozygosity in tuberous sclerosis and sporadic lymphangiomyomatosis. Am J Respir Crit Care Med. 2001;164(8):1537–1540. | |
Harknett EC, Chang WY, Byrnes S, et al. Use of variability in national and regional data to estimate the prevalence of lymphangioleiomyomatosis. Q J Med. 2011;104(11):971–979. | |
Silversteen EF, Ellis K, Wolff M, Jaretzki A. Pulmonary lymphangiomyomatosis. Am J Roentgenol. 1974;120(4):832–850. | |
Corrin B, Liebow AA, Friedman PJ. Pulmonary lymphangioleiomyomatosis: a review. Am J Pathol. 1975;79(2):348–382. | |
Taylor JR, Ryu J, Colby TV, Raffin TA. Lymphangioleiomyomatosis: clinical course in 32 patients. N Engl J Med. 1990;323(18):1254–1260. | |
Cohen MM, Pollock-BarZiv S, Johnson SR. Emerging clinical picture of lymphangioleiomyomatosis. Thorax. 2005;60(10):875–879. | |
Oprescu N, McCormack FX, Byrnes S, Kinder BW. Clinical predictors of mortality and cause of death in lymphangioleiomyomatosis: a population-based registry. Lung. 2013;191(1):35–42. | |
Dwyer JM, Hickie JB, Garvan J. Pulmonary tuberous sclerosis. Q J Med. 1971;40(157):115–125. | |
Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in tuberous sclerosis. Mayo Clin Proc. 1991;66(8):792–796. | |
Castro M, Shepherd CW, Gomez MR, Lie JT, Ryu JH. Pulmonary tuberous sclerosis. Chest. 1995;107(1):189–195. | |
Hancock E, Tomkins S, Sampson J, Osborne J. Lymphangioleiomyomatosis and tuberous sclerosis. Respir Med. 2002;96(1):7–13. | |
Costello LC, Hartman TE, Ryu JH. High frequency of pulmonary lymphangioleiomyomatosis in women with tuberous sclerosis complex. Mayo Clin Proc. 2000;75(6):591–594. | |
Moss J, Avila N, Barnes PM, et al. Prevalence and clinical characteristics of lymphangioleiomyomatosis (LAM) in patients with tuberous sclerosis complex. Am J Respir Crit Care Med. 2001;164(4):669–471. | |
Franz DN, Brody A, Meyer C, et al. Mutational and radiographic analysis of pulmonary disease consistent with lymphangioleiomyomatosis and micronodular pneumocyte hyperplasia in women with tuberous sclerosis. Am J Respir Crit Care Med. 2001;164(4):661–668. | |
Cudzilo CJ, Szczesniak RD, Brody AS, et al. Lymphangioleiomyomatosis screening in women with tuberous sclerosis. Chest. 2013;144(2):578–585. | |
Adriaensen ME, Schaefer-Prokop CM, Duyndam DAC, Zonnenberg BA, Prokop M. Radiological evidence of lymphangioleiomyomatosis in female and male patients with tuberous sclerosis complex. Clin Radiol. 2011;66(7):625–628. | |
Avila NA, Kelly JA, Chu SC, Dwyer AJ, Moss J. Lymphangioleiomyomatosis: abdominopelvic CT and US findings. Radiology. 2000;216(1):147–153. | |
Steagall WK, Glasgow CG, Hathaway OM, et al. Genetic and morphologic determinants of pneumothorax in lymphangioleiomyomatosis. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L800–L808. | |
Avila NA, Dwyer AJ, Rabel A, Moss J. Sporadic lymphangioleiomyomatosis and tuberous sclerosis complex with lymphangioleiomyomatosis, comparison of CT features. Radiology. 2007;242(1):277–285. | |
Meraj R, Wikenheiser-Brokamp KA, Young LR, McCormack FX. Lymphangioleiomyomatosis: new concepts in pathogenesis, diagnosis, and treatment. Semin Respir Crit Care Med. 2012;33(5):486–497. | |
Crooks DM, Pacheco-Rodriguez G, DeCastro RM, et al. Molecular and genetic analysis of disseminated neoplastic cells in lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2004;101(50):17462–17467. | |
Cai X, Pacheco-Rodriguez G, Fan QY, et al. Phenotypic characterization of disseminated cells with TSC2 loss of heterozygosity in patients with lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2010;182(11):1410–1418. | |
Mitani K, Kumasaka T, Takemura H, et al. Cytologic, immunocytochemical and ultrastructural characterization of lymphangioleiomyomatosis cell clusters in chylous effusions of patients with lymphangioleiomyomatosis. Acta Cytol. 2009;53(4):402–409. | |
Bittmann I, Rolf B, Amann G, Löhrs U. Recurrence of lymphangioleiomyomatosis after single lung transplantation, new insights into pathogenesis. Human Pathol. 2003;34(1):95–98. | |
Karbowniczek M, Astrinidis A, Balsara BR, et al. Recurrent lymphangioleiomyomatosis after transplantation. Am J Respir Crit Care Med. 2003;167(7):976–982. | |
Hayashi T, Kumasaka T, Mitani K, et al. Prevalence of uterine and adnexal involvement in pulmonary lymphangioleiomyomatosis: a clinicopathologic study of 10 patients. Am J Surg Pathol. 2011;35(12):1776–1785. | |
Rosner M, Hanneder M, Siegel N, Valli A, Hengstschläger M. The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat Res. 2008;658(3):234–246. | |
Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cel. 2010;40(2):310–322. | |
Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cystoskeleton. Curr Biol. 2004;14(14):1296–1302. | |
Huang J, Manning BD. A complex interplay between Akt, TSC2 and the mTOR complexes. Bioch Soc Trans. 2009;37(P1):217–222. | |
Berger U, Khaghani A, Pomerance A, Yacoub MH, Coombes RC. Pulmonary lymphangioleiomyomatosis and steroid receptors. Am J Clin Pathol. 1990;93(5):609–614. | |
Ohori NP, Yousem SA, Sonmez-Alpan E, Colby TV. Estrogen and progesterone receptors in lymphangioleiomyomatosis, epithelioid hemangioendothelioma, and sclerosing hemangioma of the lung. Am J Clin Pathol. 1991;96(4):529–535. | |
Logginidou H, Ao X, Russo I, Henske EP. Frequent estrogen and progesterone receptor immunoreactivity in renal angiomyolipomas from women with pulmonary lymphangioleiomyomatosis. Chest. 2000;117(1):25–30. | |
Brunelli A, Catalini G, Fianchini A. Pregnancy exacerbating unsuspected mediastinal lymphangioleiomyomatosis and chylothorax. Int J Gynaecol Obstet. 1996;52(3):289–290. | |
Yano S. Exacerbation of pulmonary lymphangioleiomyomatosis by exogenous oestrogen used for infertility treatment. Thorax. 2002;57(12):1085–1086. | |
Taveira-DaSilva AM, Stylianou MP, Hedin CJ, Hathaway O, Moss J. Decline in lung function in patients with lymphangioleiomyomatosis treated with or without progesterone. Chest. 2004;126(6):1867–1874. | |
Howe SR, Gottardis MM, Everitt JI, Walker C. Estrogen stimulation and tamoxifen inhibition of leiomyoma cell growth in vitro and in vivo. Endocrinology. 1995;136(11):4996–5003. | |
Yu J, Astrinidis A, Howard S, Henske EP. Estradiol and tamoxifen stimulate LAM-associated angiomyolipoma cell growth and activate both genomic and nongenomic signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L694–L700. | |
Yu JJ, Robb VA, Morrison TA, et al. Estrogen promotes the survival and pulmonary metastasis of tuberin-null cells. Proc Natl Acad Sci U S A. 2009;106(8):2635–2640. | |
Glassberg MK, Elliot SJ, Fritz J, et al. Activation of the estrogen receptor contributes to the progression of pulmonary lymphangioleiomyomatosis via matrix metalloproteinase-induced cell invasiveness. J Clin Endocrinol Metab. 2008;93(5):1625–1633. | |
Henske EP, McCormack FX. Lymphangioleiomyomatosis – a wolf in sheep’s clothing. J Clin Invest. 2012;122(11):3807–3816. | |
Matsui K, Takeda K, Yu Z-X, Travis WD, Moss J, Ferrans VJ. Role for activation of matrix metalloproteinases in the pathogenesis of pulmonary lymphangioleiomyomatosis. Arch Pathol Lab Med. 2000;124(2):267–275. | |
Krymskaya VP, Shipley JM. Lymphangioleiomyomatosis. A complex tale of serum response factor-mediated tissue inhibitor of metalloproteinase-3 regulation. Am J Respir Cell Mol Biol. 2003;28(5):546–550. | |
Papakonstantinou E, Dionyssopoulos A, Aletras AJ, Pesintzaki C, Minas A, Karakiulakis G. Expression of matrix metalloproteinases and their endogenous tissue inhibitors in skin lesions from patients with tuberous sclerosis. J Am Acad Dermatol. 2004;51(4):526–533. | |
Zhe X, Yang Y, Jakkaraju S, Schuger L. Tissue inhibitor of metalloproteinase-3 downregulation in lymphangioleiomyomatosis. Am J Respir Cell Mol Biol. 2003;28(4):504–511. | |
Odajima N, Betsuyaku T, Nasuhara Y, Inoue H, Seyama K, Nishimura M. Matrix metalloproteinases in blood from patients with LAM. Respir Med. 2009;103(1):124–129. | |
Goncharova EA, Goncharov DA, Fehrenbach M, et al. Prevention of alveolar destruction and airspace enlargement in a mouse model of pulmonary lymphangioleiomyomatosis (LAM). Sci Transl Med. 2012;4(154):154ra134. | |
Yu J, Henske EP. mTOR activation, lymphangiogenesis, and estrogen-mediated cell survival: the “perfect storm” of pro-metastatic factors in LAM pathogenesis. Lymphat Res Biol. 2010;8(1):43–49. | |
Seyama K, Kumasaka T, Souma S, et al. Vascular endothelial growth factor-D is increased in serum of patients with lymphangioleiomyomatosis. Lymphat Res Biol. 2006;4(3):143–152. | |
Glasgow CG, Avila NA, Lin JP, Stylianou MP, Moss J. Serum vascular endothelial growth factor-D levels in patients with lymphangioleiomyomatosis reflect lymphatic involvement. Chest. 2009;135(5):1293–1300. | |
Young L, Lee HS, Inoue Y, et al. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med. 2013;1(4):445–452. | |
Crausman RS, Jennings CA, Mortensen RL, Ackerson LM, Irvin CG, King TE Jr. Lymphangioleiomyomatosis: the pathophysiology of diminished exercise capacity. Am J Respir Crit Care Med. 1996; 153(4 Pt 1):1368–1376. | |
Taveira-DaSilva AM, Stylianou MP, Hedin CJ, et al. Maximal oxygen uptake and severity of disease in lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2003;168(12):1427–1431. | |
Sobonya RE, Quan SF, Fleishman JS. Pulmonary lymphangioleiomyomatosis: quantitative analysis of lesions producing airflow limitation. Hum Pathol. 1985;16(11):1122–1228. | |
Burger CD, Hyatt RE, Stats BA. Pulmonary mechanics in lymphangioleiomyomatosis. Am Rev Respir Dis. 1991;143(5 Pt 1):1030–1033. | |
Baldi BG, Albuquerque AL, Pimenta SP, Salge JM, Kairalla RA, Carvalho CR. Exercise performance and dynamic hyperinflation in lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2012;186(4):341–348. | |
Taveira-DaSilva AM, Hathaway OM, Sachdev V, Shizukuda Y, Birdsall CW, Moss J. Pulmonary artery pressure in lymphangioleiomyomatosis: an echocardiographic study. Chest. 2007;132(5):1573–1578. | |
Crausman RS, Lynch DA, Mortensen RL, et al. Quantitative CT predicts the severity of physiologic dysfunction in patients with lymphangiomyomatosis. Chest. 1996;109(1):131–137. | |
Avila NA, Chen CC, Chu SC, et al. Pulmonary lymphangioleiomyomatosis: correlation of ventilation-perfusion scintigraphy, chest radiography, and CT with pulmonary function tests. Radiology. 2000;214(2):441–446. | |
Avila NA, Dwyer AJ, Murphy-Johnson DV, Brooks P, Moss J. Lymphangioleiomyomatosis: correlation of qualitative and quantitative thin-section CT with pulmonary function tests and assessment of dependence on pleurodesis. Radiology. 2002;223(1):189–197. | |
Paciocco G, Uslenghi E, Bianchi A, et al. Diffuse cystic lung diseases. Correlation between radiologic and functional status. Chest. 2004;125(1):135–142. | |
Schmithorst VJ, Altes TA, Young LR, et al. Automated algorithm for quantifying the extent of cystic change on volumetric chest CT: initial results in lymphangioleiomyomatosis. AJR Am J Roentgenol. 2009;192(4):1037–1044. | |
Yao J, Taveira-DaSilva AM, Colby TV, Moss J. Computed tomography grading of lung disease in lymphangioleiomyomatosis. AJR Am J Roentgenol. 2012;199(4):787–793. | |
Bissler JJ, Kingswood JC. Renal angiomyolipomata. Kidney Int. 2004;66(3):924–934. | |
Avila NA, Dwyer AJ, Moss J. Imaging features of lymphangioleiomyomatosis: diagnostic pitfalls. AJR Am J Roentgenol. 2011;196(4):982–986. | |
Avila NA, Dwyer AJ, Murphy-Johnson DV, Brooks P, Moss J. Sonography of lymphangioleiomyoma in lymphangioleiomyomatosis: demonstration of diurnal variation in lesion size. A J R Am J Roentgenol. 2005;184(2):459–464. | |
Avila NA, Bechtle J, Dwyer AJ, Ferrans VJ, Moss J. Lymphangioleiomyomatosis: CT of diurnal variation of lymphangioleiomyomas. Radiology. 2001;221(2):415–421. | |
Harari S, Torre O, Cassandro R, Taveira-DaSilva AM, Moss J. Bronchoscopic diagnosis of Langerhans cell histiocytosis and lymphangioleiomyomatosis. Respir Med. 2012;106(9):1286–1292. | |
Meraj R, Wikenheiser-Brokamp KA, Young LR, Byrnes S, McCormack FX. Utility of transbronchial lung biopsy in the diagnosis of lymphangioleiomyomatosis. Front Med. 2012;6(4):395–405. | |
Johnson SR, Cordier JF, Lazor R, et al. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J. 2010;35(1):14–26. | |
Johnson SR, Tattersfield AE. Decline in lung function in lymphangioleiomyomatosis: relation to menopause and progesterone treatment. Am J Respir Crit Care Med. 1999;160(2):628–633. | |
Hayashida M, Seyama K, Inoue Y, et al. Respiratory Failure Research Group of the Japanese Ministry of Health, Labor, and Welfare. The epidemiology of lymphangioleiomyomatosis in Japan: a nationwide cross-sectional study of presenting features and prognostic factors. Respirology. 2007;12(4):523–530. | |
Matsui K, Beasley MB, Nelson WK, et al. Prognostic significance of pulmonary lymphangioleiomyomatosis histologic score. Am J Surg Path. 2001;25(4):479–484. | |
Taveira-DaSilva AM, Hedin CJ, Stylianou MP, et al. Reversible airflow obstruction, proliferation of abnormal smooth muscle cells and impairment of gas exchange as predictors of outcome in lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2001;164(6):1072–1076. | |
Taveira-Dasilva AM, Steagall WK, Rabel A, et al. Reversible airflow obstruction in lymphangioleiomyomatosis. Chest. 2009;136(6):1596–1603. | |
Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):31–35. | |
McCormack FX, Inoue Y, Moss J, et al. National Institutes of Health Rare Lung Diseases Consortium; MILES Trial Group. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364(17):1595–1606. | |
Taveira-DaSilva AM, Hathaway O, Stylianou M, Moss J. Changes in lung function and chylous effusions in patients with lymphangioleiomyomatosis treated with sirolimus. Ann Intern Med. 2011;154(12):797–805. | |
Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817–824. | |
Yu J, Parkhitko AA, Henske EP. Mammalian target of rapamycin signaling and autophagy: roles in lymphangioleiomyomatosis therapy. Proc Am Thorac Soc. 2010;7(1):48–53. | |
Goncharova EA, Goncharova DA, Li H, et al. mTORC2 is required for proliferation and survival of TSC2-null cells. Mol Cell Biol. 2011;31(12):2484–2498. | |
Hammes SR, Krymskaya VP. Targeted approaches towards understanding and treating pulmonary lymphangioleiomyomatosis. Horm Canc. 2012;4(2):70–77. | |
Atochina-Vasserman EN, Goncharov DA, Volgina AV, Milavec M, James ML, Krymskaya VP. Statins in lymphangioleiomyomatosis. Simvastatin and atorvastatin induce differential effects on tuberous sclerosis complex 2-null cell growth and signaling. Am J Respir Cell Mol Biol. 2013;49(5):704–709. | |
Lee N, Woodrum CL, Nobil AM, Rauktys AE, Messina MP, Dabora SL. Rapamycin weekly maintenance dosing and the potential efficacy of combination of sorafenib plus rapamycin but not atorvastatin or doxycycline in tuberous sclerosis preclinical models. BMC Pharmacol. 2009;9:8. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.