")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 20
Clinical Features and Prognostic Analysis of MuSK-Antibody-Positive Myasthenia Gravis versus Double-Seropositive Myasthenia Gravis: A Single-Center Study from Central South China
Authors He T, Chen K, Li Y, Luo Z, Luo M , Yang H
Received 2 December 2023
Accepted for publication 13 March 2024
Published 29 March 2024 Volume 2024:20 Pages 725—735
DOI https://doi.org/10.2147/NDT.S450651
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Taro Kishi
Ting He,1,* Kangzhi Chen,1,* Yi Li,1 Zhaohui Luo,1 Mengchuan Luo,2 Huan Yang1
1Department of Neurology, Xiangya Hospital, Central South University, Changsha, 410008, People’s Republic of China; 2Department of Geriatrics, Xiangya Hospital, Central South University, Changsha, 410008, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Huan Yang, Department of Neurology, Xiangya Hospital, Central South University, 87 Xiangya Road, Changsha, 410008, People’s Republic of China, Email [email protected]
Purpose: To decipher the discrepancies between muscle-specific kinase antibody-positive myasthenia gravis (MuSK-MG) and double-seropositive myasthenia gravis (DSP-MG), and to determine prognostic factors for minimal manifestation status (MMS) achievement in MG patients with MuSK autoantibodies (MuSK-Ab).
Patients and Methods: A total of 34 MG patients seropositive for MuSK-Ab were enrolled in this study. The demographic and clinical features were compared between MuSK-MG (n = 28) and DSP-MG (n = 6) patients, and factors affecting MMS induction in all patients with MuSK-Ab were identified using Cox regression analysis.
Results: Compared to MuSK-MG patients, those with DSP-MG had similar clinical characteristics, except that they had a lower frequency of bulbar muscle involvement at nadir (50% vs 92.9%; P = 0.029) and higher proportions of comorbidities with diabetes mellitus (33.3% vs 0%; P = 0.027) and thymic abnormalities (33.3% vs 0%; P = 0.027). Higher MG Activities of Daily Living (MG-ADL) scores (HR = 0.16, 95% CI: 0.037– 0.7, P = 0.015) and axial muscle involvement at nadir (HR = 0.39, 95% CI: 0.16– 0.94, P = 0.035) were negative prognostic factors for MMS achievement in patients with MuSK-Ab regardless of acetylcholine receptor antibody (AChR-Ab) positivity. Multivariable Cox regression analysis further established higher MG-ADL scores at the nadir (HR = 0.19, 95% CI: 0.04– 0.94; P = 0.042) as an independent risk factor for MMS achievement.
Conclusion: DSP-MG was comparable to MuSK-MG and could be considered a single entity in our cohort. In all MG patients with MuSK-Ab, a higher MG-ADL score at nadir may herald a lower chance of MMS achievement, with no observed potential effect of AChR-Ab presence.
Keywords: double-seropositive myasthenia gravis, muscle-specific kinase antibody, acetylcholine receptor antibody, minimal manifestation status, myasthenia gravis activities of daily living
Introduction
Myasthenia gravis (MG) is a typical autoimmune disorder involving the dysfunction of acetylcholine signaling at the neuromuscular junction (NMJ) that incurs muscle weakness, where functional autoantibodies directed against nicotinic acetylcholine receptor (AChR), muscle-specific kinase (MuSK), lipoprotein-receptor-related protein 4 (LRP4) and other postsynaptic proteins represent the culprits.1 Most patients with MG are positive for AChR autoantibodies (AChR-Ab), while MuSK autoantibodies (MuSK-Ab) have been detected in AChR-Ab-seronegative individuals with a wide range of prevalence of 0% to 70% possibly due to the diversity of cohorts and detection methods.2 MuSK is indispensable for NMJ formation and facilitates AChR clustering in response to nerve-terminal-derived agrin,3 which binds to LRP4 that forms a heterotetrametric complex with MuSK to enhance the phosphorylation and activation of MuSK.4 Collagen Q (ColQ), the collagenic tail subunit of acetylcholinesterase (AChE) that regulates AChR clustering, is also anchored via interaction with MuSK.5 Thus, the presence of MuSK-Ab could interfere with the signaling cascade for neuromuscular transmission and further the normal activity of the nerve-innervated muscle contraction.
MuSK-Ab mainly recognize the extracellular Ig-like domains, especially Ig1-2 of MuSK, and the majority of them are of the IgG4 subclass with myasthenogenic activity.6–8 In contrast to dominance of IgG1 and IgG3 subclasses in AChR-MG that mediate pathogenic effects via complement activation, antigenic modulation and direct blockade of AChR, the IgG4 subclass of MuSK-Ab seems to play roles greatly by disrupting the interactions among the postsynaptic proteins.9 Intriguingly, MuSK-IgG4 may undergo Fab arm exchange, a unique process that endows the autoantibody with bispecific Fab fragments and functional monovalency, which was demonstrated to exacerbate the pathogenicity of MuSK-Ab.10,11 In addition, MuSK-Ab are more likely to be produced by short-lived plasmablasts (SLPB) whereas AChR-Ab stem largely from long-lived plasma cells (LLPC).9 As a consequence, the discrepancies in immunopathology between AChR-Ab and MuSK-Ab account for the heterogeneity in clinical manifestations to a great extent.
Generally, MuSK-Ab-positive MG (MuSK-MG) patients are primarily females with predominant involvement of bulbar muscles yet less weakness of limb muscles, as well as greater proneness to respiratory crises and worse prognosis in the status of myasthenic crisis.12,13 Atrophy of facial and tongue muscles is markedly observed in MuSK-MG, potentially arising from intrinsic predisposition or higher doses of steroids for being resistant to the therapy.14 As for neurophysiological features, repetitive compound muscle action potentials (R-CMAPs) that signify cholinergic hyperactivity are prevalent in MuSK-MG, which potentially mirrors the interrupted interactions between MuSK and ColQ as well as lower response rates to anticholinesterases.12,15 Histopathological findings revealed that thymic hyperplastic alterations were milder with a lower frequency of germinal centers in MuSK-MG,16 and thymectomy is also considered unfavorable for MuSK-MG.17 In contrast, anti-CD20 therapies such as rituximab have been proved to be rather clinically beneficial for MuSK-MG,18,19 especially when compared with AChR-MG,20 which is in concordance with the notion that CD20+ SLPB is the linchpin for secreting MuSK-Ab.9 Besides, plasma exchange (PE) seems to be more effective and recommended than intravenous immunoglobulin (IVIg) as a rescue therapy for MuSK-MG,21 probably owing to the deviation of the immunoregulatory effects of IVIg from the pathogenic mechanisms of MuSK-IgG4.22
Though extensive studies have described the clinical characteristics and prognostic factors of MuSK-MG, they are still not fully elucidated in China due to the low frequency of only 3.8% in MG patients seronegative for AChR-Ab.23 More recently indirect evidence from the SCREAM study and a multicenter retrospective study on MuSK-MG in China indicated that positivity rate of MuSK-Ab among MG patients could be even lower as roughly estimated to be 2.4%–3.5%.24,25 Several studies from different parts of China dedicated to deciphering the features of the subsets exhibited disparities in patterns of disease onset, progression or prognosis, possibly arising from regional divergence and small sample size.25–28 Inasmuch as the even rarer cases of double-seropositive MG (DSP-MG) with concurrent presence of AChR-Ab and MuSK-Ab, a few existing reports have argued that DSP-MG may be similar to MuSK-MG.29,30 However, it remains controversial whether DSP-MG represents virtually a distinct phenotype, considering that there have been only dozens of cases reported worldwide and transition from single-seropositive MG to DSP-MG exists.30–34 Thus, expanding the pool of MuSK-MG and DSP-MG will undoubtedly facilitate the understanding of these entities. In this study, we on the one hand investigated the differences between MuSK-MG and DSP-MG patients in a single center from central south China, and on the other hand analyzed the clinical features and outcomes of patients with MuSK-Ab seropositivity irrespective of the serological status of AChR-Ab, with the aim of adding weight to the repertoire of MuSK-MG and DSP-MG and reflecting on the clustering and subgrouping of heterogenous or parallel MG patients.
Subjects and Methods
Patient Enrollment
We retrospectively screened all MG patients who tested positive for MuSK-Ab and were diagnosed at the Department of Neurology, Xiangya Hospital, from June 2017 to June 2022. The diagnosis of MG was defined as fatigable muscle weakness together with seropositivity for associated autoantibodies, responses to acetylcholinesterase inhibitors, or abnormal findings on electrophysiological evaluation via the low-frequency repetitive nerve stimulation (RNS).1 As for the serological tests, the concentration of MuSK-Ab > 0.05 nmol/L via radioimmunoprecipitation assay (RIPA) and the concentration of AChR-Ab > 0.45 nmol/L via enzyme-linked immunosorbent assay (ELISA) were regarded as positive, respectively. MG patients with MuSK-Ab positivity were defined as MuSK-Ab positivity regardless of the serological status of other antibodies, whereas MuSK-MG patients were defined as seropositive for MuSK-Ab without the presence of AChR-Ab. In addition, DSP-MG patients referred to those with concurrent MuSK-Ab and AChR-Ab in the serum. We retrieved all patients with definite MG diagnoses and being seropositive for MuSK-Ab tested in the Da’an Clinical Laboratory Center (Guangzhou, China). Of the recorded 41 MG patients with MuSK-Ab, 34 were eventually included, of which five were lost to follow-up and two refused follow-up. This study was carried out in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Xiangya Hospital (approval number: 202309200). Due to the retrospective nature of the study and no identifiable patient information was provided, the patients’ informed consents were exempted.
Data Collection
Demographic and clinical data, including MG-associated conditions at onset, at nadir, and at endpoint, as well as comorbid diseases and immunosuppressive therapies were collected from medical records or structured telephone interviews. The baseline was set as the time point for a definite diagnosis based on the determined serological status. MG Activities of Daily Living (MG-ADL) was applied in this study to evaluate the severity of symptoms at nadir, which largely assessed the conditions of patients in terms of talking, chewing, swallowing, breathing, impairment of ability to brush teeth or comb hair, impairment of ability to arise from a chair, eyelid droop, and double vision.35 Each item ranges from 0 to 3 for a total score range of 0 to 24 with a higher score indicating the worse living ability.35 As for the prognostic analysis, the outcome was set as achieving minimal manifestation status (MMS), where the patients had no symptoms or functional limitations from MG but some weakness upon examination of certain muscles, or with the MG-ADL score being 0 or 1 for at least one year.21,35,36
Statistical Analysis
Quantitative data with normal distribution are expressed as mean plus or minus one standard deviation (SD), and group differences were assessed using the t-test. Otherwise, quantitative data are presented as median (interquartile range [IQR]), and differences among groups were compared using the Mann-Whitney U-test. Categorical variables are presented as numbers and percentages and were compared using Fisher’s exact test. Survival analysis was performed on 34 MG patients with MuSK-Ab who were followed up for at least 1 year. Continuous variables were dichotomized for ease of clinical utility, where the maximally selected rank statistics was applied to determine the optimal cutoff point for the MuSK concentration, MG-ADL scores at nadir, and time from onset to maximal worsening using the “maxstat” R package while restricting the proportion of the lesser subset to no lower than 25%.37 Kaplan-Meier survival curves were employed to determine the proportional hazards (PH) assumption of variables of interest. The Log rank test was used to compare the MMS distributions between subgroups with distinct clinical features if the PH assumption was not violated. When Kaplan-Meier survival curves crossed, the two-stage procedure was performed using the “TSHRC” R package with the number of bootstrap samples being 1000.38 For conservative estimation, significance levels of the two stages were both 0.025 with the significance level of the whole procedure set as 0.05.38 The variables of interest with a P-value less than 0.05 which met the PH hypothesis were included in the univariable and multivariable Cox regression analyses by “survival” and “survminer” R packages to identify the independent and potential risk factors that contribute to MMS achievement of MG patients. Statistical analyses were performed using SPSS version 26 (IBM Corp., Armonk, N.Y., USA) or R version 4.3.0 (R Foundation for Statistical Computing, Vienna, Austria). Statistical significance was set at P < 0.05.
Results
Demographic and Clinical Characteristics of MuSK-MG Patients
A total of 34 patients with MuSK-Ab were eventually included, with a median disease duration at enrollment of 16 months (IQR, 4.00–57.25 months), consisting of 28 (82.4%) patients seropositive for MuSK-Ab without AChR-Ab positivity (MuSK-MG) and six (17.6%) seropositive for both MuSK-Ab and AChR-Ab (DSP-MG). The overall demographic and clinical features of the patients are summarized in Table 1. For MuSK-MG, 71.4% (20/28) of the patients were female and 60.7% (17/28) of them were early-onset (<50 years). The median MuSK-Ab concentration was 0.44 nmol/L (IQR, 0.27–0.67 nmol/L) and a few of individuals combined with seropositive anti-titin antibodies (titin-Ab) and anti-ryanodine receptor antibodies (RyR-Ab). Regarding ancillary examinations for diagnosis, 86.4% (19/22) were positive for neostigmine tests and 60.0% (12/20) showed decremental responses in the RNS test. There were 35.7% (10/28) patients complicating other chronic diseases and hypertension was the most common (17.9%). In addition, 22.2% (6/27) patients had thyroid antibodies but rarely had thymic abnormalities.
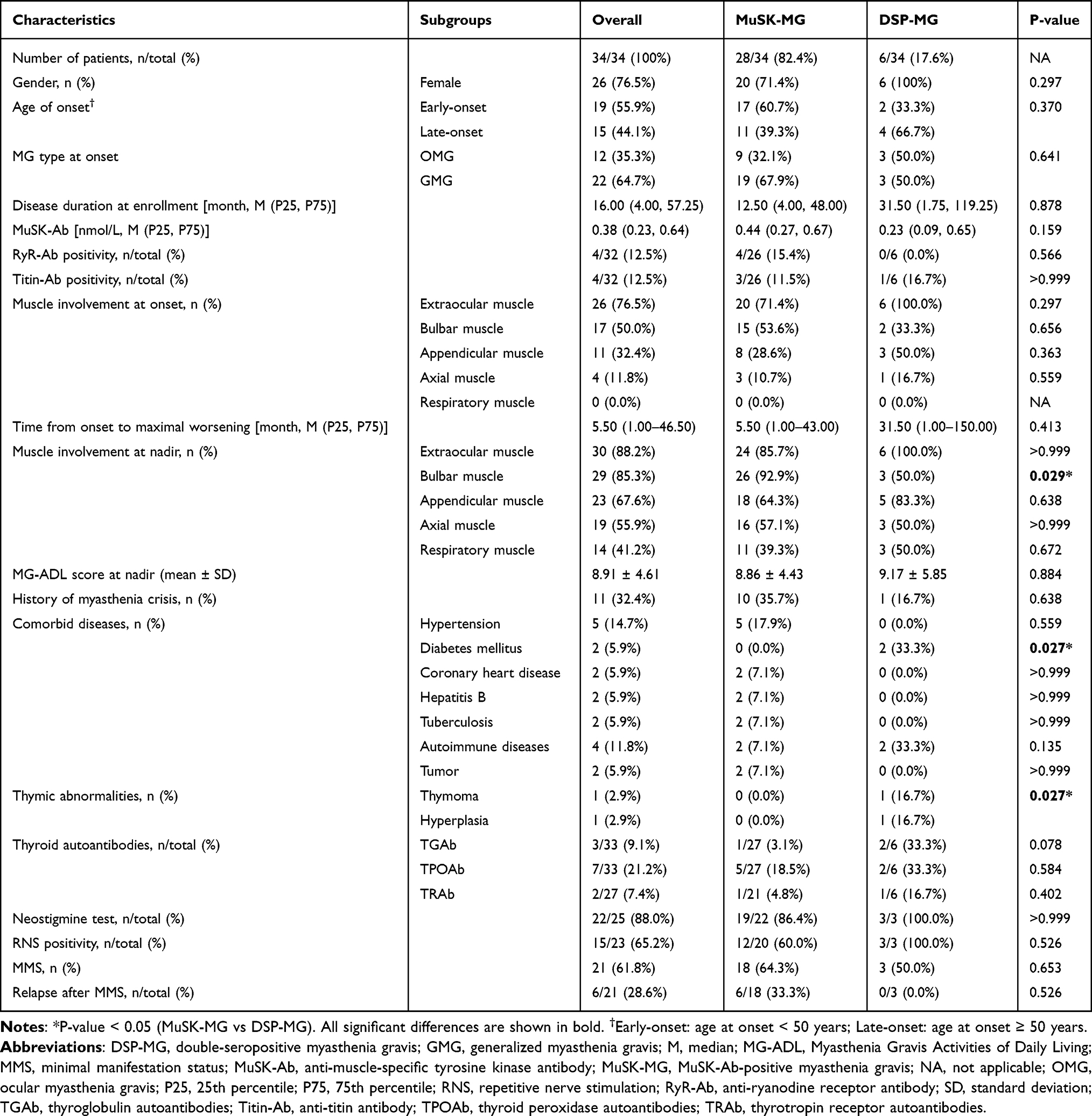 |
Table 1 Demographic and Clinical Characteristics of MuSK-MG and DSP-MG Patients |
At disease onset, the majority of patients (67.9%) were categorized as the generalized MG (GMG), most commonly involving the extraocular muscle (71.4%) and bulbar muscle (53.6%). The median duration from onset to the worst condition was 5.5 months (IQR, 1.00–43.00 months), which reflected the relatively rapid progression of the disease. At nadir, the bulbar (92.9%), appendicular (64.3%), axial (57.1%), and respiratory muscle (39.3%) were extensively involved, with a mean (SD) MG-ADL score recorded being 8.86 (4.43). In addition, 10 (35.7%) patients experienced at least one episode of myasthenic crisis.
The therapeutic regimens for all the patients are shown in Table 2. Twenty-six (92.9%) patients received steroids, with tacrolimus being the most commonly used non-steroid immunosuppressant. Eighteen (64.3%) patients received rescue therapies, especially plasma exchange, but only 5 (17.9%) patients received anti-CD20 therapies, including rituximab and ofatumumab. Overall, 18 (64.3%) patients reached MMS with the median time to MMS achievement being 10.5 months (IQR, 1.75–20.25 months), whereas 6 (33.3%) of them experienced relapses after maintenance of MMS for no less than 1 year. Throughout the course of the disease, 10 MuSK-MG patients were retested for MuSK-Ab, and only one patient exhibited a negative conversion of MuSK-Ab, whereas MuSK-Ab became seropositive again after disease relapse.
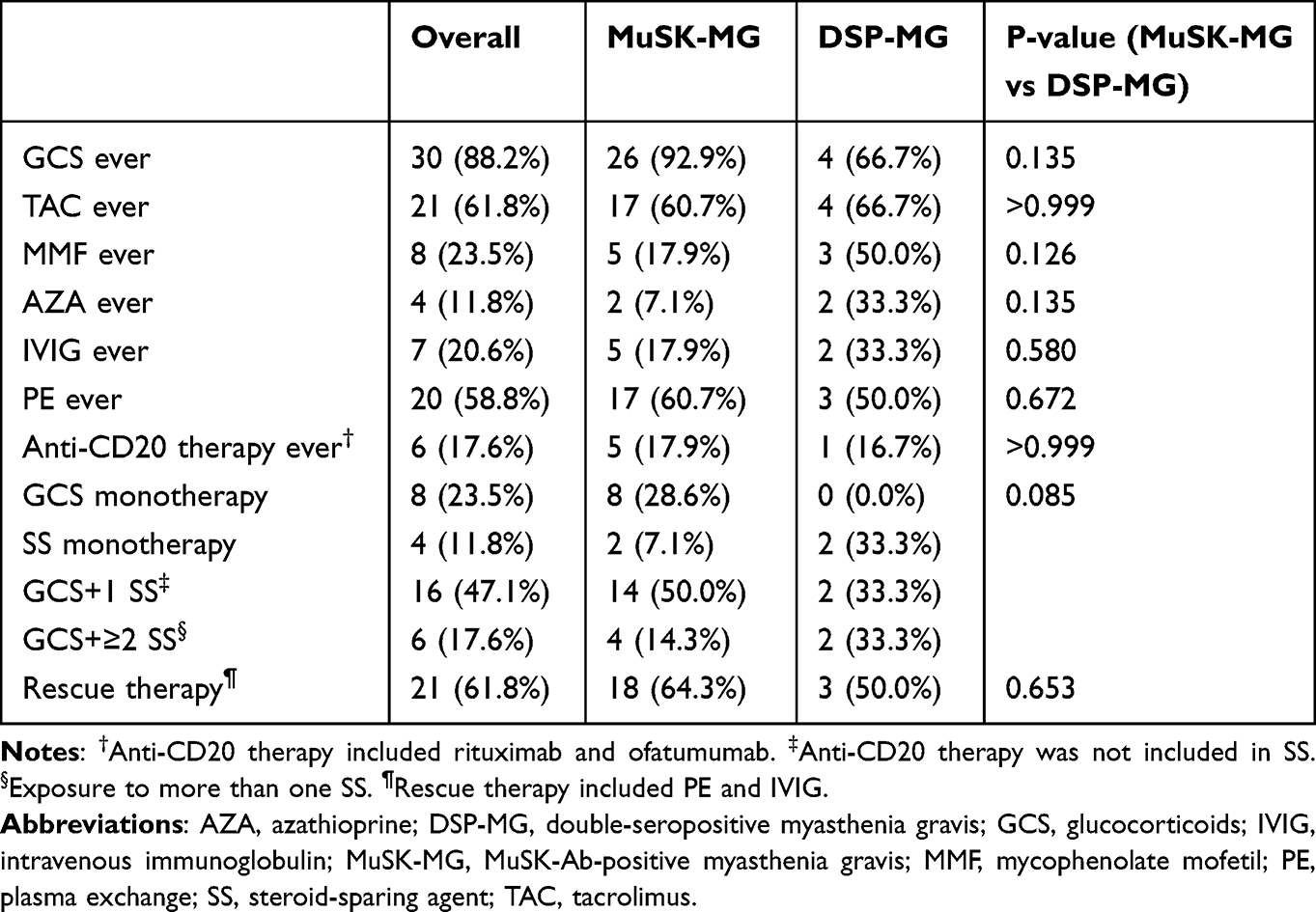 |
Table 2 Treatment Modalities in MuSK-MG and DSP-MG Patients |
Discrepancies Between MuSK-MG and DSP-MG
Considering that DSP-MG may be a subtype of MuSK-MG, we compared the characteristics of these two subsets. Although there was no statistically significant difference in the age of onset or sex composition between the two groups, early onset (60.7%) and late onset (66.7%) were slightly dominant in MuSK-MG and DSP-MG, respectively. Besides, all of the DSP-MG were female in our cohort. Compared with MuSK-MG patients, DSP-MG patients were significantly less likely to have bulbar muscle involvement at nadir (50% vs 92.9%; P = 0.029), but more likely to have thymoma or hyperplasia (33.4% vs 0%; P = 0.027). Intriguingly, diabetes mellitus was more common in DSP-MG (33.3% vs 0%; P = 0.027). In addition, DSP-MG patients seemed to have lower concentrations of MuSK-Ab despite with no statistical difference (0.23 nmol/L vs 0.44 nmol/L; P = 0.159). Two patients with DSP-MG were retested for MuSK-Ab, and one patient with thymoma turned negative for MuSK-Ab. Steroids, conventional non-steroid immunosuppressants, anti-CD20 therapies, and rescue therapies did not differ significantly between the two groups (Table 2). Furthermore, the proportion of patients who achieved MMS was similar between the two groups (50% vs 64.3%; P = 0.653). Thus, the presence of AChR antibodies did not affect the rate of MMS achievement, nor did most of the clinical phenotypes of MG patients with MuSK-Ab.
Factors Affecting MMS Achievement Among MG Patients with MuSK-Ab
Considering the similarities between MuSK-MG and DSP-MG, we included all MG patients with MuSK-Ab as a whole to identify potential factors affecting the induction of MMS. Among the 34 MG patients with MuSK-Ab, 21 (61.8%) achieved MMS, and the median time to achieve MMS was 11 months (IQR, 3.5–20.5 months). To facilitate the interpretation of the results, maximally selected rank statistics was used to determine the cut-off values for continuous variables. Specifically, the optimal cut-off points for the MG-ADL score at nadir was 12 (high MG-ADL score >12 vs low MG-ADL score ≤12), time from onset to maximal worsening 1 month, and the MuSK-Ab concentration 0.61 nmol/L (Supplementary Figure 1). Analysis of Kaplan-Meier survival curves revealed that MuSK-Ab concentration, MG-ADL score at nadir, and axial muscle involvement at nadir met the PH assumption. The Log rank test showed that Kaplan-Meier survival curves of the MG-ADL score at nadir (P = 0.006) and axial muscle involvement at nadir (P = 0.033) were different between groups, whereas the two-stage procedure suggested that the achievement of MMS was also affected by the combination with diabetes mellitus (P = 0.041) (Figure 1 and Table 3). However, owing to the violence of the PH assumption and the low frequency of diabetes mellitus in the cohort, only the MG-ADL score at nadir and axial muscle involvement at nadir were included in the Cox regression analyses. In the univariable Cox analysis, high MG-ADL score at nadir (HR = 0.16, 95% CI: 0.037–0.7, P = 0.015) and axial muscle involvement at nadir (HR = 0.39, 95% CI: 0.16–0.94, P = 0.035) were potential risk factors for MMS achievement (Figure 2A and B). Further multivariable Cox analysis involving the above two indicators demonstrated that only high MG-ADL score at nadir (HR = 0.19, 95% CI: 0.04–0.94; P = 0.042) was an independent risk factor for MMS achievement in MG patients with MuSK-Ab (Figure 2C).
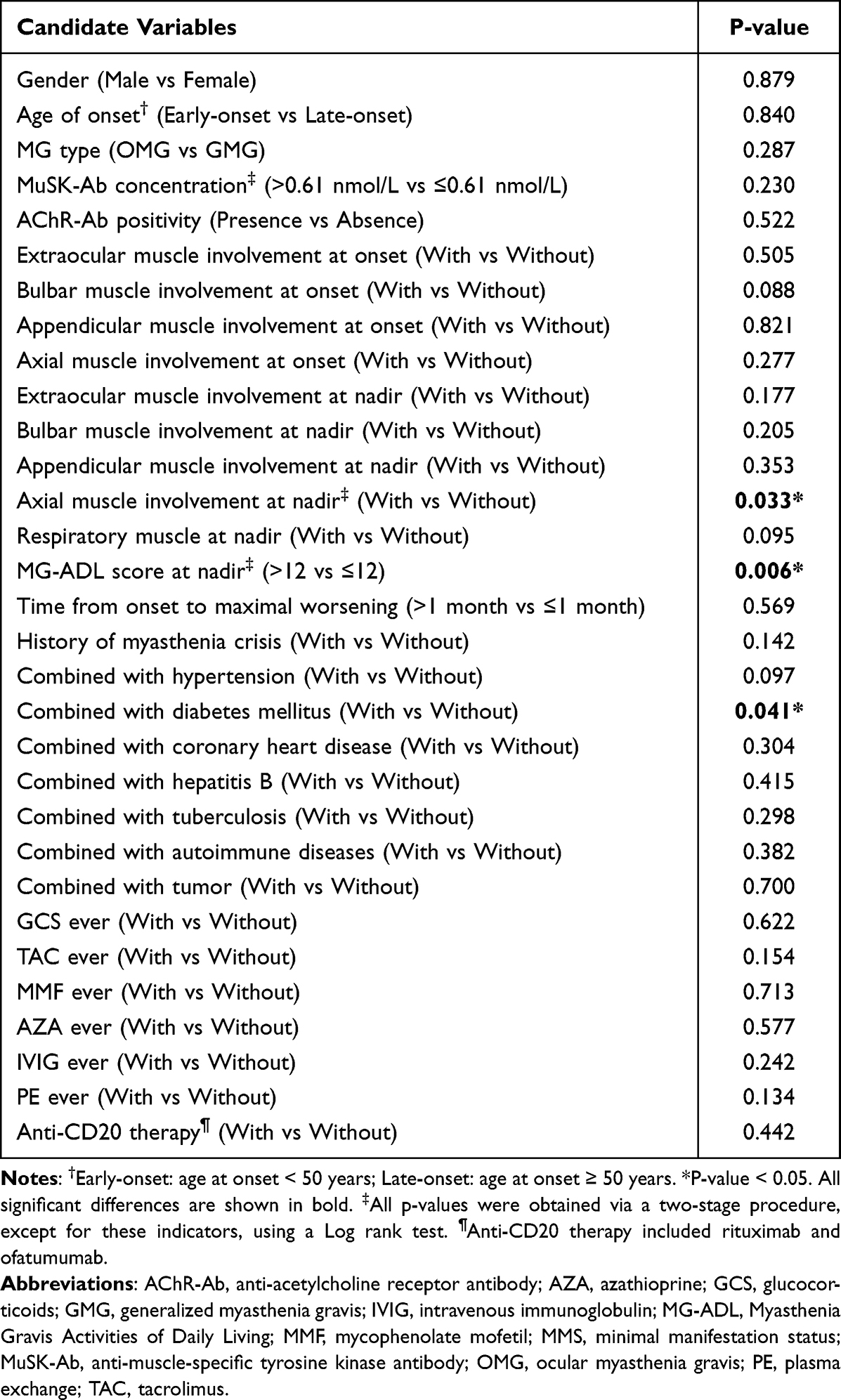 |
Table 3 Univariable Analysis for MMS Achievement in MG Patients with MuSK-Ab |
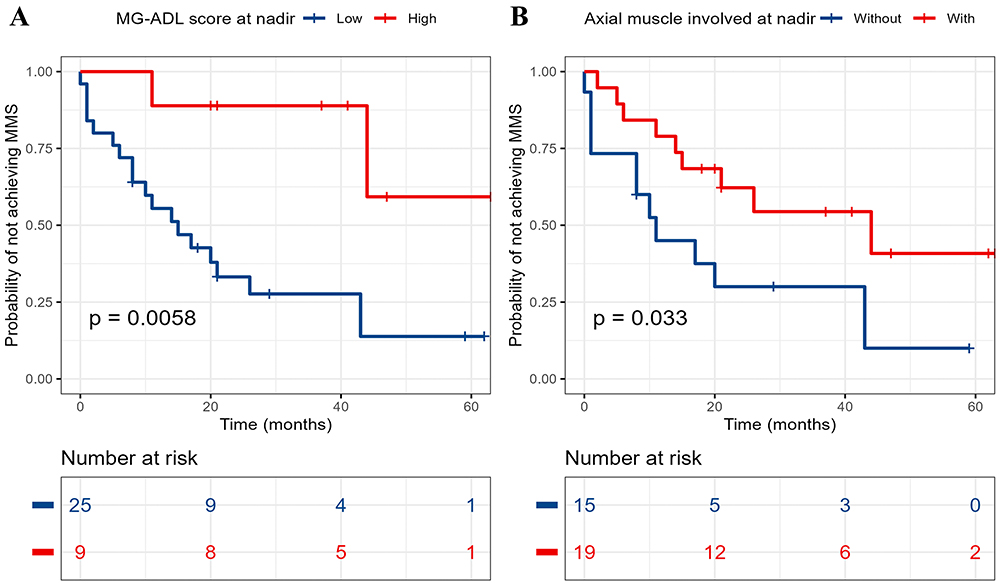 |
Figure 1 Kaplan-Meier curves of MG-ADL score at nadir (A) and axial muscle involved at nadir (B) for MMS achievement in MG patients with MuSK-Ab. MG-ADL, Myasthenia Gravis Activities of Daily Living; MMS, minimal manifestation status. |
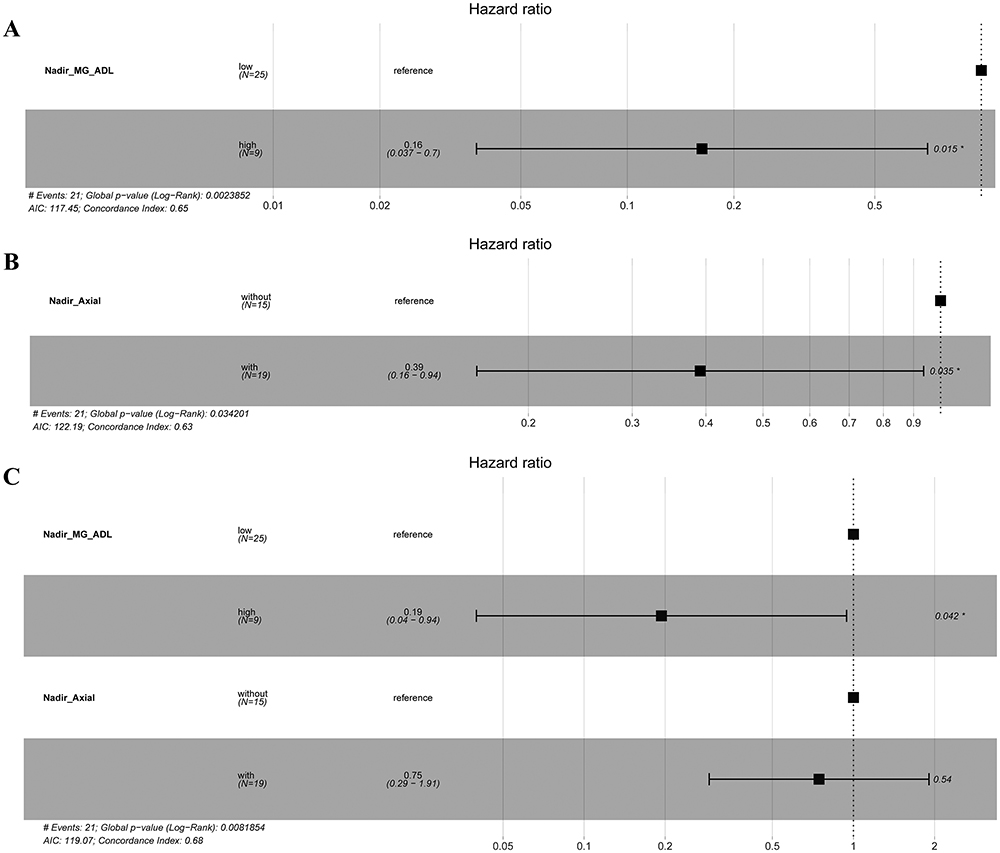 |
Figure 2 Forest plots of hazard ratios estimated from univariable and multivariable Cox proportional hazards regression models including 34 patients with MuSK-Ab. (A and B) In univariable Cox regression analysis, the MG-ADL score at nadir and axial muscle involved at nadir negatively influenced on the MMS achievement. (C) In multivariable Cox regression analysis, the MG-ADL score at nadir was identified as the independent prognostic factor for MMS achievement. Nadir_MG_ADL, MG-ADL score at nadir; Nadir_Axial, axial muscle involved at nadir; *P-value < 0.05. |
Discussion
In MG, individuals with distinct subtypes of autoantibodies represent specific entities, where AChR-Ab and MuSK-Ab accounted for most of cases. Because of the intrinsic heterogeneity between AChR-Ab and MuSK-Ab in terms of either their production or effector function, patients with AChR-MG and MuSK-MG differ in muscle involvement,12 electrophysiological properties,15 thymic abnormalities,16 and responses to therapies.12,17,18 In a few instances, co-occurrence of both AChR-Ab and MuSK-Ab could be observed, and only dozens of cases have been reported worldwide.30 By and large, our study indicated that the clinical characteristics of MuSK-MG are comparable to those of DSP-MG, although there are still certain discrepancies among them. Remarkably, all DSP-MG patients in our cohort were female, yet more than half of them had late-onset disease. Female predominance has been unraveled universally in most of the already existing studies on either MuSK-MG13 or DSP-MG,30 while the age of onset in previously reported DSP-MG seemed to be lower than ours.30 As for symptoms at onset, a high frequency of extraocular impairment was seen in both MuSK-MG and DSP-MG with moderate involvement of bulbar and limb muscles, which was consistent with the patterns in already existing DSP-MG cases.30 Actually, ocular involvement, including diplopia and ptosis, is also not uncommon in MuSK-MG either at onset or over a long disease course, although bulbar and respiratory symptoms are often emphasized.26,27,39 Half of the DSP-MG patients in our cohort exhibited bulbar and respiratory involvement at nadir, although an even larger proportion of bulbar dysfunction was observed in MuSK-MG. However, only one case of DSP-MG experienced myasthenic crisis, indicating relatively mild conditions in our patients.
A seemingly distinguishable feature of DSP-MG from MuSK-MG is the thymus pathology. In our cohort, none of the MuSK-MG patients exhibited thymic abnormalities, while thymoma and thymic hyperplasia were observed in two DSP-MG patients, respectively. In addition, our findings demonstrated that DSP-MG patients were more likely to have comorbid diabetes mellitus, which may restrict the use of glucocorticoids (GCS) and affect the outcome of MG. However, the underlying mechanism and definite clinical significance of the comorbidity with diabetes mellitus in this subset require further investigation, although it may occur by chance due to the small sample size. Most of our patients received GCS with steroid-sparing agents as maintenance therapy, whereas nearly half of the MuSK-MG and DSP-MG patients received rescue therapies. In response to these treatments, 64.5% and 50.0% of MuSK-MG and DSP-MG patients achieved MMS during the follow-up. We did not observe substantial differences in treatment modalities and prognosis, as assessed by MMS, between MuSK-MG and DSP-MG. However, the potential use of novel therapies for MG in the setting of concurrent autoantibodies should be considered. For instance, anti-CD19 monoclonal antibodies such as inebilizumab depleting a wider range of B cells and antibody-producing cells,40 neonatal Fc receptor (FcRn) blockers such as efgartigimod clearing IgG of multiple subclasses,41 and telitacicept suppressing the activities of both B-lymphocyte stimulator (BLyS) and a proliferation-inducing ligand (APRIL) that sustain B cell development and survival,42 could all be promising agents for treating DSP-MG or even MG complicating other B-cell-mediated autoimmune disorders like stiff-person syndrome.43 Overall, the similarities between MuSK-MG and DSP-MG led to the notion that DSP-MG may be a subtype of MuSK-MG,30 though the low prevalence of DSP-MG and potential selection bias may render it plausibly arbitrary.
Notably, the methods of diagnostic assay may affect the positivity of MuSK-Ab and AChR-Ab and mask the detection rate of these autoantibodies in MG. In our cohort, AChR-Ab and MuSK-Ab were determined using ELISA and RIPA, respectively. RIPA44 for AChR-Ab, and ELISA30 and cell-based assay (CBA)32 for MuSK-Ab have also been reported in other cases. The discrepancies in reference values among laboratories and risks of false positives or negatives could also lead to inconsistency in analyses. In addition, the serological status of MuSK-Ab is likely to be initially ignored when AChR-Ab positivity is detected in certain cases. Therefore, inaccurate determination of the autoantibody profile could interfere with the understanding of the clinical characteristics of DSP-MG. AChR-Ab and MuSK-Ab could be present simultaneously at onset45 or appear in sequence.32 The pathogenesis underlying the occurrence of both AChR-Ab and MuSK-Ab or the switch from AChR-MG to DSP-MG remains unknown. Presumably, thymectomy may be a suspicious evil, as the co-existence of AChR-Ab and MuSK-Ab as well as the conversion from AChR-MG to DSP-MG has been reported in thymectomized patients,32,33,46 although it is possible that thymectomy precedes serological and immunological changes merely by coincidence. None of the patients in our cohort underwent thymectomy indicates that there should be alternative mechanisms for the onset of DSP-MG. Environmental triggers could also be involved, as exemplified by a case of patient induced by D-penicillamine, who was free of symptoms and autoantibodies after drug discontinuation.44 Anyhow, seroconversion of autoantibodies may reflect epitope spreading caused by enigmatic triggers. The mechanisms by which AChR-Ab and MuSK-Ab cause pathologies in DSP-MG have not been deciphered. Analysis of subunit specificity of AChR-Ab in a DSP-MG patient revealed the presence of autoantibodies against β1 and γ subunits rather than the dominant α1 subunit, potentially explaining the resemblance between MuSK-MG and DSP-MG.47 There lies as well the possibility that the pathological effects of MuSK-Ab could interfere with those of AChR-Ab or vice versa to some extent, and thus the phenotype of DSP-MG may still vary among patients.
Regarding the clinical characteristics and outcomes of MuSK-MG in China, several efforts have been made to depict the regional features among these patients from multiple perspectives. In parallel to other cohorts, MuSK-MG could present with acute onset and rapid progression, primarily in female patients with a great proportion of bulbar and respiratory dysfunction.25,27,28 In contrast, the age of onset seemed to be later in the Chinese population with MuSK-MG,26 and very-late-onset patients especially exhibited limb, bulbar, and respiratory involvement in the early phase of the disease.25 A higher frequency of comorbid autoimmune diseases in MuSK-MG has been found in the northeast area,26 while musculus deltoideus was the most responsive to RNS within Northwest patients.27 In addition to low-dose rituximab,19,27 patients responded well to tacrolimus treatment combined with GCS,26,27 which has been applied in the majority of our patients as well.
However, the percentage of MuSK-MG patients with favorable outcomes was quite different among the cohorts,27,28 and factors affecting the prognosis of MuSK-MG have not been fully unraveled except that taking non-steroid immunosuppressants was found to be associated with a lower risk of relapse.48 In our analyses on MG patients with MuSK-Ab, axial muscle involvement at nadir and higher maximum MG-ADL score were less prone to MMS achievement, while the latter was still significant when adjusted for axial involvement at nadir. Axial muscles, such as the neck muscles, are usually reported to be relatively less severely affected or even neglected in MuSK-MG patients.25,28 Thus, it possibly implies that the disease condition tends to be severe when there exists axial involvement that is not easily manifested, which further affects the prognosis of patients. MG-ADL is an effective scale in evaluating the disease status of MG, which particularly reflects the perception of MG burden on the routine life by patients themselves and is applicable for determining a state of minimal symptoms.25 Besides, fluctuation of symptoms is not evident in MuSK-MG,26 rendering it more reliable to represent the disease status of patients with MG-ADL. Hence, higher MG-ADL scores at maximal worsening reasonably heralded poor outcomes, as demonstrated in our results. This is also consistent with the finding that short-term response to treatment assists in predicting the long-term outcomes of MG patients.49
Overall, our study further supplemented the knowledge of MuSK-MG as well as DSP-MG with experience from a single center in central south China, where we found that general clinical features, including female dominance, frequent involvement of ocular and bulbar muscles at onset and nadir, and no thymic abnormalities were typical in MuSK-MG, and that DSP-MG was akin to MuSK-MG despite slight discrepancies. In addition, the prognostic analysis of MG patients with seropositive MuSK-Ab revealed that a severe condition represented by higher MG-ADL scores at maximal worsening was associated with failure of MMS induction. These results could facilitate the recognition of DSP-MG and assessment of MuSK-MG in clinical practice. However, the limitations of the small sample size and single outcome variable may restrict extrapolation from our conclusions, which calls for evidence leaning on larger-scale cohorts in the future.
Funding
This study was supported by the National Natural Science Foundation of China (Grand No. 82171399 to Huan Yang; Grand No. 82101474 to Mengchuan Luo).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren J. Myasthenia gravis. Nat Rev Dis Primers. 2019;5(1):30. doi:10.1038/s41572-019-0079-y
2. Vincent A, Leite MI. Neuromuscular junction autoimmune disease: muscle specific kinase antibodies and treatments for myasthenia gravis. Curr Opinion in Neurol. 2005;18(5):519–525. doi:10.1097/01.wco.0000180660.57801.3f
3. DeChiara TM, Bowen DC, Valenzuela DM, et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85(4):501–512. doi:10.1016/S0092-8674(00)81251-9
4. Kim N, Stiegler AL, Cameron TO, et al. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135(2):334–342. doi:10.1016/j.cell.2008.10.002
5. Kawakami Y, Ito M, Hirayama M, et al. Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology. 2011;77(20):1819–1826. doi:10.1212/WNL.0b013e318237f660
6. Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nature Med. 2001;7(3):365–368. doi:10.1038/85520
7. McConville J, Farrugia ME, Beeson D, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. 2004;55(4):580–584. doi:10.1002/ana.20061
8. Klooster R, Plomp JJ, Huijbers MG, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain. 2012;135(Pt 4):1081–1101. doi:10.1093/brain/aws025
9. Fichtner ML, Jiang R, Bourke A, Nowak RJ, O’Connor KC. Autoimmune pathology in myasthenia gravis disease subtypes is governed by divergent mechanisms of immunopathology. Front Immunol. 2020;11:776. doi:10.3389/fimmu.2020.00776
10. Vergoossen DLE, Plomp JJ, Gstöttner C, et al. Functional monovalency amplifies the pathogenicity of anti-MuSK IgG4 in myasthenia gravis. Proc Natl Acad Sci USA. 2021;118(13). doi:10.1073/pnas.2020635118
11. Koneczny I, Stevens JAA, De Rosa A, et al. IgG4 autoantibodies against muscle-specific kinase undergo Fab-arm exchange in myasthenia gravis patients. J Autoimmun. 2017;77:104–115. doi:10.1016/j.jaut.2016.11.005
12. Evoli A, Tonali PA, Padua L, et al. Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain. 2003;126(Pt 10):2304–2311. doi:10.1093/brain/awg223
13. König N, Stetefeld HR, Dohmen C, et al. MuSK-antibodies are associated with worse outcome in myasthenic crisis requiring mechanical ventilation. J Neurol. 2021;268(12):4824–4833. doi:10.1007/s00415-021-10603-9
14. Farrugia ME, Robson MD, Clover L, et al. MRI and clinical studies of facial and bulbar muscle involvement in MuSK antibody-associated myasthenia gravis. Brain. 2006;129(Pt 6):1481–1492. doi:10.1093/brain/awl095
15. Modoni A, Mastrorosa A, Spagni G, Evoli A. Cholinergic hyperactivity in patients with myasthenia gravis with MuSK antibodies: a neurophysiological study. Clin Neurophysiol. 2021;132(8):1845–1849. doi:10.1016/j.clinph.2021.04.019
16. Leite MI, Ströbel P, Jones M, et al. Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Ann Neurol. 2005;57(3):444–448. doi:10.1002/ana.20386
17. Clifford KM, Hobson-Webb LD, Benatar M, et al. Thymectomy may not be associated with clinical improvement in MuSK myasthenia gravis. Muscle and Nerve. 2019;59(4):404–410. doi:10.1002/mus.26404
18. Hehir MK, Hobson-Webb LD, Benatar M, et al. Rituximab as treatment for anti-MuSK myasthenia gravis: multicenter blinded prospective review. Neurology. 2017;89(10):1069–1077. doi:10.1212/WNL.0000000000004341
19. Zhou Y, Yan C, Gu X, et al. Short-term effect of low-dose rituximab on myasthenia gravis with muscle-specific tyrosine kinase antibody. Muscle and Nerve. 2021;63(6):824–830.
20. Di Stefano V, Lupica A, Rispoli MG, Di Muzio A, Brighina F, Rodolico C. Rituximab in AChR subtype of myasthenia gravis: systematic review. J Neurol Neurosurg. 2020;91(4):392–395. doi:10.1136/jnnp-2019-322606
21. Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology. 2016;87(4):419–425. doi:10.1212/WNL.0000000000002790
22. Dalakas MC. IgG4-mediated neurologic autoimmunities: understanding the pathogenicity of IgG4, ineffectiveness of IVIg, and long-lasting benefits of anti-B cell therapies. Neurol Neuroimmunol Neuroinflam. 2022;9(1):e1116.
23. Yeh JH, Chen WH, Chiu HC, Vincent A. Low frequency of MuSK antibody in generalized seronegative myasthenia gravis among Chinese. Neurology. 2004;62(11):2131–2132. doi:10.1212/01.WNL.0000128042.28877.C3
24. Li Z, Zhang C, Chang T, et al. A multicentre, prospective, double-blind study comparing the accuracy of autoantibody diagnostic assays in myasthenia gravis: the SCREAM study. Lancet Reg Health West Pac. 2023;38:100846. doi:10.1016/j.lanwpc.2023.100846
25. Zhou Y, Chen J, Li Z, et al. Clinical features of myasthenia gravis with antibodies to MuSK based on age at onset: a multicenter retrospective study in China. Front Neurol. 2022;13:879261. doi:10.3389/fneur.2022.879261
26. Zhang Z, Guan Y, Han J, Li M, Shi M, Deng H. Regional features of MuSK antibody-positive myasthenia gravis in Northeast China. Front Neurol. 2020;11:516211. doi:10.3389/fneur.2020.516211
27. Zhao S, Zhang K, Ren K, et al. Clinical features, treatment and prognosis of MuSK antibody-associated myasthenia gravis in Northwest China: a single-centre retrospective cohort study. BMC Neurol. 2021;21(1):428. doi:10.1186/s12883-021-02439-7
28. Huang Q, Li F, Zhao S. Spotlight on MuSK positive myasthenia gravis: clinical characteristics, treatment and outcomes. BMC Neurol. 2022;22(1):73. doi:10.1186/s12883-022-02593-6
29. Seth V, Kushwaha S, Bapat P, Rajashekar K, Grover D. Is double-seropositive myasthenia gravis a distinct subtype? Acta Neurologica Belgica. 2023;123(1):251–252. doi:10.1007/s13760-021-01759-2
30. Zhang J, Chen Y, Chen J, et al. AChRAb and MuSKAb double-seropositive myasthenia gravis: a distinct subtype? Neurol Sci. 2021;42(3):863–869. doi:10.1007/s10072-021-05042-3
31. Li M, Ren L, Zhang Y, et al. Clinical characteristics of AChRAb and MuSKAb double seropositive myasthenia gravis patients. Clin Neurol Neurosurg. 2018;172:69–73. doi:10.1016/j.clineuro.2018.06.041
32. Zouvelou V, Zisimopoulou P, Psimenou E, Matsigkou E, Stamboulis E, Tzartos SJ. AChR-myasthenia gravis switching to double-seropositive several years after the onset. J Neuroimmunol. 2014;267(1–2):111–112. doi:10.1016/j.jneuroim.2013.12.012
33. Jordan B, Schilling S, Zierz S. Switch to double positive late onset MuSK myasthenia gravis following thymomectomy in paraneoplastic AChR antibody positive myasthenia gravis. J Neurol. 2016;263(1):174–176. doi:10.1007/s00415-015-7982-2
34. Lu Y, Ran H, Yang W, et al. AChR myasthenia gravis switching to MuSK or double antibody positive myasthenia gravis in two children and literature review. Neuromusc disord. 2020;30(7):534–538. doi:10.1016/j.nmd.2020.03.012
35. Muppidi S, Silvestri NJ, Tan R, Riggs K, Leighton T, Phillips GA. Utilization of MG-ADL in myasthenia gravis clinical research and care. Muscle and Nerve. 2022;65(6):630–639. doi:10.1002/mus.27476
36. Jaretzki A, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology. 2000;55(1):16–23. doi:10.1212/WNL.55.1.16
37. Hothorn T, Lausen B. Maximally selected rank statistics in R. R News. 2002;2(1):3–5.
38. Qiu P, Sheng J. A two-stage procedure for comparing hazard rate functions. J Royal Stat Soci Ser B. 2008;70(1):191–208. doi:10.1111/j.1467-9868.2007.00622.x
39. Evoli A, Alboini PE, Iorio R, Damato V, Bartoccioni E. Pattern of ocular involvement in myasthenia gravis with MuSK antibodies. J Neurol Neurosurg. 2017;88(9):761–763. doi:10.1136/jnnp-2017-315782
40. Frampton JE. Inebilizumab: first Approval. Drugs. 2020;80(12):1259–1264. doi:10.1007/s40265-020-01370-4
41. Howard JF, Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, Phase 3 trial. Lancet Neurol. 2021;20(7):526–536. doi:10.1016/S1474-4422(21)00159-9
42. Zhang Z, Wang Z, Du X, Huang X, Zhang Y. Refractory generalized myasthenia gravis treated successfully with telitacicept: two cases report. J Neurol. 2024;271(1):584–588. doi:10.1007/s00415-023-12036-y
43. Di Stefano V, Alonge P, Rini N, et al. Efgartigimod beyond myasthenia gravis: the role of FcRn-targeting therapies in stiff-person syndrome. J Neurol. 2024;271(1):254–262. doi:10.1007/s00415-023-11970-1
44. Poulas K, Koutsouraki E, Kordas G, Kokla A, Tzartos SJ. Anti-MuSK- and anti-AChR-positive myasthenia gravis induced by d-penicillamine. J Neuroimmunol. 2012;250(1–2):94–98. doi:10.1016/j.jneuroim.2012.05.011
45. Zouvelou V, Psimenou E. AChR-and MuSK-positive myasthenia gravis: double trouble. J Neuroimmunol. 2020;348:577364. doi:10.1016/j.jneuroim.2020.577364
46. Rajakulendran S, Viegas S, Spillane J, Howard RS. Clinically biphasic myasthenia gravis with both AChR and MuSK antibodies. J Neurol. 2012;259(12):2736–2739. doi:10.1007/s00415-012-6661-9
47. Zouvelou V, Michail M, Belimezi M, Zisimopoulou P. Subunit specificity of the acetylcholine receptor antibodies in double seropositive myasthenia gravis. Muscle and Nerve. 2021;63(4):E36–E37. doi:10.1002/mus.27177
48. Tan Y, Shi J, Huang Y, et al. Long-term efficacy of non-steroid immunosuppressive agents in anti-muscle-specific kinase positive myasthenia gravis patients: a prospective study. Front Neurol. 2022;13:877895. doi:10.3389/fneur.2022.877895
49. Tomschik M, Hilger E, Rath J, et al. Subgroup stratification and outcome in recently diagnosed generalized myasthenia gravis. Neurology. 2020;95(10):e1426–e1436. doi:10.1212/WNL.0000000000010209
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.