")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
Clinical Features and Long-Term Outcomes of Anti-Leucine-Rich Glioma-Inactivated 1 Encephalitis: A Multi-Center Study
Authors Qiao S, Wu H , Liu L, Wang M, Zhang R, Han T, Liu X
Received 17 November 2020
Accepted for publication 11 January 2021
Published 25 January 2021 Volume 2021:17 Pages 203—212
DOI https://doi.org/10.2147/NDT.S292343
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yuping Ning
Shan Qiao,1 Huai-kuan Wu,1 Ling-ling Liu,2 Mei-ling Wang,3 Ran-ran Zhang,4 Tao Han,5 Xue-wu Liu4
1Department of Neurology, The First Affiliated Hospital of Shandong First Medical University, Jinan, Shandong Province 250014, People’s Republic of China; 2Department of Neurology, Liaocheng People’s Hospital, Liaocheng, Shandong Province 252000, People’s Republic of China; 3Department of Neurology, Binzhou Medical University Hospital, Binzhou, Shandong Province 256603, People’s Republic of China; 4Department of Neurology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong Province 250012, People’s Republic of China; 5Department of Neurology, Shandong Provincial Hospital, Jinan, Shandong Province, People’s Republic of China
Correspondence: Xue-wu Liu
Department of Neurology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, No. 107 Wen Hua Xi Road, Jinan 250012, Shan Dong, People’s Republic of China
Tel + 86 0531-82169114
Email [email protected]
Purpose: To describe the clinical manifestation, immunotherapy, and long-term outcomes of anti-leucine-rich glioma-inactivated 1 (LGI1) encephalitis.
Patients and Methods: This study was a retrospective analysis of 117 patients with a diagnosis of anti-LGI1 encephalitis identified from the databases of multiple clinical centers between September 2014 and December 2019. The clinical features, ancillary test results, and details of long-term outcomes were evaluated.
Results: Among the 117 patients with anti-LGI1 encephalitis, 69.2% (81/117) were male and 30.8% (36/117) were female. The median age of all patients at the onset of the disease was 57 years (interquartile range [IQR], 52– 67). The median time from symptom onset to diagnosis was 8.7 weeks (IQR, 4.2– 25). The main clinical features identified were seizures, cognitive impairment, and mental and behavioral abnormalities. Of the 117 patients, 109 were treated with immunotherapy. Symptoms including memory, mental ability, and behavior improved in all 109 patients after 3– 5 days of treatment. The median time of follow-up for the treated patients was 33 months (IQR, 17– 42). Of the treated patients, 16.2% (19/117) experienced a relapse, with a median delay of 5 months (IQR, 2.1– 17) between onset and the first relapse. There were no mortalities over the follow-up period.
Conclusion: The long-term outcome of patients with anti-LGI1 encephalitis was mostly favorable, although some patients continued to experience cognitive dysfunction. Early recognition is important for prompt initiation of immunotherapy that can improve clinical symptoms of anti-LGI1 encephalitis.
Keywords: anti-LGI1 encephalitis, autoimmune epilepsy, follow-up, immunotherapy, relapse
Introduction
Autoimmune encephalitis (AE) refers to a group of inflammatory diseases of the central nervous system triggered by the immune system. Research on autoimmune diseases has broadened the spectrum of AE subtypes.1–3 Some subtypes of AE have complex clinical manifestations that make diagnosis difficult. Leucine-rich glioma inactivated 1 (LGI1) is associated with voltage-gated potassium channels (VGKCs). Antibodies against LGI1, which are associated with AE, co-precipitate LGI1 and VGKCs.4,5 Since first described in 2010, an increasing number of cases of anti-LGI1 encephalitis have been reported.6,7
There are differences in the incidence, clinical features, and treatment strategies for anti-LGI1 encephalitis cases among different countries.4,5,8,9 In 2017, China proposed a consensus to improve the identification and management of AE among Chinese patients.10 However, the clinical characteristics of anti-LGI1 encephalitis and long-term prognosis among Chinese patients are limited because few reports of anti-LGI1 encephalitis have included a sufficiently large sample size, and AE-related antibody analysis is not routinely performed. To improve the understanding of the clinical course of anti-LGI1 encephalitis and to provide experiential knowledge for clinical diagnosis and treatment, we undertook a retrospective analysis of the clinical characteristics, treatment regimen, and long-term outcomes of 117 patients diagnosed with anti-LGI1 encephalitis in China.
Patients and Methods
The study sample included 117 patients who were positive for anti-LGI1 antibodies in their serum and/or cerebrospinal fluid (CSF) and who were subsequently diagnosed with anti-LGI1 encephalitis according to published diagnostic criteria.4,5,10 Patients were identified from the databases of the following five clinical centers: Qilu Hospital of Shandong University, Shandong Provincial Hospital Affiliated to Shandong University, The First Affiliated Hospital of Shandong First Medical University, Affiliated Hospital of Binzhou Medical College, and Liaocheng People’s Hospital between September 2014 and December 2019. The inclusion criteria were as follows: (1) acute or subacute onset of one or more of the major groups of manifestations including psychosis, seizures, memory deficit, speech disturbance; (2) serum and/or CSF testing positive for anti-LGI1 antibodies; and (3) reasonable exclusion of other disorders. The exclusion criteria included incomplete clinical data and loss to follow-up. This study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of Qilu Hospital of Shandong University (NO. KYLL-202,008-044). A written informed consent was obtained from all the participants and their legal guardians.
Clinical features, cognitive testing, laboratory findings (CSF and serum analyses), video electroencephalograms (VEEGs), cranial magnetic resonance imaging (MRI), tumor screenings, and therapy details were retrieved from patients’ medical records. Serum and CSF samples in the 117 patients were sent to the Golden Field Testing Center for evaluation between September 2014 and December 2019. Auto-antibodies to the N-methyl-D-aspartate receptor (NMDAR), LGI1, the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors AMPA1 and AMPA2, contactin-associated protein-like 2 (CASPR2), and the γ-aminobutyric acid receptor-B (GABABR) in the serum and CSF were assessed by indirect immunofluorescence tests. Indirect immunofluorescence was performed according to the manufacturer’s instructions (Euroimmun, Germany). For the 117 patients, screening for tumors included tumor marker tests, chest computed tomography (CT) imaging, and abdominal and pelvic ultrasounds. After completion of treatment, patients were followed-up every 3 months for the first year and every 6 months thereafter. Clinical outcomes were evaluated using the modified Rankin Scale (mRS).
Statistical analyses were performed using GraphPad Prism Software 8.0 (GraphPad Software, Inc., California, USA). Categorical variables were described as percentages; continuous variables with a normal distribution were presented as means and standard deviations. Non-normal data were presented as medians and ranges. Student’s t-test was used to compare the mRS score at symptom onset and 6 months after immunosuppressive therapy. P-values < 0.05 were considered significant.
Results
Clinical Characteristics
Among the 117 patients with anti-LGI1 encephalitis, 69.2% (81/117) were male and 30.8% (36/117) were female. The median onset age of all patients was 57 years (interquartile range [IQR], 52–67). In the study, the annual number of confirmed cases trended upward with improvements in immunofluorescence detection methods (Figure 1A). The majority of patients were 40 to 70 years old (79.5%, 93/117). The distribution of gender and age of patients with anti-LGI1 encephalitis is showed in Figure 1B. The most common clinical manifestations of anti-LGI1 AE were seizures (87.2%, 102/117), cognitive impairments (70.1%, 82/117), and insomnia (52.1%, 61/117) (Table 1). Details of the common initial symptoms in these 117 patients are shown in Figure 2A. The median time from symptom onset until diagnosis was 8.7 weeks (IQR, 4.2–25).
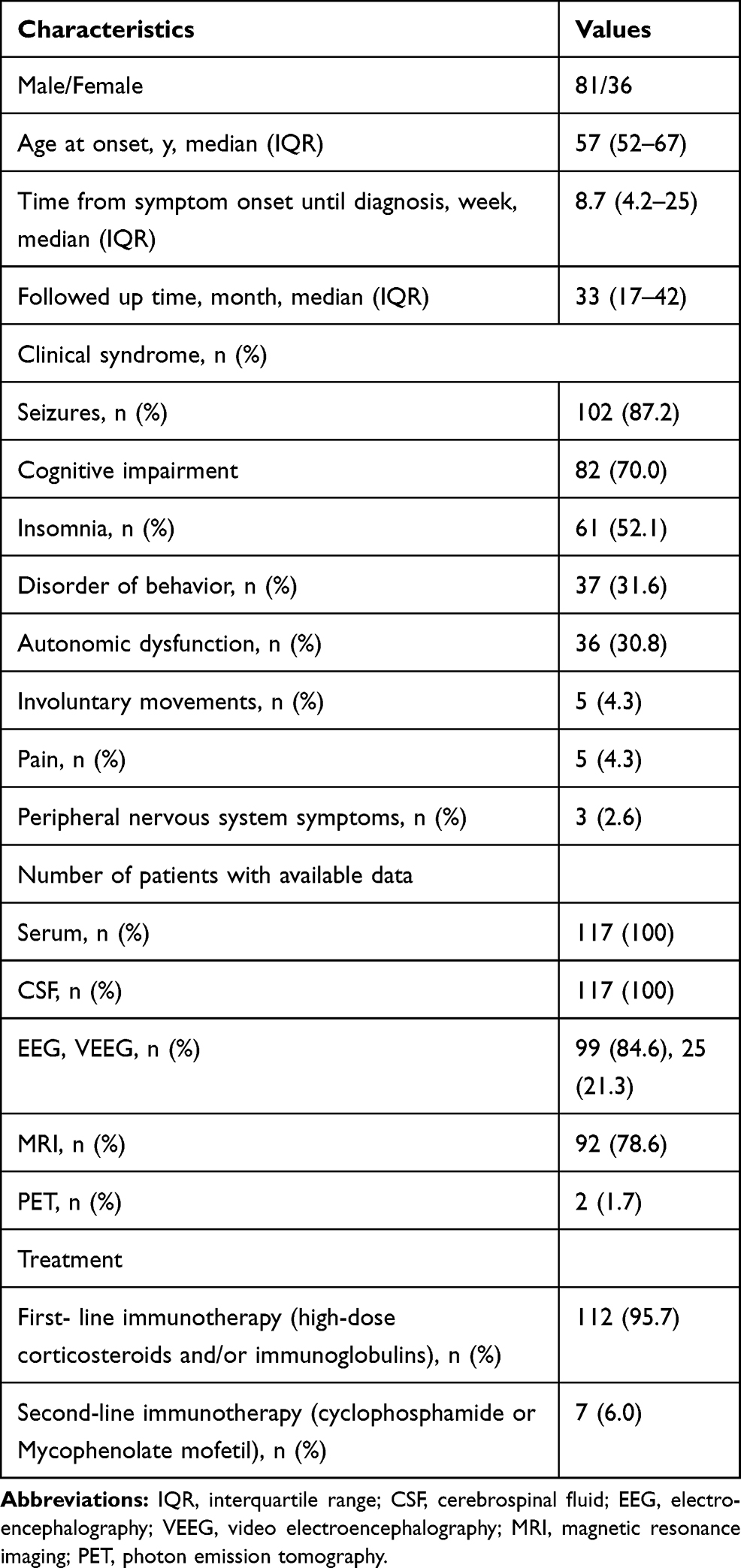 |
Table 1 Clinical Features of Patients (n=117) |
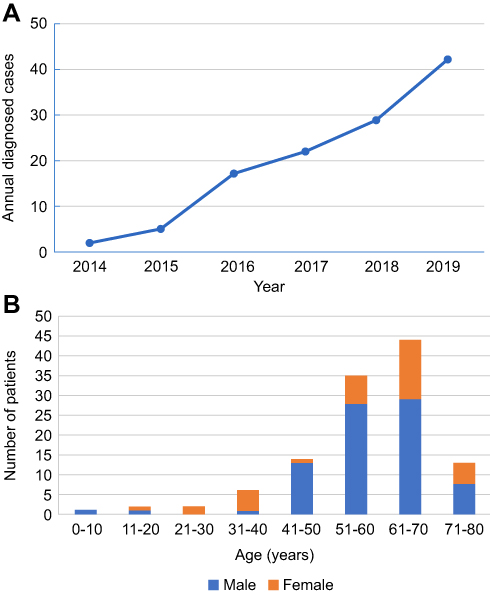 |
Figure 1 (A) Trends in the numbers of annually diagnosed cases of anti-leucine-rich glioma-inactivated 1 (LGI1) encephalitis. (B) Distribution of gender and age of patients with anti-LGI1 encephalitis. |
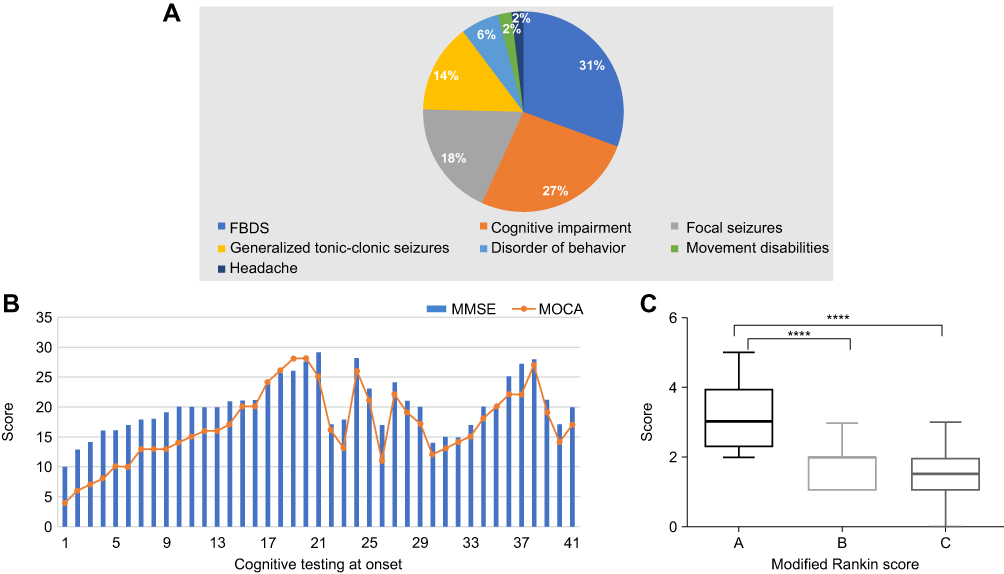 |
Figure 2 (A) Initial symptoms in 117 patients with anti–leucine-rich glioma-inactivated 1 (LGI1) encephalitis. (B) Assessment of cognitive function of 42 patients at the time of initial admission to the hospital. MMSE, Mini-Mental State Examination; MOCA, Montreal Cognitive Assessment Scale; FBDS, faciobrachial dystonic seizure. (C) Modified Rankin Scale (mRS) score at onset and follow-up. A, mRS score at onset; B, mRS score at 6 months after immunosuppressive therapy; C, mRS score at the last follow-up. ****P < 0.0001. |
Seizures occurred in 87.2% (102/117) of the patients. Some patients (35.9%, 42/117) presented with faciobrachial dystonic seizures (FBDSs). Among them, 36 patients started with FBDSs. FBDSs always involved the arm, and in 69.0% (29/42) of the patients, the ipsilateral hemiface was involved as well. Patients with FBDSs had short, sudden, and primarily tonic contractions of the upper limbs, accompanied by an ipsilateral hemifacial grimacing and dystonic posture that lasted for 0.3–5 seconds, occurring 5–80 times per day. None of the patients experienced prodromal symptoms or loss of consciousness. In patients with FBDS, the hemi-trunk (16.7%, 7/42) and a lower limb (14.3%, 6/42) were involved. Focal seizures affected 35.0% (41/117) of the patients, and generalized tonic-clonic seizures affected 16.2% (19/117) of the patients (with 32% [6/19] affected by status epilepticus). The characteristics of 117 patients are shown in Table 2.
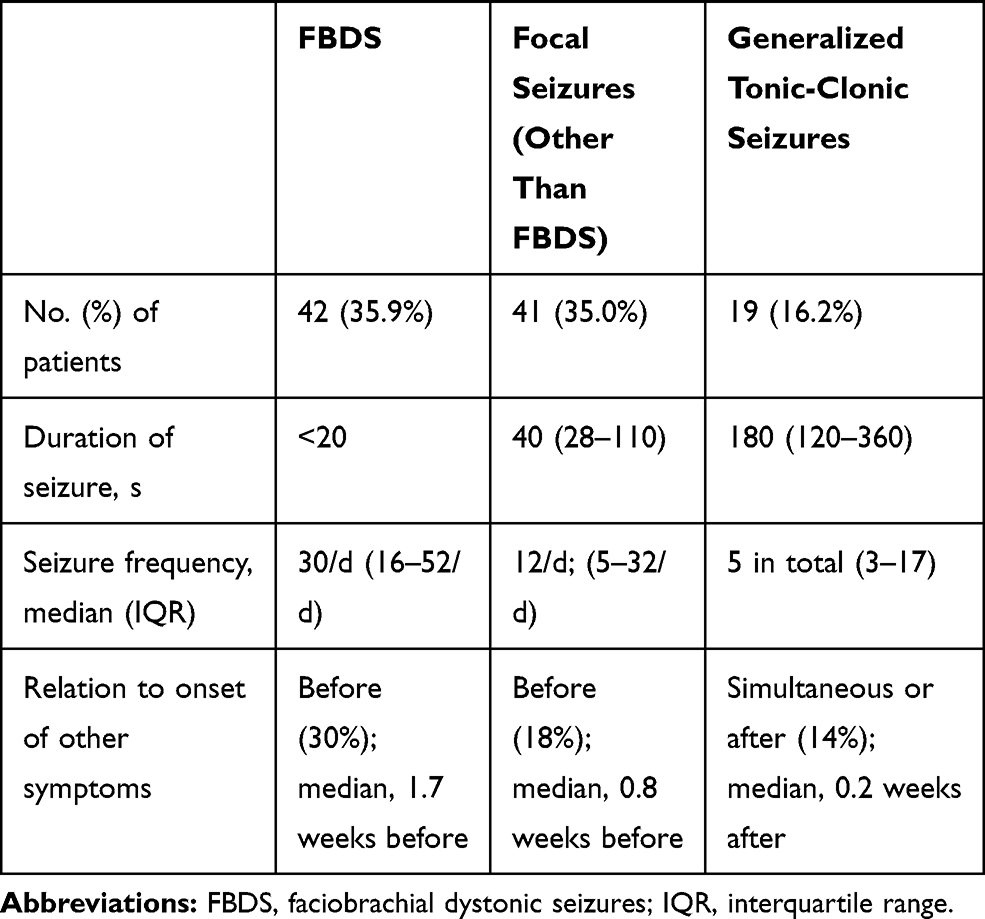 |
Table 2 Seizure Characteristics of the 117 Patients |
Cognitive impairments were diagnosed in 70.1% (82/117) of the patients based on a neurological examination at admission. Memory deficits were found in 81% (66/82) of the cognitively impaired patients, and anterograde memory deficits were found in 62% (51/82) of the cognitively impaired patients. Five patients presented with recent memory loss, and 16 patients showed a decrease in response capacity. In 41 patients, the median Mini-Mental State Examination (MMSE) score was 20 (IQR, 17–22), and the median score on the Montreal Cognitive Assessment Scale (MOCA) was 15 (IQR, 13–21) (Figure 2B). Immunosuppressive therapy resulted in improved cognitive function.
Other common symptoms included insomnia (52.1%, 61/117), behavioral disorders (31.6%, 37/117), and autonomic dysfunction (30.8%, 36/117). Patients with sleep disorders were usually characterized as having reduced sleep time and increased dreams. Chorea was found in 4.3% (5/117) of the patients. Five cases started with chronic headaches. Three patients suffered from peripheral nervous system dysfunction. One patient’s electromyogram showed extensive peripheral neuropathy, mainly involving axons. Another two patients showed peripheral neuropathy in both lower limbs, and motor and sensory fibers were involved (Table 1). There was no incidence of neoplastic disease.
Ancillary Test Results
Antibody titers in 117 patients ranged from 1:1 to 1:100 in the serum and from 1:1 to 1:320 in the CSF. Of the 117 patients with anti-LGI1 encephalitis, 75 patients were positive for anti-LGI1 antibodies in both the serum and CSF, 17 were only positive for anti-LGI1 antibodies in the CSF, and 25 were only positive for anti-LGI1 antibodies in the serum (Supplement 1). Increased CSF pressure (205–220 mm H2O) was found in 4.3% (5/117) of the patients. White blood cell counts were predominantly normal or slightly elevated. White blood cell counts were elevated in 14.5% (17/117) of the patients (median, 80%; range, 56–90%). Protein levels (primarily IgG and IgA) were increased in 36.7% (43/114) of the patients (median, 590 mg/L; range, 49–900 mg/L). Glucose and chloride levels were normal, and oligoclonal band measurements were negative. Hyponatremia was present in 65.0% (76/117) of the patients, and hypokalemia was present in 21.4% (25/117) of the patients. Both hyponatremia and hypokalemia were present in 19.7% (23/117) of the patients. Thyroid function indices of 31.6% (37/117) patients were abnormal, and 8 of these 37 (22%) patients showed elevated serum thyroid peroxidase antibodies (TPO Ab). Serum tumor markers were mildly abnormal in 27.4% (32/117) of the patients. Rubella virus/cytomegalovirus and herpes simplex virus IgG were detected in the serum of 9.4% (11/117) of the patients.
Brain MRIs were performed in 78.6% (92/117) of the patients. MRIs were abnormal in 32.5% (38/117) of the patients (20 patients had abnormal bilateral hippocampal signals, 14 showed an abnormal signal in the left hippocampus, 2 showed an abnormal signal in the left basal ganglia, and 2 had abnormal signals in the right occipital lobe and corpus callosum). MRIs were repeated on 13% (12/92) of the patients during or after immunotherapy. The results showed that lesion areas in six of these patients decreased or disappeared, four patients showed no significant changes, and two patients showed an increase in the size of the lesions compared with the time of onset or since new lesions appeared. However, cognitive function and the frequency of seizures in all 92 patients showed varying degrees of improvement. Two patients underwent positron emission tomography (PET) examinations of the brain with 18F-fluorodeoxyglucose (18F-FDG) (one patient had PET-CT results from outside the hospital, showing hypermetabolism in the right medial temporal lobe but not a brain MRI at the same stage). A PET examination of another patient revealed bilateral hypermetabolism in the basal ganglia and medial temporal lobe (Figure 3E–H). At the same time, a craniocerebral MRI scan of this patient showed abnormal bilateral signals in the hippocampus and the occipital lobe (Figure 3A–D).
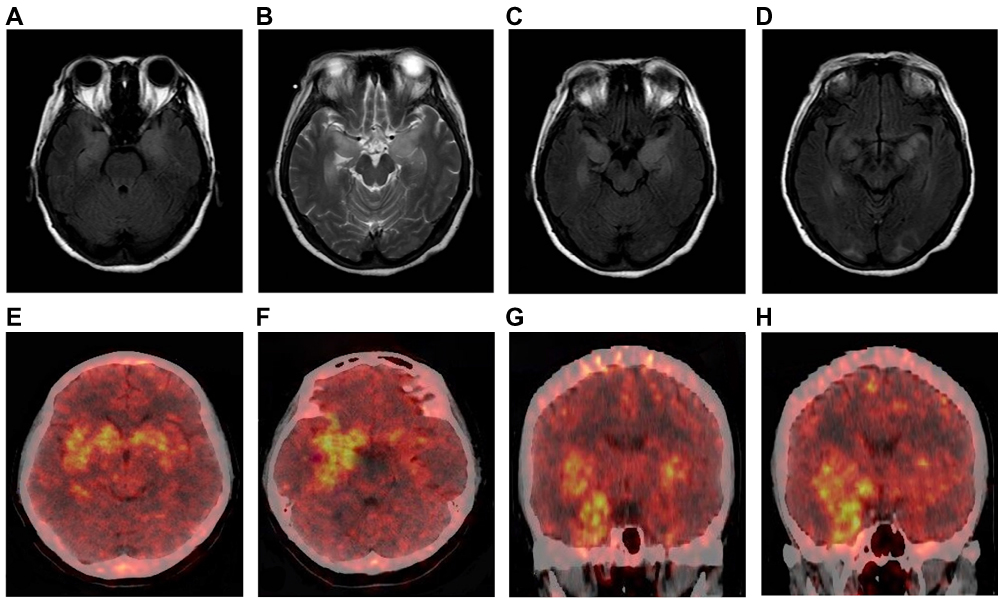 |
Figure 3 Brain magnetic resonance imaging and positron emission tomography (PET) scans of patients with anti-leucine-rich glioma-inactivated 1 (LGI1) encephalitis. Axial fluid-attenuated inversion recovery (A, C, D) and T2-weighted (B) images. Bilateral high signal in the medial temporal lobe (A–C). Bilateral abnormal signals in the medial temporal lobe and occipital lobe (D). PET images show bilateral hypermetabolism in the basal ganglia and medial temporal cortex (more significant on the right) (E–H). |
An electroencephalogram (EEG) was conducted for 99 of the 117 patients. Video-electroencephalograms (VEEGs) were performed for 25 patients, with the monitoring time ranging from 8 h to 36 h. EEG abnormalities were detected in 61 of the 99 patients, most showing generalized slow waves, sometimes accompanied by paroxysmal sharp waves. Sharp waves were commonly detected in the frontal and temporal lobes, which were considered to be epileptiform discharges (Figure 4). In 10 patients who had a VEEG, 42 FBDSs were recorded (median, 3 per patient; IQR, 2–7) with no significant abnormal discharge detected during the corresponding EEG. Thirty-two FBDSs involved the ipsilateral hemiface and an arm, of which six involved the legs. Four events involved only the face, four involved only an arm, and two events exclusively involved the legs. FBDSs occurred during wakefulness (n = 21), drowsiness (n = 5), and sleep (n = 18).
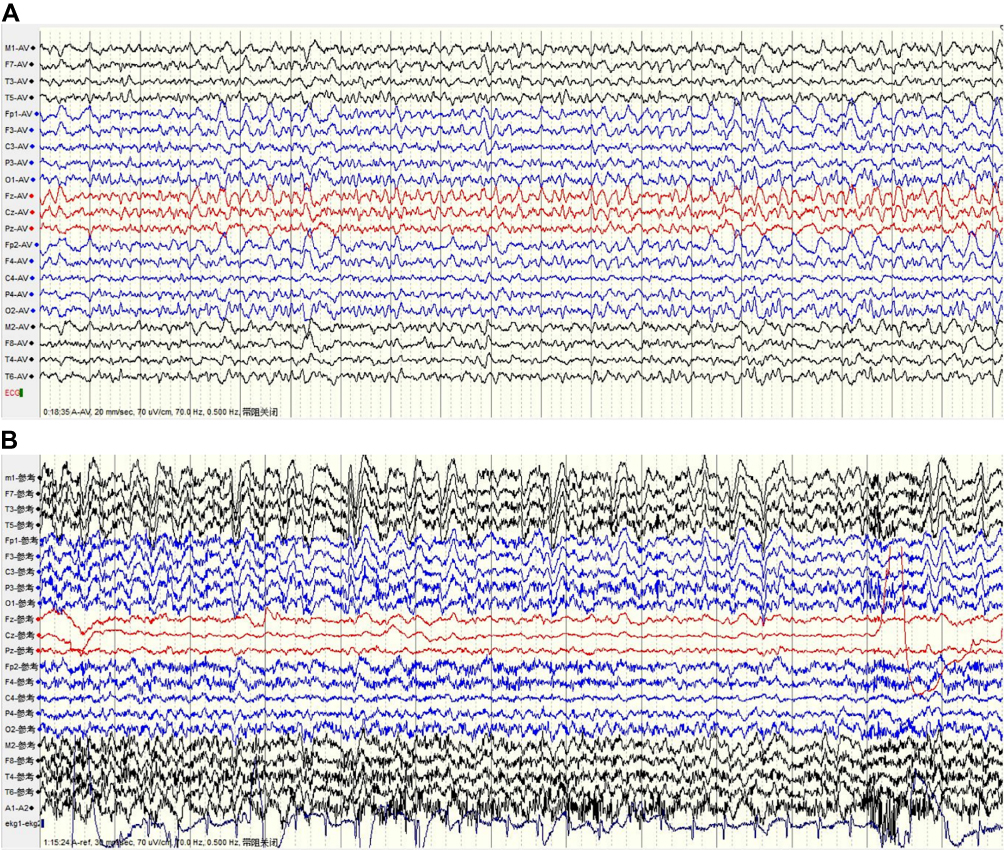 |
Figure 4 Anti-leucine-rich glioma-inactivated 1 (LGI1) encephalitis video electroencephalogram (EEG). (A) The EEG shows slow background rhythm as well as slow and sharp waves in both frontal areas (Fp1\F3\Fp2\F4). (B) Monitoring the faciobrachial dystonic seizures (FBDSs), amplitudes of the bilateral cerebral hemispheres were asymmetrical with diffuse slow waves in the left cerebral hemisphere, and no epileptiform discharges were detected. |
Treatment Outcomes
Of the 117 patients, 109 patients were treated with immunotherapy. Thirty-four patients received both intravenous immunoglobulin (IVIG; 0.4 g/kg/d for 5 days) and steroids: intravenous methylprednisolone (500–1000 mg [15–30 mg/kg/d] for 3–5 days); dexamethasone (intravenous infusion, 10 mg/d for 7–14 days), or oral prednisone (1 mg/kg/d). Seven of the 34 patients received second-line treatment (mycophenolate, cyclophosphamide, or azathioprine) because of a relapse. Except for patients with a relapse, steroids were maintained for 6 months. Details of the immunotherapy and its strategy are shown in Table 3 and Supplement 2. After 3–5 days of immunotherapy, the clinical manifestations were alleviated in all the patients who demonstrated improvements in memory, mental ability, language function, and behavior. The neurological mRS score of the patients decreased from 3.19 ± 0.13 (range, 1–5) before immunotherapy to 1.71 ± 0.09 (range, 1–3) after 6 months of immunotherapy. The mRS score was 1.404± 0.16 at the last follow-up (Figure 2C). The 112 patients with seizures received single or a combination of 2 or 3 antiepileptic drugs (AEDs), including levetiracetam (1000–1500 mg/day), valproate acid (600–1000 mg/day), carbamazepine (300–600 mg/day), and oxcarbazepine (600–1200 mg/day). Details are shown in Table 4. Antiepileptic drugs were maintained for at least 1 year after the patients were seizure-free and had a normal EEG.
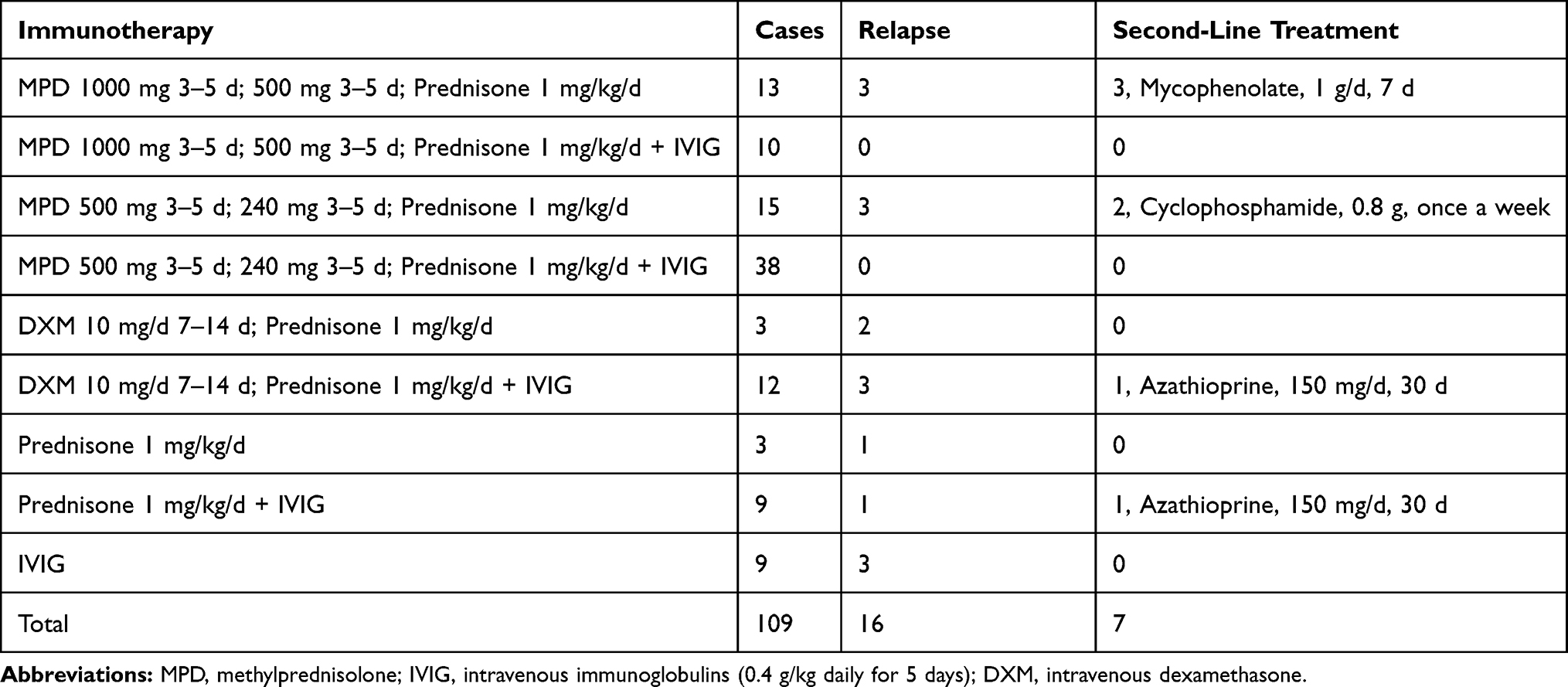 |
Table 3 Treatment and Follow-Up |
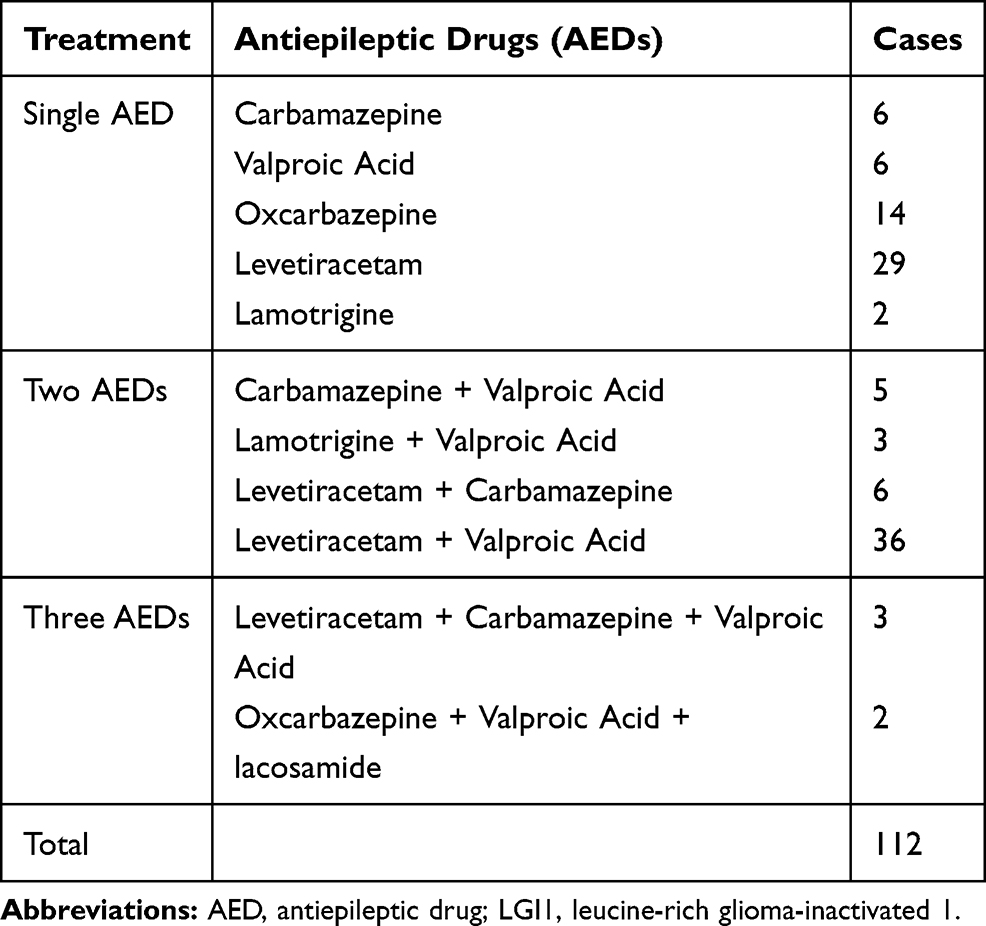 |
Table 4 Details of the Anti-Epileptic Therapy for Patients with Anti-LGI1 Encephalitis |
Relapse
Patients with AE who had improved or stabilized clinical symptoms for more than 2 months, reappearance of symptoms, or aggravation of the original symptoms (increased Rankin score of 1 point or more) were considered to have relapsed.10 Over a median follow-up of 33 months (IQR, 17–42), there were no mortalities, and 16.2% (19/117) of patients experienced a relapse. The median delay from disease onset to the first relapse was 5 months (IQR, 2.1–17). Of the patients who experienced a relapse, 16 patients had received immunotherapy at the first onset, while 3 other patients did not receive immunotherapy at their first onset. Twelve patients were positive for anti-LGI1 antibodies in both the CSF and serum, three patients were positive for anti-LGI1 antibodies in the serum but negative for anti-LGI1 antibodies in the CSF, and four patients were only positive for anti-LGI1 antibodies in the CSF (Supplement 1). The most common symptoms of relapse manifested as impaired cognitive function, seizures, and increased sleep. Of the 19 relapsed patients, 12 had slow-wave delivery at the time of onset in their EEG, and 5 of them had sharp waves. Notably, 6 of the 19 patients who relapsed showed abnormal signal lesions in brain MRI scans at the first onset. Three of these six patients revealed new abnormal signals of hippocampus T2-FLAIR at relapse. The remaining 16 patients showed no obvious abnormalities in brain MRI scans at relapse.
Discussion
Anti-LGI1 encephalitis is a rare neuroinflammatory brain condition. Since first reported in 2010, an increasing number of patients who are positive for anti-LGI1 antibodies have been described.11–15 However, there have been few large-scale epidemiological surveys of anti-LGI1 encephalitis in China. In this study, we provide detailed medical information for 117 Chinese patients who were positive for anti-LGI1 antibodies from 5 clinical centers. We report incidence rates and long-term outcomes.
With regard to age, the majority of patients in our study group were 40–70 years old (79.5%, 93/117), with a higher percentage of men than women, consistent with reported data on other autoimmune diseases.4,16,17 A total of 16.2% (19/117) of patients experienced a relapse, consistent with previous studies.1,8,11 One study on the long-term follow-up of patients with anti-LGI1 encephalitis in the United States reported that 67% (14/21) of patients who were followed up for >2 years had a favorable outcome.4 These data are consistent with a good prognosis for anti-LGI1 encephalitis, although some differences may be related to race, region, and the number of cases of patients included in the study. Currently, research on the relationship between anti-LGI1 antibody titer and disease is still lacking. More patients were positive for anti-LGI1 antibodies in the serum than those who were positive for anti-LGI1 antibodies in CSF in our study. Studies investigating larger sample sizes are needed for further exploration.
In recent years, the coexistence of AE and other autoimmune diseases has attracted the attention of researchers, although the clinical significance and mechanisms have not been clarified.18–20 In a retrospective cohort study, researchers enrolled 517 patients with AE, among whom 45 were affected by one or more types of autoimmune diseases including Hashimoto’s thyroiditis (HT), and skin or mucosal lesions such as vitiligo and bullous pemphigoid.21 In our study, 37 patients showed indications of abnormal thyroid function. There were also patients with anti-LGI1 encephalitis combined with other autoimmune diseases (1 patient with psoriasis and 1 patient with Hashimoto’s thyroiditis). This may provide clues for the pathogenesis of anti-LGI1 encephalitis. Examinations of anti-thyroid antibodies might be useful in clarifying an underlying immune correlation.
Immunotherapy is thought to be effective in the treatment of anti-LGI1 encephalitis.22–24 First-line immunotherapy may consist of high intravenous steroid (methylprednisolone), IVIG, and/or plasma exchange. Second-line therapies (rituximab, mycophenolate, cyclophosphamide, and azathioprine) are used in refractory cases or as a maintenance therapy to prevent relapse. A small number of patients are prone to relapse after treatment.25 Studies have reported on the favorable effects of immunotherapy in decreasing seizure frequency and improving cognition.4,11 In a retrospective study, Sonderen et al reported that first-line treatment was considered effective in 80% of patients including cognitive improvements in 42%.4 Helena et al retrospectively analyzed the clinical information of 76 patients with anti-LGI1 encephalitis. They believed most patients with LGI1 antibody-associated cognitive deficits respond well to immunotherapy, with 35% of patients returning to their baseline cognitive functions after immunotherapy.11 In our study, 109 patients who received immunotherapy had an improvement in memory, mental ability, and behavior to varying degrees, consistent with previous studies.
Immunotherapy plays a key role in the treatment of anti-LGI1 encephalitis-related seizures. In a retrospective study on anti-LGI1 AE,17 immunotherapy was found to be more effective than AEDs in 19 treated patients, and immunotherapy combined with AEDs was considered more effective than either alone. A clinical trial by Sarosh et al in 2011 reported that immunotherapies were associated with a remarkable reduction in the frequency of FBDS in patients with anti-LGI1 AE.22 Among 27 patients receiving immunotherapy (including steroid therapy, IVIG, plasma exchange, and rituximab), 14 patients showed a greater than 50% reduction in seizure frequency.22 In our study, after 3–5 days of immunotherapy, seizures were alleviated in all the patients, consistent with these findings. These results indicate the importance of immunotherapy for anti-LGI1 encephalitis. We found that 48 patients who received high-dose methylprednisolone combined with IVIG treatment did not relapse during the follow-up period. Several patients who did not receive immunotherapy and received only steroids or IVIG experienced relapse. This suggests that methylprednisolone combined with IVIG treatment can reduce recurrence (Table 3), although these results require additional evidence and long-term follow-up.
Relapses were not common in our cohort. The definition of relapse in our study was based on observations of clinical symptoms. Monitoring of AE-related antibodies is not routinely performed during follow-up. Accurate indicators for relapse should be identified by future studies. In our cohort, only 3 of 19 patients showed abnormal signals in MRI scans at relapses, indicating that relapses are not always synchronized with MRI scans. However, it should be noted that 6 of the 19 patients who relapsed showed abnormal signal lesions in brain MRI scans at disease onset. This suggests that patients with abnormal signal lesions on brain MRI scans at disease onset may be at risk of recurrence in later stages of the disease. In addition, we observed that a large number of slow waves or intermittent spikes in EEG were detected at onset in 12 of 19 patients who relapsed. This suggests the importance of EEG monitoring for disease recurrence. Most patients in our study experienced their first relapse within 1 year. However, prior studies have suggested that AE relapse could occur years after the initial episode.4,11 This underlines the need for long-term follow-up in assessing outcomes. Although the patient medical records we obtained are reliable and were evaluated by a physician, this study is limited as a retrospective study. Our understanding of anti-LGI1 encephalitis will improve with expanded sample sizes and extended follow-up times.
Conclusion
We describe the clinical characteristics, immunotherapy, and long-term outcomes of patients with anti-LGI1 encephalitis in China. Early adequate immunotherapy has positive implications for the improvement of clinical symptoms of anti-LGI1 encephalitis. The outcome for patients with anti-LGI1 encephalitis is mostly favorable, although cognitive dysfunction does persist in some patients. The treatment of severe, refractory, and/or recurrent anti-LGI1 encephalitis has yet to be investigated. Although recurrence was uncommon in our study, physicians should be aware that disease relapse is possible, even years after disease onset. Therefore, long-term follow-up is essential. Further research, using more robust study designs and extended follow-up periods, is necessary.
Acknowledgments
We are grateful to all the participants in this study. We thank Editage and TopEdit for their linguistic assistance for this manuscript and the reviewers at the Neuropsychiatric Disease and Treatment for their hard work and consideration.
Funding
This research was supported by grants from the National Natural Science Foundation (NO.81873786).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med. 2018;378:840–851. doi:10.1056/NEJMra1708712
2. Giordano A, Fazio R, Gelibter S, et al. Diagnosing autoimmune encephalitis in a real-world single-centre setting. J Neurol. 2019;267:449–460. doi:10.1007/s00415-019-09607-3
3. Schubert J, Brämer D, Huttner HB, et al. Management and prognostic markers in patients with autoimmune encephalitis requiring ICU treatment. Neurol Neuroimmunol Neuroinflamm. 2019;6:e514. doi:10.1212/NXI.0000000000000514
4. van Sonderen A, Thijs RD, Coenders EC, et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology. 2016;87:1449–1456. doi:10.1212/WNL.0000000000003173
5. Griffith SP, Malpas CB, Alpitsis R, O’Brien TJ, Monif M. The neuropsychological spectrum of anti-LGI1 antibody mediated autoimmune encephalitis. J Neuroimmunol. 2020;345:577271. doi:10.1016/j.jneuroim.2020.577271
6. Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9(8):776–785. doi:10.1016/S1474-4422(10)70137-X
7. Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain. 2010;133:2734–2748. doi:10.1093/brain/awq213
8. Dubey D, Britton J, McKeon A, et al. Randomized placebo‐controlled trial of intravenous immunoglobulin in autoimmune LGI1/CASPR2 epilepsy. Ann Neurol. 2019;87:313–323. doi:10.1002/ana.25655
9. Sato M, Kishida D, Miyazaki D, Sekijima YA. Patient with limbic encephalitis associated with anti-leucine-rich glioma-inactivated 1 (LGI1) antibody presenting with slowly progressive cognitive impairment and fluctuating striatal lesions. Intern Med. 2019;58:287–291. doi:10.2169/internalmedicine.1082-18
10. Association NBoCM. Chinese expert consensus on the diagnosis and management of autoimmune encephalitis. Chin J Neurol. 2017;50:91–98.
11. Arino H, Armangué T, Petit-Pedrol M, et al. Anti-LGI1-associated cognitive impairment: presentation and long-term outcome. Neurology. 2016;87:759–765. doi:10.1212/WNL.0000000000003009
12. Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15:391–404. doi:10.1016/S1474-4422(15)00401-9
13. Steriade C, Mirsattari SM, Murray BJ, Wennberg R. Subclinical temporal EEG seizure pattern in LGI1-antibody-mediated encephalitis. Epilepsia. 2016;57:e155–e160. doi:10.1111/epi.13436
14. Dalmau J, Geis C, Graus F. Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the central nervous system. Physiol Rev. 2017;97:839–887. doi:10.1152/physrev.00010.2016
15. Hermetter C, Fazekas F, Hochmeister S. Systematic review: syndromes, early diagnosis, and treatment in autoimmune encephalitis. Front Neurol. 2018;9:706. doi:10.3389/fneur.2018.00706
16. Husari KS, Dubey D. Autoimmune epilepsy. Neurotherapeutics. 2019;16:685–702. doi:10.1007/s13311-019-00750-3
17. Li LH, Ma CC, Zhang HF, et al. Clinical and electrographic characteristics of seizures in LGI1-antibody encephalitis. Epilepsy Behav. 2018;88:277–282. doi:10.1016/j.yebeh.2018.08.019
18. C L X, Liu L, W Q Z, et al. Anti-N-methyl-D-aspartate receptor encephalitis with serum anti-thyroid antibodies and IgM antibodies against Epstein-Barr virus viral capsid antigen: a case report and one year follow-up. BMC Neurol. 2011;11(149):2–7. doi:10.1186/1471-2377-11-2
19. Guan W, Fu Z, Zhang H, et al. Non-tumor-associated Anti-N-Methyl-D-aspartate (NMDA) receptor encephalitis in chinese girls with positive anti-thyroid antibodies. J Child Neurol. 2015;30(12):1582–1585. doi:10.1177/0883073815575365
20. Ma XY, Yin QQ, Zeng ZL, et al. Thyroid function and autoimmune indications in patients with Anti-N-Methyl-D-aspartate receptor encephalitis. Neuroimmunomodulation. 2018;25(2):110–117. doi:10.1159/000492179
21. Zhao J, Wang CC, Xu XL, et al. Coexistence of autoimmune encephalitis and other systemic autoimmune diseases. Front Neurol. 2019;10:1142. doi:10.3389/fneur.2019.01142
22. Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69:892–900. doi:10.1002/ana.22307
23. Pradhan S, Das A, Mani VE. Immunotherapy in autoimmune encephalitis - A need for “presumptive” diagnosis and treatment. Neurol India. 2018;66:1584–1589. doi:10.4103/0028-3886.246277
24. Wang ML, Cao XY, Liu QX, et al. Clinical features of limbic encephalitis with LGI 1 antibody. Neuropsychiatr Dis Treat. 2017;13:1589–1596. doi:10.2147/NDT.S136723
25. Steiner J, Prüss H, Köhler S, Frodl T, Hasan A, Falkai P. Autoimmune encephalitis with psychosis: warning signs, step-by-step diagnostics and treatment. World J Biol Psychiatry. 2018;22:241–254.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.