Back to Journals » Infection and Drug Resistance » Volume 16
Clinical Evaluation of Metagenomic Next-Generation Sequencing and Identification of Risk Factors in Patients with Severe Community-Acquired Pneumonia
Authors Lu D ![]() , Abudouaini M, Kerimu M, Leng Q, Wu H
, Abudouaini M, Kerimu M, Leng Q, Wu H ![]() , Aynazar A, Zhong Z
, Aynazar A, Zhong Z
Received 17 May 2023
Accepted for publication 29 July 2023
Published 9 August 2023 Volume 2023:16 Pages 5135—5147
DOI https://doi.org/10.2147/IDR.S421721
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Dongmei Lu,1 Maidina Abudouaini,1,2 Munire Kerimu,2 Qiuping Leng,1 Hongtao Wu,1 Amar Aynazar,1,2 Zhiwei Zhong1,2
1Center of Pulmonary and Critical Care Medicine, People’s Hospital of Xinjiang Uygur Autonomous Region, Urumqi, People’s Republic of China; 2Department of Public Health, Xinjiang Medical University, Urumqi, People’s Republic of China
Correspondence: Dongmei Lu, People’s Hospital of Xinjiang Uygur Autonomous Region, Urumqi, People’s Republic of China, Tel +18997982968, Email [email protected]
Purpose: Severe community-acquired pneumonia (SCAP) is the leading cause of death among patients with infectious diseases worldwide. This study aimed to evaluate the effectiveness of metagenomic next-generation sequencing (mNGS) through detecting pathogens in bronchoalveolar lavage fluid (BALF) and identifying risk factors for recovery in SCAP patients.
Patients and Methods: This prospective study recruited 158 SCAP patients admitted to respiratory intensive care unit that were randomly divided into control and study groups, with receiving conventional tests and the same conventional tests plus mNGS, respectively. The diagnostic efficiency of mNGS was evaluated by comparing with conventional tests. Furthermore, univariate and multivariate logistic regression analyses were performed to determine the independent risk factors for recovery in SCAP patients, and a nomogram prediction model was established based on these factors.
Results: Within the study group, the pathogen detection rate was significantly higher with mNGS than that with conventional tests (84.81% vs 45.57%, P < 0.001), with a positive coincidence rate of 94.44%. Acinetobacter baumannii (21.52%, 17/79), Candida albicans (17.72%, 14/79), and Klebsiella pneumonia (15.19%, 12/79) were the top three common pathogens detected by mNGS. Of note, the improvement rate of patients in the study group was significantly higher than that in the control group. The further analysis revealed that the increased levels of interleukin-6, blood urea nitrogen, procalcitonin, the longer length of hospital stay, and bacterial infection were independent risk factors for recovery of SCAP patients, while mNGS detection status was a protective factor. The predictive model showed a good performance for the modeling and validation sets.
Conclusion: Early mNGS exhibited a superior diagnostic efficiency to conventional tests in SCAP patients, which can reduce the risk of death in SCAP patients. Moreover, the clinical factors could also be used for the management and prognosis prediction of SCAP patients.
Keywords: severe community-acquired pneumonia, metagenomic next-generation sequencing, pathogen, risk factor
Introduction
Infection is one of the major causes of death in patients with acute and critical illnesses. Especially, lower respiratory tract infections (LRTIs) are the most common infectious diseases worldwide, with a high mortality rate.1,2 As estimated, there are about 10 million new cases of LRTIs in China each year.3,4 Pneumonia, typically caused by LRTIs, is the leading cause of illness and death in adults, resulting in more than one million hospitalizations and a high intensive care unit (ICU) admission rate in the United States per year.5 It is noteworthy that the mortality rate of severe community-acquired pneumonia (SCAP) is as high as 20–50%, and its high long-term morbidity is a serious challenge for ICU survivors,6 with at least 3 million deaths worldwide in 2016.1 In China, more than 20% of ICU patients are admitted due to sepsis with up to 35.5% 90-day mortality; actually, 68.2% of these sepsis cases are secondary to SCAP.7 The high mortality is mainly attributable to the increased rate of treatment failure due to antibiotic resistance8 and the lack of measures that can identify the relevant pathogens in a timely and accurate manner.9,10
Due to fundamental diseases, invasive operations, compromised immune function, and overuse of wide-spectrum antibiotics in patients, especially elderly patients, the types of pathogens and antibiotic resistance in the lower respiratory tract have also been changing significantly and continuously, seriously affecting the clinical outcomes and threatening patients’ health. Because of the low sensitivity, efficiency, and spectrum of detectable pathogens, current conventional microbial tests are always time-consuming and have a low positive detection rate of pathogens (~10%). Moreover, the detection of mixed infections and unknown pathogen infections is almost impossible with these conventional tests, resulting in the inability to identify the exact pathogens and provide useful clinical guidance, despite at least 60% of all cases are systematical screened with conventional tests;9 actually, only about 38% of adult patients with community-acquired pneumonia are confirmed with specific pathogens.10,11 Specifically, in ICU, the diagnosis of LRTIs is particularly complicated, as the non-infectious inflammatory state often overlaps with clinical characteristics of infection,12 and patients with a severely immunocompromised system may present symptoms of non-specific pulmonary infection. Unclear etiological diagnosis can lead to patients using unnecessary antibiotics, further resulting in the risks of systemic toxicity, double infection, or even multi-drug resistance, which may cause public health concerns.13 In other cases, without an etiological diagnosis, the treatment may be delayed, or the clinicians may assume that the symptoms are caused by non-infectious inflammatory conditions and empirically administer corticosteroids, which may further aggravate occult infections.14 Thus, early and accurate etiological detection in LRTIs is critical for timely and precise treatment; however, there is a lack of effective detection methods currently and a new etiological diagnostic method needs to be developed urgently.
One of the promising methods is the metagenomics next-generation sequencing (mNGS) technique, which can unbiasedly detect almost all pathogens from the complicated clinical samples with high sensitivity, broad pathogen spectrum and short turnaround time, especially for the detection of unknown, rare, and atypical etiologies.15,16 Moreover, mNGS can obtain transcriptional biomarkers of diseases based on comprehensive detection of pathogens, airway microbiome, and host immune cells simultaneously.17–19 Up to date, many studies have explored the microbiological diagnostic performance of mNGS in LRTIs using bronchoalveolar lavage fluid (BALF) or sputum samples, which reveal a superior pathogen-positive detection rate compared with conventional tests, especially in atypical pathogens and mixed infections.20–22 Considering a wide range of potential pathogens and the complexity of host background in ICU patients, although several scholars have been attracted to explore the clinical application of mNGS in these populations;23,24 however, the clinical application evaluation of mNGS in SCAP with a larger cohort is still necessary across different geographical areas.
Thus, this study aims to evaluate the effectiveness of mNGS through detecting pathogens in BALF from SCAP patients using both metagenomics extended-spectrum pathogenic microorganism sequencing (a specific version of mNGS) and conventional culture methods. We also aim to identify risk factors for recovery in SCAP patients. In summary, this study is of great importance in assisting clinicians to formulate an accurate and timely treatment plan for SCAP patients.
Materials and Methods
Study Subjects
A total of 158 SCAP patients admitted to the respiratory ICU of the People’s Hospital of Xinjiang Uygur Autonomous Region from October 2019 to August 2021 were recruited in this prospective cohort study. Inclusion criteria were made according to the 2007 IDSA/ATS SCAP diagnosis and treatment guidelines, including the presence of a major criterion (invasive mechanical ventilation or septic shock with a need for vasopressors) or of 3 minor criteria from a list of 9 clinical radiobiological signs (respiratory rate ≥ 30 breaths/min, artery oxygen partial pressure (PaO2)/fraction of inspiration O2 (FiO2) ≤ 250 mmHg, multilobar infiltrates, confusion/disorientation, blood urea nitrogen (BUN) ≥ 20 mg/dL, white blood cell (WBC) count < 4000 cells/mm3, platelet (PLT) count < 100,000 cells/mm3, core temperature < 36°C, and hypotension requiring aggressive fluid resuscitation). Exclusion criteria were as follows: 1) non-infectious pneumonia; 2) severe immunosuppression; 3) active tuberculosis or end-stage disease or a written “do not revive” order; 4) a history of antibiotic or other drug allergies; 5) no microbial culture results obtained; 6) collected BALF samples failed the quality control for mNGS; and 7) lost to follow-up. The recruited patients were randomly divided into the study (mNGS + conventional tests) and control (only conventional tests) groups at a 1:1 ratio (79 cases in each group) using the random number table method. Specifically, in the study group, sputum, blood, and BALF samples were collected for conventional tests (culture and Xpert), and BALF samples were collected for mNGS; treatment plans for the patients in this group were made according to the results from mNGS analysis. In contrast, for patients in the control group, the collected sputum, blood and BALF samples were applied for only conventional tests; treatment plans for the patients in this group were made according to results from conventional microbiological tests. Of note, all the samples were collected before any empirical antibiotic was used.
Clinical Data Collection
Clinical information for these patients, including demographics, comorbidities, levels of serum inflammatory markers such as interleukin-6 (IL-6) and procalcitonin (PCT), and C-reactive protein (CRP), Acute Physiology and Chronic Health Evaluation (APACHEII) score, length of hospital stay (days), and outcomes, were collected.
Specimen Collection
BALF was sampled at the patients’ bedsides while the patients were under local anesthesia by bronchoscopy physicians with more than 5 years’ experience. Bronchoscopy and bronchoalveolar lavage were performed via nasal or artificial airway entry, and local anesthesia was performed by applying 2% lidocaine to the airway mucosal surface. Briefly, after the segmental bronchus of the lung with severe lesions was chosen and the top of the bronchoscopy was embedded in the corresponding bronchial opening, 60–120 mL of warm (~37°C) saline was injected for lavage, with suction performed at 100 mmHg negative pressure. About 40–60% of recycled lavage fluid (BALF, at least 10 mL) was collected into a sterile spiral tube, which was then equally divided into 2 tubes. One tube of BALF sample was used for routine bacterial and fungal culture, and acid-fast staining of smear for detection of tuberculosis. Another tube of BALF sample was sent to BGI Genomics (Shenzhen, China) for DNA/RNA extraction and mNGS. Sputum was sampled (at least 1 mL) using the disposable sputum collector if the artificial airway intubation was established; otherwise, sputum was sampled by patient-initiated cough. The collected sputum samples were submitted for routine bacterial and fungal culture, and acid-fast staining of smear for detection of tuberculosis.
mNGS Processing
mNGS was carried out according to the following procedures: DNA/RNA preparation, DNA reverse transcription (for RNA), library construction, sequencing, data processing, and result interpretation. Briefly, 3–5 mL of BALF was centrifuged at 4000 r/min for 10 min at 4°C. The pellet was used for DNA extraction using the TIANamp Micro DNA Kit [DP316, TIANGEN BIOTECH (Beijing) Co., Ltd.] according to the manufacturer’s instruction. RNA was extracted following the manufacturer’s operational manual, using TIANMicrobe magnetic beads method pathogenic microorganism DNA/RNA extraction kit (NG550-01). For RNA enrichment, after mixing 33 μL of the extracted nucleic acid sample with 7 μL of the enrichment reaction mixture, the mixture was incubated on a PCR machine at 37°C for 10 min, and then performed magnetic bead purification to remove DNA from nucleic acids, thereby improving the concentration of RNA content. The enriched nucleic acid was subjected to fragmentation reaction, one-strand synthesis and two-strand synthesis to form double-stranded DNA, then purified by magnetic beads, and the purified DNA was used for DNA library construction. Then, DNA libraries were constructed through DNA-fragmentation, end-repair, adapter-ligation and PCR amplification. Agilent 2100 was used for quality control of the DNA libraries. Quality qualified libraries were pooled, DNA Nanoball (DNB) was made and sequenced by MGISEQ-2000 platform.
The raw sequencing data were cleaned by removing low-quality (Base Call Quality score < 20) and short fragments (<35 bp). Reads that were mapped to the human reference genome by BWA (BWA: http://bio-bwa.sourceforge.net/) were also removed. The resulting reads were classified by simultaneously aligning to Pathogens metagenomics Database (PMDB), consisting of bacteria, fungi, viruses and parasites. The classification reference databases were downloaded from NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/).
Determination of mNGS Results
The mNGS results were determined according to the diagnostic criteria of Miao et al.25 Briefly, for bacteria (mycobacteria excluded), viruses, and parasites, microbes with a coverage rate scored 10-fold higher than that of any other microbes at species level were considered as clinically significant pathogens. Fungi (species level) with a coverage rate scored 5-fold higher than that of any others were considered as potential pathogens due to difficult DNA extraction. Mycobacterium tuberculosis (MTB) with at least one read was mapped at genus or species level was considered positive, while nontuberculous mycobacteria with the mapped read number at either the species or genus level was in the top 10 of the list of bacteria were considered positive due to the low possibility for contamination and low yield rate.
Statistical Analysis
All statistical analyses were performed using SPSS Statistics (version 24.0) and R (version 4.1.2). Continuous variables were expressed as mean ± standard deviation (SD) or median (interquartile range, IQR). Categorical variables were represented as frequency or percentages (%). Univariate analysis (independent sample t-test and Chi-square χ2 test) was performed to identify the statistically significant risk factors affecting the recovery of SCAP patients; these significant factors were then applied to the multivariate logistic regression analysis to screen out independent risk factors for recovery in SCAP patients. Of note, considering the sample size, a less strict P value (P < 0.10) instead of P < 0.05 was considered statistically significant. Subsequently, the independent risk factors were used to construct the nomogram prediction model using the “rms” R package. Totally, 59 patients randomly selected from the two groups were served as a modeling set, and the rest 20 from the two groups as a validation set, the calibration curve is established by the Bootstrap method to validate the reliability of the predictive model. The receiver operating characteristic (ROC) curve of independent risk factors was drawn, and the C-index and the area under each curve (AUC) were calculated. The decision curve analysis was used to evaluate the predictive efficacy of the nomogram prediction model.
Results
Patient Characteristics and Outcomes
Totally, 158 patients with SCAP were recruited in this study, including 79 patients who underwent mNGS combined with conventional tests (mNGS group) and the rest 79 patients receiving only conventional tests (control group). No significant differences were found regarding factors including age, gender, comorbidities, laboratory examination results (lactic acid, IL-6, D-dimer, PCT, CRP, BNP, ALT, AST, PLT, CCr, BUN, and PaO2), and APACHII score between these two groups (Table 1).
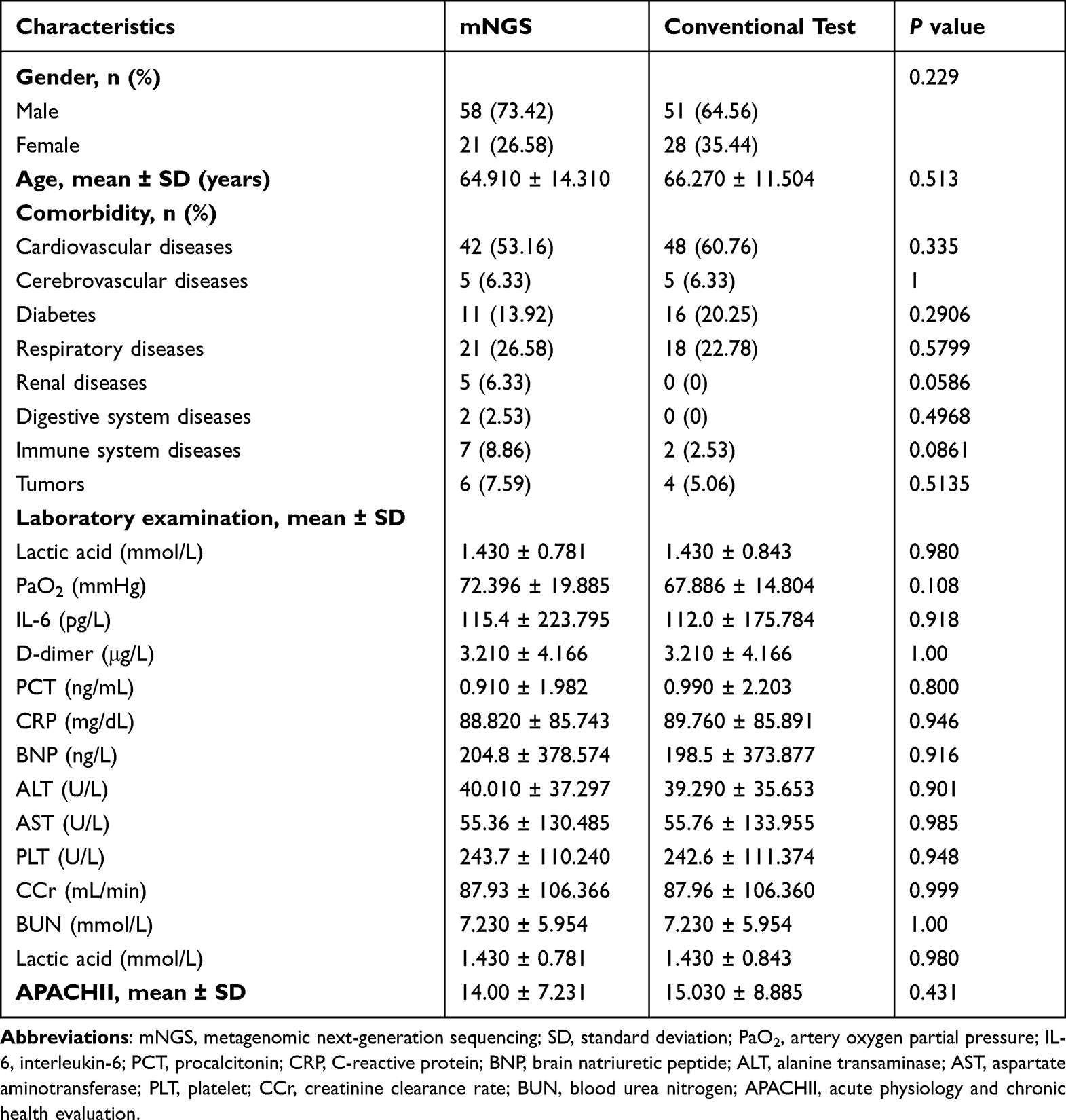 |
Table 1 Clinical Characteristics of the Patients in the Two Groups |
Diagnostic Efficiency of mNGS Compared to Conventional Tests
Within the mNGS group, mNGS identified 67 SCAP patients with positive results out of the 79 patients (84.81%, 67/79), while conventional tests showed 34 cases with positive results (45.57%, 36/79), indicating a significantly elevated positive detection rate by mNGS compared with conventional tests (P < 0.01). It should be recognized that conventional tests in this study consisted of culture and Xpert, which could only identify part of bacteria, fungi and MTB, but not viruses and other special pathogens. In detail, the positive detection rates of BALF culture, sputum culture, and blood culture were 6.46% (13/79), 37.98% (30/79), and 6.3% (5/79), respectively, which were all lower than that of mNGS. We then evaluated the coincidence rate between mNGS and conventional tests, and the results showed a positive coincidence rate of 94.44% (34/36) and a negative coincidence rate of 23.26% (10/43) (Figure 1A). Considering mNGS was performed using BALF samples, the coincidence rate of mNGS with BALF culture were focused, and the results revealed that the positive and negative coincidence rates of mNGS with BALF culture were 100% (13/13) and 18.18% (12/66), respectively (Figure 1B).
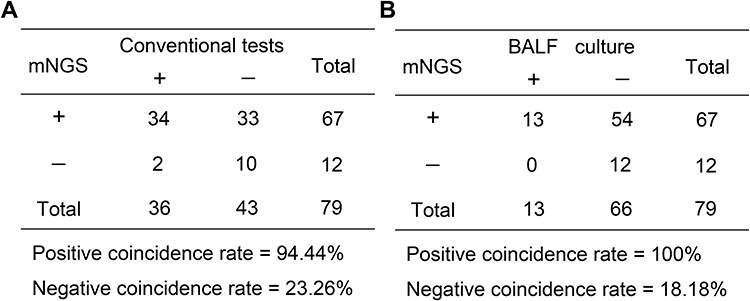 |
Figure 1 Comparison of diagnostic efficiency of metagenomics next-generation sequencing (mNGS) with the conventional tests. Contingency tables for the mNGS with the conventional tests (A) and culture (B). |
Pathogen Characteristics Detected by mNGS Compared to Conventional Tests
In total, 52 kinds of pathogenic microorganisms were detected by mNGS, including bacteria (31, 59.62%), fungi (9, 17.31%), viruses (10, 19.23%), Chlamydia (1, 1.92%), and mycoplasma (1, 1.92%) (Figure 2A). Regarding the pattern of pathogen infection identified by mNGS, 23 of 67 (34.33%) positive cases were detected as single pathogen infections, and 44 cases (65.67%) were detected as multiple pathogen infections (Figure 2B). Among the 23 cases with single pathogen infection, the frequency of bacterial infection was 65.22% (15/23), of fungal infection was 1.74% (5/23), of viral infection was 4.35% (1/23), and of Chlamydial infection was 4.35% (1/23). Among the 44 cases with mixed infections, bacterial-fungal infection was the most common pattern (15, 34.09%), followed by bacterial–fungal–viral infection (9, 20.45%), bacterial–bacterial infection (8, 18.18%), and bacterial–viral infection (6, 13.64%). In addition, the frequency of fungal–viral, bacterial–chlamydial, and fungal–fungal infection was 4.55% (2/44), 4.55% (2/44), and 2.27% (1/44), respectively.
 |
Figure 2 Pathogen characteristics of patients with severe community-acquired pneumonia (SCAP) based on metagenomics next-generation sequencing (mNGS) compared with conventional tests. (A) Distribution of pathogens of patients with SCAP in the mNGS group; (B) Type of infection of patients with SCAP in the mNGS group. The pie plot on the left side shows the number of cases and the proportion of pathogen species identified using mNGS, and the pie plot on the right side shows the number of cases and the detailed proportion of mixed infections identified using mNGS. (C) Comparison of bacterial spectrum by mNGS and conventional tests; (D) Comparison of fungal spectrum by mNGS and conventional tests; (E) Proportion of viruses, chlamydia and mycoplasma identified using mNGS. |
Based on mNGS results, bacteria were found in 56 (70.89%) patients, mainly including Acinetobacter baumannii (17), Klebsiella pneumoniae (12), Corynebacterium striatum (9), and Enterococcus faecium (9) (Figure 2C). Acinetobacter baumannii (16) and Klebsiella pneumoniae (10) were the most frequently detectable bacteria by culture, which was similar to the mNGS results (Figure 2C). The remaining bacteria detected in the culture were Stenotrophomonas maltophilia (3), Escherichia coli (3), Corynebacterium striatum (1), and Pseudomonas aeruginosa (1) (Figure 2C). Fungi were identified in 33 (43.42%) patients by mNGS, with the dominating fungi of Candida albicans (14), Aspergillus fumigatus (7), Pneumocystis jirovecii (5) and Candida glabrata (5) (Figure 2D). Consistent with mNGS results, Candida albicans (8) was the most identified fungus by culture, and the others were Candida glabrata (3) and Candida parapsilosis (1) (Figure 2D). All bacteria and fungi detected by culture could be identified by mNGS. Moreover, the most detected viruses by mNGS were Human alphaherpesvirus 1 and Human gammaherpesvirus 4, with the both frequencies of 7.59% (6/79), followed by Human betaherpesvirus 7 (4), rhinovirus (2), Human betaherpesvirus 5 (2), and human coronavirus (2) (Figure 2E). In addition, Chlamydia psittaci were detected in 4 (5.06%) patients, and Mycoplasma was only identified in one patient (Figure 2E). In addition, for MTB infection, Xpert showed that 4 of 79 (5%) patients were positive, of which 2 were also positive detected by mNGS of BALF. Meanwhile, 2 of the 4 positive MTB patients identified by mNGS were not detected by Xpert.
Prognostic Outcome of mNGS Group Compared to Control Group
Compared with the control group, the average length of hospital stay in the mNGS group was slightly reduced, but without statistical significance between these two groups (Table 2). Of note, the improvement rate and mortality rate of patients in the mNGS group were significantly higher and lower, respectively, than those in the control group (Table 2, P < 0.05).
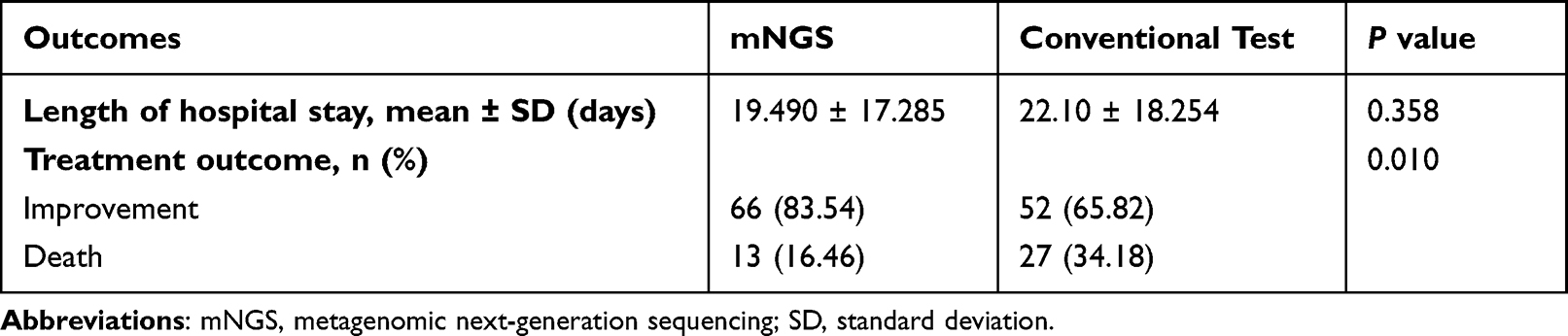 |
Table 2 Outcomes of the Patients in the Two Groups |
Prognostic Risk Factors for Recovery in SCAP Patients
Among the 158 SCAP patients included, 118 (74.68%, 118/158) were improved and 40 (25.32%, 40/158) died. To explore risk factors contributing to death in SCAP patients, univariate and multivariate logistic regression analyses were performed. Univariate analysis showed that the inflammation-related indicators (IL-6, PCT, D-dimer, and BUN), the prognostic indicators (APACHII and PaO2) and the length of hospital stay (P < 0.10), but not factors including gender, age, levels of lactic acid, CRP, BNP, ALT, AST, PLT, and CCr, and mixed infection were significantly different between the improved/cured and death groups. In addition, mNGS detection status and bacterial infection were both statistically significant between these two groups (P < 0.10) (Table 3). Subsequently, the above 9 factors with significant difference were further applied to the logistic regression analysis for further screening, and the results revealed that the increased levels of IL-6 (P < 0.016, OR: 1.003; 95% CI: 1.001, 1.006), PCT (P = 0.060; OR: 1.205; 95% CI: 0.993, 1.464) and BUN (P = 0.096; OR: 1.061; 95% CI: 0.990, 1.138), the longer length of hospital stay (P = 0.002; OR: 1.052; 95% CI: 1.018, 1.087), and bacterial infection (P = 0.002; OR: 3.366; 95% CI: 1.571, 7.209) were positively correlated with poor prognosis of SCAP patients. However, mNGS detection status could reduce the risk of death (P = 0.006; OR: 0.237; 95% CI: 0.085, 0.666) (Table 4).
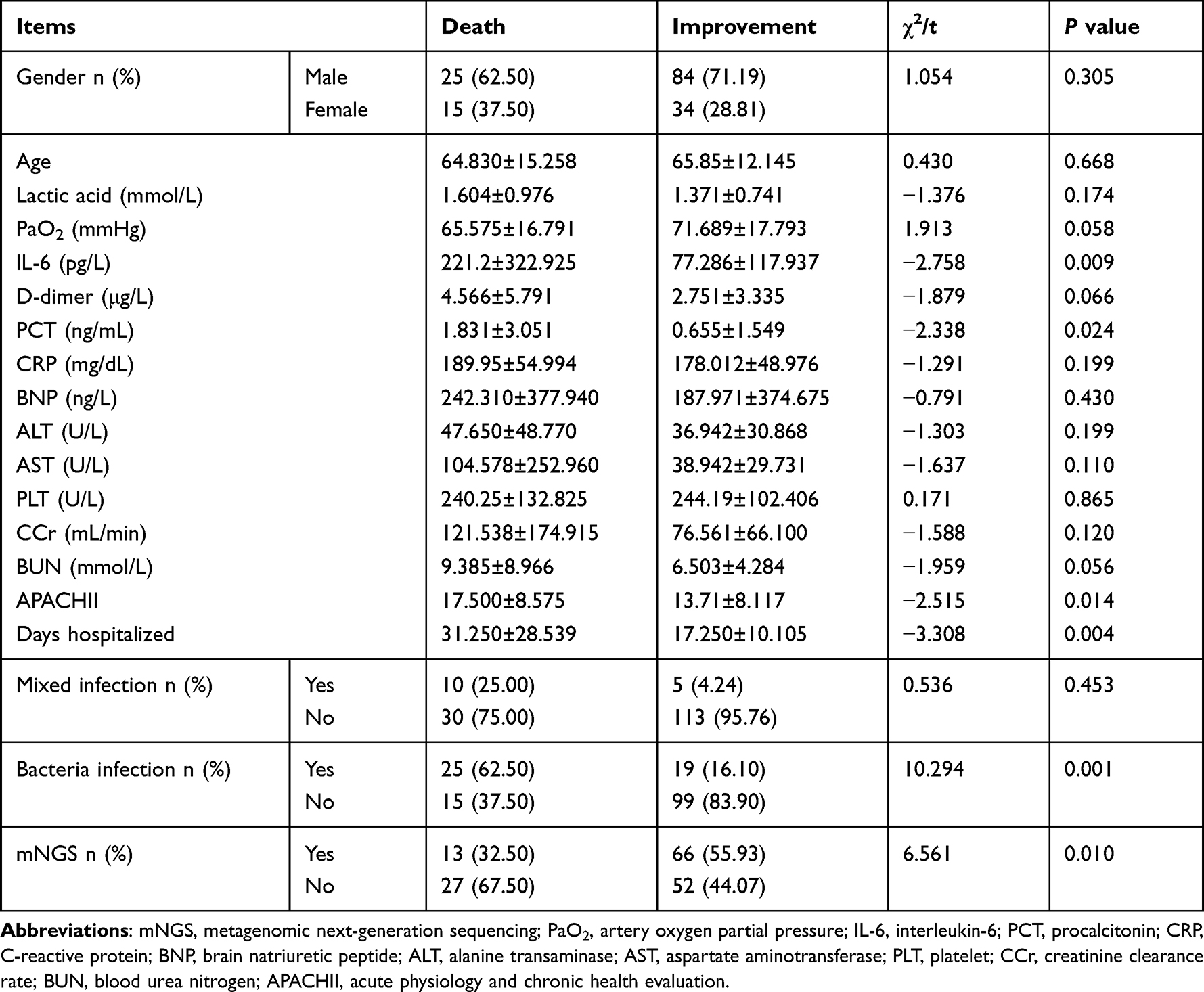 |
Table 3 Univariate Analysis of Risk Factors for Recovery in SCAP Patients |
 |
Table 4 Multivariate Logistic Regression Analysis of Risk Factors for Recovery in SCAP Patients |
Subsequently, we further constructed a nomogram model for predicting the risk of death in SCAP patients using the above six independent risk or protective factors. Specifically, the left breakpoints of each scoring line segment corresponded to 0 point, and the right breakpoints corresponded to 100, 45, 32, 84, 12, 16 scores and with a total score of 289 points for all variables. As shown in Figure 3A, the higher the total score of the nomogram model, the higher the risk of death in SCAP patients. Meanwhile, the calibration curve of the modeling set does not deviate significantly from the reference line (Figure 3B), indicating a good consistency between the predicted and actual outcomes. The ROC curve of the nomogram was also generated, with the AUC values of the two groups in the modeling and validation sets being 0.8291 [95% CI (0.7439–0.9142)] and 0.9211[95% CI (0.8342–1)], respectively, showing high predictive effectiveness of the nomogram model (Figures 3C and D).
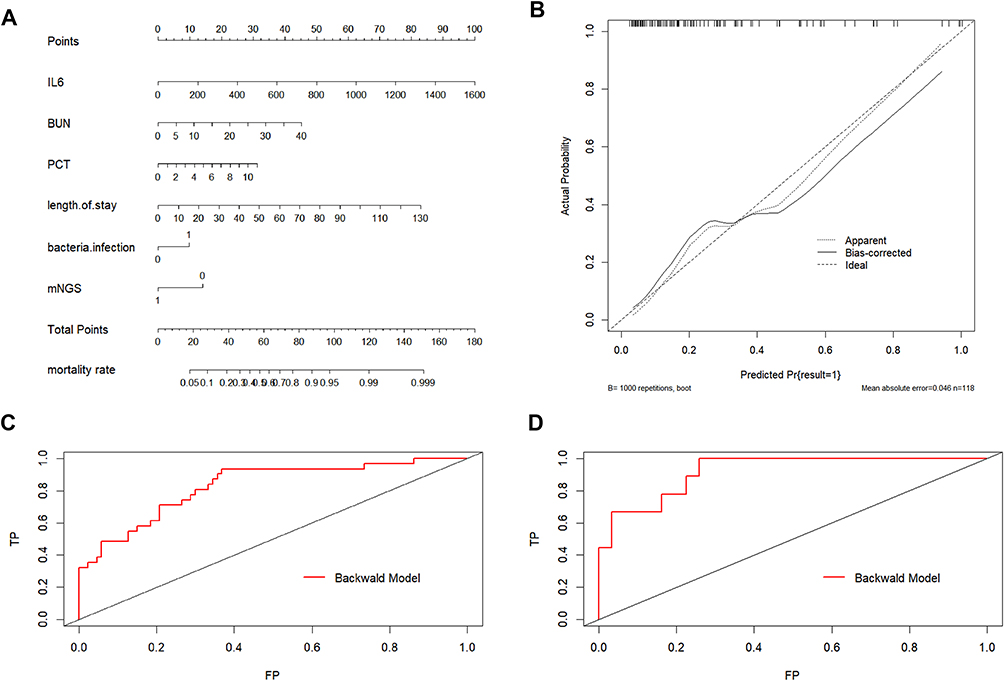 |
Figure 3 Prognostic risk factors for recovery in patients with severe community-acquired pneumonia (SCAP). (A) Nomogram prediction of death risk in SCAP patients; (B) Calibration curves of the modeling set for the nomogram in predicting the death risk of SCAP patients (modeling set); (C) ROC curve of the modeling set illustrated using the nomogram model in predicting the death risk of SCAP patients; (D) ROC curve of the validation set illustrated using the nomogram model for predicting the death risk in SCAP patients. |
Relationship Between Specific Bacterial Infections and Outcomes of SCAP Patients
In order to further explore whether several specific bacterial infections were related to outcomes of SCAP patients, 2 most commonly identified bacteria, Acinetobacter baumannii and Klebsiella pneumoniae, were selected for further analysis. The positive rate of Acinetobacter baumannii was 30% (12/40) in the death cases and 9.32% (11/118) in the improved/cured cases, showing a significant difference (P = 0.001). The positive rate of Klebsiella pneumoniae in the death cases was 10% (4/40) and 1.69% (2/118) in the improved/cured cases, also showing a significant difference (P = 0.018).
Discussion
The current study systematically compared the effectiveness of mNGS with conventional tests in identifying pathogens in SCAP patients and found that mNGS has advantages in detecting pathogens in SCAP patients from several aspects.
First, we showed that mNGS has a higher detection rate, a higher detection sensitivity as well as a wider spectrum of detectable pathogens, compared with conventional tests. In detail, we found that the pathogen detection rate of mNGS was 84.81%, significantly higher than that of conventional tests (45.57%, P < 0.001). This is also consistent with a previous clinical study showing that 85% of patients were identified as pathogen positive in BALF samples by mNGS, compared with only 50% by conventional tests.26 Moreover, the sensitivity of mNGS for single pathogen species is also high. For instance, we identified 70.89% of patients with bacterial infection using BALF samples, which was significantly higher than that of the conventional tests (6.46%), consistent with a previous study.27 In addition, mNGS can detect more pathogen species. The results of mNGS in our study showed that a variety of pathogens, including 31 bacteria, 10 viruses, 9 fungi, 1 Chlamydia, and 1 mycoplasma, were identified. As comparison, conventional culture of BALF samples identified only bacteria; this observation is also similar to previous case reports and studies.28–31 Detection of mixed infection is also very important in clinical practice. It is well known that SCAP is usually not caused by a single pathogen, but by a mixed infection, and various microorganisms may interact with each other and thus inhibit each other’s growth. mNGS can detect a wider spectrum of pathogen species and thus has advantages for guiding more accurate treatment. Xie et al have reported a retrospective study on the application of mNGS in the detection of SCAP pathogens,28 and found that viruses (15.4%), Acinetobacter baumannii (12.3%), and fungi (11.8%) were the most abundant species in SCAP patients, while the top three pathogens identified in our study with mNGS were Acinetobacter baumannii (21.52%), Candida albicans (17.72%), and Klebsiella pneumoniae (15.19%). Similarly, mNGS indicated that compared with SCAP patients from USA,9 the detection rate of Enterobacteriaceae (Klebsiella pneumonia 15.19%) in China was much higher. These results indicate that the distribution of etiological pathogens for SCAP patients may not always be the same across regions or countries, and empirical use of antibiotics might be risky and less effective.
Indeed, our study showed that patients in the study group who received mNGS-guided treatments had significantly lower mortality than those in the control group who received conventional method-guided or empirical treatments, indicating the significance of evidence-based treatments in clinics. Meanwhile, since respiratory viruses play an increasingly important role in community-acquired pneumonia, especially in SCAP, mNGS also has a great advantage in detecting virus-related SCAP. This has been proved in the early stage of the COVID-19 pandemic where mNGS has been utilized to rapidly and accurately identify pathogenic viruses.32 Due to the higher pathogen detection rate and sensitivity, as well as wilder spectrum of detectable pathogens, mNGS has a great advantage in guiding evidence-based treatment, which can improve curative rate while reducing side effects, compared with empiric-based treatment.
Of special note, mNGS may be of greater significance in some regions/countries around the world. For example, MTB infection has a high prevalence in China compared with that in other countries. Importantly, MTB and most non-tuberculous mycobacteria (NTM) types are difficult to be cultured leaving these patients always misdiagnosed. Recent studies have demonstrated that mNGS is able to improve the overall diagnosis accuracy and treatment outcomes for patients with MTB or NTM infections.33,34 Notably, in this study, although mNGS could effectively detect mycobacteria at a higher rate, two cases of MTB were missed by mNGS, which may be due to the inadequate amount of extracted DNA. A previous study showed that including a cell wall beading step in DNA extraction can help to break down the cell wall of mycobacteria more thoroughly,35 which may contribute to further increasing the detection sensitivity of mNGS. Overall, a combination of mNGS and Xpert detection can further maximize MTB detection rate and diagnostic accuracy.
Other than above-discussed advantages, mNGS also has its limitations. For example, the technique is relatively demanding and expensive, and thus typically, could not be implemented in community-level medical units or even in some large tertiary medical centers. Moreover, currently, there is a lack of unified and standard mNGS detection protocols and guidelines,36 making the application of mNGS even less convenient.
Furthermore, this study also evaluated the risk factors for recovery in SCAP patients. Understanding the risk factors for recovery in SCAP patients can also facilitate the treatment and management of SCAP patients. Our analyses identified IL-6, PCT, BUN, bacterial infection and hospitalization time to be the independent risk factors, while mNGS detection status to be the independent protective factor, for SCAP patients. The “cytokine storm” theory suggests that inflammatory factors, especially IL-6, play a crucial role in the development of many diseases. For instance, Tang et al reveal that IL-6 level in peripheral blood can be used as an independent factor in predicting the progression of severe COVID-19.37 These results support the notion in our study that IL-6 is an independent risk factor for SCAP patients. The importance of PCT and prolonged hospital stay as risk factors has also been supported by other studies.38 These factors can be used for the management and prognosis prediction of SCAP patients and may also be used as treatment targets, which requires more in-depth mechanistic studies.
Our study also has some limitations that need to be acknowledged. First, this is a single-tertiary medical center study with a relatively small sample size. Second, due to the lack of external validation datasets, the generalization of the nomogram to other cohorts may be unclear, and large cohorts of SCAP patients from multicenter, prospective studies with different demographic characteristics are required to further evaluate the effectiveness and generalization of our established nomogram. Finally, due to the small sample size in both the study and control groups (n = 79 for each group), data from both groups were combined to establish the nomogram, which might cause bias of the nomogram. In our future study, we will further expand the sample size, and establish the nomogram using patients from the study and control groups separately.
Conclusions
In summary, mNGS can identify a wide spectrum of pathogens in a comprehensive way using easily accessible clinical specimens from SCAP patients in a timely and accurate manner, and mNGS-guided treatment can reduce the mortality in these patients, suggesting that mNGS can be used as an important etiological diagnostic method in clinics for SCAP patients. Clinical factors, including levels of IL-6, PCT, and BUN, bacterial infection status and hospitalization time, were independent risk factors, while mNGS detection status was an independent protective factor, for SCAP patients.
Data Sharing Statement
The datasets generated during and/or analyzed during the current study are not publicly available due to the policy of People’s Hospital of Xinjiang Uygur Autonomous Region but are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
All procedures performed in the studies involving human participants were in accordance with the ethical standards of the Ethics Committee of the People’s Hospital of Xinjiang Uygur Autonomous Region and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All experimental protocols were approved by the licensing ethical committee of People’s Hospital of Xinjiang Uygur Autonomous Region. The Ethical Approval No. was KY202010101922. Written informed consent to participate in this study was provided by the participants or their legal guardians.
Acknowledgments
We express our gratitude to the patients and their families included in this study. Also, we are grateful to BGI-Shenzhen for its technical support in metagenomic next-generation sequencing.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by Natural Science Foundation Project 2020D01C098 of Xinjiang Uygur Autonomous Region. Special Project for the Construction of Innovative Environment (Talents and Bases) in Xinjiang Uygur Autonomous Region (Talents Special Plan – Tianshan Youth Program) – Training Project 2020Q045 for Outstanding Young Scientific and Technological Talents.
Disclosure
The authors report no conflicts of interest in this work.
References
1. The top 10 causes of death. Available from: www.who.int/en/news-room/fact-sheets/detail/the-top-10-causes-of-death.
2. El Bcheraoui C, Mokdad AH, Dwyer-Lindgren L, et al. Trends and patterns of differences in infectious disease mortality among US counties, 1980–2014. JAMA. 2018;319:1248–1260. doi:10.1001/jama.2018.2089
3. Troeger C, Blacker B, Khalil IA; Collaborators GBDLRI. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Infect Dis. 2018;18:1191–1210. doi:10.1016/S1473-3099(18)30310-4
4. Cookson W, Cox MJ, Moffatt MF. New opportunities for managing acute and chronic lung infections. Nat Rev Microbiol. 2018;16:111–120. doi:10.1038/nrmicro.2017.122
5. Xu J, Murphy SL, Kochanek KD, et al. Deaths: final Data for 2016. Natl Vital Stat Rep. 2018;67:1–76.
6. Chalmers JD, Taylor JK, Mandal P, et al. Validation of the Infectious Diseases Society of America/American Thoratic Society minor criteria for intensive care unit admission in community-acquired pneumonia patients without major criteria or contraindications to intensive care unit care. Clin Infect Dis. 2011;53:503–511. doi:10.1093/cid/cir463
7. Xie J, Wang H, Kang Y, et al. The epidemiology of sepsis in Chinese ICUs: a national cross-sectional survey. Crit Care Med. 2020;48:e209–e218. doi:10.1097/CCM.0000000000004155
8. Currie CJ, Berni E, Jenkins-Jones S, et al. Antibiotic treatment failure in four common infections in UK primary care 1991–2012: longitudinal analysis. BMJ. 2014;349:g5493. doi:10.1136/bmj.g5493
9. Jain S, Self WH, Wunderink RG, et al. Community-acquired pneumonia requiring hospitalization among U.S. adults. N Engl J Med. 2015;373:415–427. doi:10.1056/NEJMoa1500245
10. Jain S, Williams DJ, Arnold SR, et al. Community-acquired pneumonia requiring hospitalization among U.S. children. N Engl J Med. 2015;372:835–845.
11. Zaas AK, Garner BH, Tsalik EL, et al. The current epidemiology and clinical decisions surrounding acute respiratory infections. Trends Mol Med. 2014;20:579–588. doi:10.1016/j.molmed.2014.08.001
12. Ranzani OT, Prina E, Menéndez R, et al. New sepsis definition (Sepsis-3) and community-acquired pneumonia mortality. A validation and clinical decision-making study. Am j Resp Crit Care. 2017;196:1287–1297. doi:10.1164/rccm.201611-2262OC
13. Laxminarayan R, Duse A, Wattal C, et al. Antibiotic resistance-the need for global solutions. Lancet Infect Dis. 2013;13:1057–1098. doi:10.1016/S1473-3099(13)70318-9
14. Wilson MR, Naccache SN, Samayoa E, et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. New engl j med. 2014;370:2408–2417. doi:10.1056/NEJMoa1401268
15. Bibby K. Metagenomic identification of viral pathogens. Trends Biotechnol. 2013;31:275–279. doi:10.1016/j.tibtech.2013.01.016
16. Yozwiak NL, Skewes-Cox P, Stenglein MD, et al. Virus identification in unknown tropical febrile illness cases using deep sequencing. PLoS Negl Trop Dis. 2012;6:e1485. doi:10.1371/journal.pntd.0001485
17. Fischer N, Indenbirken D, Meyer T, et al. Evaluation of unbiased next-generation sequencing of RNA (RNA-seq) as a diagnostic method in influenza virus-positive respiratory samples. J clin microbiol. 2015;53:2238–2250. doi:10.1128/JCM.02495-14
18. Graf EH, Simmon KE, Tardif KD, et al. Unbiased detection of respiratory viruses by use of RNA Sequencing-Based metagenomics: a systematic comparison to a commercial PCR panel. J clin microbiol. 2016;54:1000–1007. doi:10.1128/JCM.03060-15
19. Wilson MR, Shanbhag NM, Reid MJ, et al. Diagnosing balamuthia mandrillaris encephalitis with metagenomic deep sequencing. Ann Neurol. 2015;78:722–730. doi:10.1002/ana.24499
20. Zheng Y, Qiu X, Wang T, et al. The diagnostic value of metagenomic next-generation sequencing in lower respiratory tract infection. Front Cell Infect Microbiol. 2021;11:694756. doi:10.3389/fcimb.2021.694756
21. Liang M, Fan Y, Zhang D, et al. Metagenomic next-generation sequencing for accurate diagnosis and management of lower respiratory tract infections. Int J Infect Dis. 2022;122:921–929. doi:10.1016/j.ijid.2022.07.060
22. Gaston DC, Miller HB, Fissel JA, et al. Evaluation of metagenomic and targeted next-generation sequencing workflows for detection of respiratory pathogens from bronchoalveolar lavage fluid specimens. J Clin Microbiol. 2022;60:e0052622. doi:10.1128/jcm.00526-22
23. Wei Y, Zhang T, Ma Y, et al. Clinical evaluation of metagenomic next-generation sequencing for the detection of pathogens in BALF in severe community acquired pneumonia. Ital J Pediatr. 2023;49:1–13. doi:10.1186/s13052-023-01431-w
24. Wu X, Li Y, Zhang M, et al. Etiology of severe community-acquired pneumonia in adults based on metagenomic next-generation sequencing: a prospective multicenter study. Infect Dis Ther. 2020;9:1003–1015. doi:10.1007/s40121-020-00353-y
25. Miao Q, Ma Y, Wang Q, et al. Microbiological diagnostic performance of metagenomic next-generation sequencing when applied to clinical practice. Clin Infect Dis. 2018;67:S231–S240. doi:10.1093/cid/ciy693
26. Chen J, Zhao Y, Shang Y, et al. The clinical significance of simultaneous detection of pathogens from bronchoalveolar lavage fluid and blood samples by metagenomic next-generation sequencing in patients with severe pneumonia. J Med Microbiol. 2021;2021:70.
27. Qu J, Zhang J, Chen Y, et al. Aetiology of severe community acquired pneumonia in adults identified by combined detection methods: a multi-centre prospective study in China. Emerg Microbes Infect. 2022;11:556–566. doi:10.1080/22221751.2022.2035194
28. Xie Y, Du J, Jin W, et al. Next generation sequencing for diagnosis of severe pneumonia: China, 2010–2018. J Infect. 2019;78:158–169. doi:10.1016/j.jinf.2018.09.004
29. Cilloniz C, Ferrer M, Liapikou A, et al. Acute respiratory distress syndrome in mechanically ventilated patients with community-acquired pneumonia. Eur Respir J. 2018;2018:51.
30. Kohl C, Brinkmann A, Dabrowski PW, et al. Protocol for metagenomic virus detection in clinical specimens. Emerg Infect Dis. 2015;21:48–57. doi:10.3201/eid2101.140766
31. Sun T, Liu Y, Cai Y, et al. A paired comparison of plasma and bronchoalveolar lavage fluid for metagenomic next-generation sequencing in critically ill patients with suspected severe pneumonia. Infect Drug Resist. 2022;15:4369–4379. doi:10.2147/IDR.S374906
32. Chen L, Liu W, Zhang Q, et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg Microbes Infect. 2020;9:313–319. doi:10.1080/22221751.2020.1725399
33. Zhou X, Wu H, Ruan Q, et al. Clinical evaluation of diagnosis efficacy of active mycobacterium tuberculosis complex infection via metagenomic next-generation sequencing of direct clinical samples. Front Cell Infect Microbiol. 2019;9:351. doi:10.3389/fcimb.2019.00351
34. Huang H, Deng J, Qin C, et al. Disseminated coinfection by mycobacterium fortuitum and talaromyces marneffei in a Non-HIV case. Infect Drug Resist. 2021;14:3619–3625. doi:10.2147/IDR.S316881
35. Epperson LE, Strong M. A scalable, efficient, and safe method to prepare high quality DNA from mycobacteria and other challenging cells. J Clin Tuberc Other Mycobact Dis. 2020;19:100150. doi:10.1016/j.jctube.2020.100150
36. Qu JM, Liu HX. Application and value of molecular diagnostic techniques in the detection of lower respiratory tract infections. Zhonghua Jie He He Hu Xi Za Zhi. 2019;42:486–489. doi:10.3760/cma.j.issn.1001-0939.2019.07.003
37. Tang J, Lin J, Zhang E, et al. Serum IL-6 and procalcitonin are two promising novel biomarkers for evaluating the severity of COVID-19 patients. Medicine. 2021;100:e26131. doi:10.1097/MD.0000000000026131
38. Rodriguez A, Reyes LF, Monclou J, et al. Relationship between acute kidney injury and serum procalcitonin (PCT) concentration in critically ill patients with influenza infection. Med Intensiva. 2018;42:399–408. doi:10.1016/j.medin.2017.12.004
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Clinical Utility and Therapeutic Strategy Value of Metagenomic Next-Generation Sequencing in Pulmonary Infection Among Cancer Patients
Cao Y, Huang J, Wu W, Xu Z, Wang C, Wu X, Zhan C, Xing J, Liu J, Zhu M, Ma S
Infection and Drug Resistance 2026, 19:568562
Published Date: 13 February 2026
Metagenomic Next-Generation Sequencing for Brain Abscess: Improved Pathogen Detection, Targeted Antimicrobial Therapy, and Association with Fewer Surgical Interventions
Li X, Fan M, Yue J, Xie J, Zhang Y, Lu X, Liu L, Li X, Huang Y
Infection and Drug Resistance 2026, 19:617362
Published Date: 14 July 2026