")
Back to Archived Journals » Biosimilars » Volume 5
Clinical efficacy and safety of biosimilar epoetin: focus on epoetin zeta
Received 1 January 2015
Accepted for publication 2 April 2015
Published 9 September 2015 Volume 2015:5 Pages 65—73
DOI https://doi.org/10.2147/BS.S50456
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Shein-Chung Chow
Ashraf Mikhail , Christopher Brown
Renal Unit, Morriston Hospital, Wales, UK
Abstract: Biosimilars have been developed for several biologic therapeutic agents, including erythropoiesis-stimulating agents. Biosimilars cannot be assumed to be completely identical to the reference product. Several regulatory bodies have issued stringent guidelines to regulate the licensing of biosimilars. These guidelines, although share a unified aim of ensuring the safety and efficacy of biosimilars, show several differences. Such differences may reflect the difficulties facing regulatory bodies in defining a biosimilar, identifying sensitive means to assess equivalence in efficacy, and designing robust methodologies to monitor long-term safety. This review will discuss some of the aspects of differences in licensing requirements for biosimilars, comparing the European Medicines Agency guidelines and the American Food and Drug Administration guidelines. The pathway adopted by the manufacturer of a biosimilar (epoetin zeta) to gain licensing within the European market will be assessed, analyzing its compliance with the European Medicines Agency guidelines for the approval process. Since many patients are likely to be switched from original drugs to biosimilars in future, there is a need to establish strict guidelines on interchangeability and substitution of biosimilars and original products and to make it an integral part of the pre-registration assessment of any biosimilar in future. Eventually, long-term, observational post-marketing data will provide further reassurance on safety and tolerability of biosimilars.
Keywords: biosimilars, erythropoiesis-stimulating agents, epoetin zeta, biologic therapeutic agents
Introduction
It has been over 6 years since the first biosimilar erythropoietin (EPO) has been approved for marketing within the European Union (EU). Since then several other biosimilars (EPO preparations and others) have been licensed in Europe. While some preparations are widely used in clinical practice nowadays, others found it difficult to penetrate a highly competitive marketing environment. Despite this long duration of clinical experience, and the increasing number of patients receiving biosimilar preparations, controversies regarding safety, tolerability, and efficacy of biosimilars continue to draw the attention of clinicians, stakeholders, and patient groups. To regulate the marketing of biosimilars in clinical environment, regulatory bodies have provided extensive and strict guidelines for the approval of biosimilars. Some inconsistencies and differences in these guidelines may make it difficult to spread the use and maximize the benefits of these low cost preparations in wider clinical practice. This paper will discuss some of the differences in the current regulatory approval processes focusing mainly on the safety and efficacy of biosimilars. The second part of this paper will discuss how these guidelines have influenced clinical trial design of one of the earliest biosimilars to be licensed in Europe: epoetin zeta.
Safety and efficacy of biosimilars: regulatory approval process
It is difficult to provide an accurate tool to evaluate the safety and efficacy of biosimilars. This has been clearly demonstrated in the complexity and the relative inconsistencies of the guidelines issued by different international regulatory bodies for the approval of biosimilars. The European Medicines Agency (EMA) was the first regulatory authority that issued guidelines for biosimilars in 2005.1 This framework was replicated in local specific guidance issued by some non-EU countries like Japan, Korea, and the People’s Republic of China. The World Health Organization (WHO) has also issued a guideline as a general framework, establishing regulatory requirements for the approval of biosimilars.2 In February 2012, US Food and Drug Administration (FDA) released draft guidance documents for biosimilars,3 and in 2014 issued another draft document on “clinical pharmacology data required to support a demonstration of biosimilarity to a reference product”.4 The following part of this review article will compare the EU with the US regulatory approaches to evaluate the potential differences with respect to their implications for characterization of the safety and efficacy profile of a proposed biosimilar product.
Definition of a biosimilar
Although manufacturers of biosimilars have to conduct and establish their own strategies to develop the respective biosimilar drug, they do not have access to the full development dossier of the reference product; in fact, they only have access to the finished reference product. This knowledge gap faced by biosimilar developers cannot be addressed through any of the existing regulatory guidelines. While the EMA concentrated on the origin of biosimilars in their definition “biotechnologically derived proteins”, the FDA definition acknowledged the challenge in defining biologics in general as “a mixture of 100 closely related, complex biological substances that, in aggregate, make up the active component”.4 Nevertheless, both definitions make it clear that the manufacturing process of any biosimilar is far more complicated than small molecular weight drugs. As a result, any minor changes to the original manufacturing process may result in alterations to the secondary, tertiary, and quaternary structure of the molecule, which may have an impact on the efficacy and safety of the drug. Thus, the demonstration of high similarity to the reference product regarding quality, efficacy, and safety is necessary and has to be demonstrated using a set of comprehensive comparability exercises.
Selection of reference product
In principle, only a reference product licensed in the EU or the US is accepted for any kind of testing during the approval process of a biosimilar drug. Furthermore, the same product has to be used throughout the entire development program.1 The EMA guidelines have also allowed the use of non-EU reference products in certain cases only for distinct nonclinical and clinical investigations. Such results may be bridged for final evaluation.1 Under this approach, it is the applicant’s responsibility to establish that the batches sourced outside the EU area are representative of the reference medicinal product authorized in the EU through an extensive analytical comparison. For the FDA approval, the type of bridging data needed should include data from analytical studies (eg, structural and functional data) that directly compares all three products (ie, the proposed biosimilar product, the US-licensed reference product, and the non-US-licensed product) and is likely to also include pharmacokinetic (PK) and, if appropriate, pharmacodynamic (PD) study data for all three products.4
Clinical pharmacology for approval of biosimilar epoetin
PK data
The EU guidelines suggest a comparison between the similar biological medicinal product and the reference product in a single dose crossover study for the routes of administration applied for, usually including both subcutaneous (SC) and intravenous (IV) administration. Healthy volunteers are considered an appropriate study population. The FDA requires data on peak concentration (Cmax), lowest concentration measured following dosing (Cmin), concentration prior to the next dose during multiple dosing (C through steady state), and area under the plasma/blood concentration–time curve (AUC).
PD data
For the demonstration of efficacy for both routes of administration, the EMA suggests one of two options:5
- Two separate clinical trials: either the combination of a “correction phase” study using SC epoetin (eg, in a pre-dialysis population) and a “maintenance phase” study using IV epoetin (eg, in a hemodialysis population); or alternatively, both the SC and the IV study may be performed in the maintenance setting if appropriately justified.
- A comparable efficacy for one route of administration in a comparative clinical trial in addition to comparative single dose and multiple dose PK/PD bridging data in an epoetin-sensitive population (eg, healthy volunteers) for the other route of administration.
For the FDA,4 while data from a single or multiple studies may be acceptable, the guidelines set a clear definition of criteria needed for an acceptable biomarker of any study (Table 1). For certain biologics, it is difficult to identify a reliable PD marker; in this case, the FDA guidelines accept derived PK parameters as the primary basis for evaluating biosimilarity, and any residual PD marker may be used to augment the PK data.
 | Table 1 FDA criteria of an acceptable biomarker for a biosimilar evaluation |
Definition of a biological drug that requires approval under biosimilar approval guidelines
An important difference in the licensing pathway for certain biosimilar preparations between both areas is due to the licensing of the reference biologic preparation: In the US, for historical reasons, certain biologicals are regulated under the Federal Food, Drug, and Cosmetic (FD&C) Act and are subject to an abbreviated approval process for generic versions of conventional drugs while still protecting the patent rights of brand-name drug manufacturers.7 Such biological products include insulin, glucagon, somatropin, and low molecular weight heparins. In contrast, the EU regulation does not allow for the use of classical generics application procedures for marketing authorization of biosimilars for these classes of biologicals.
These differences, as well as others are summarized in Table 2.
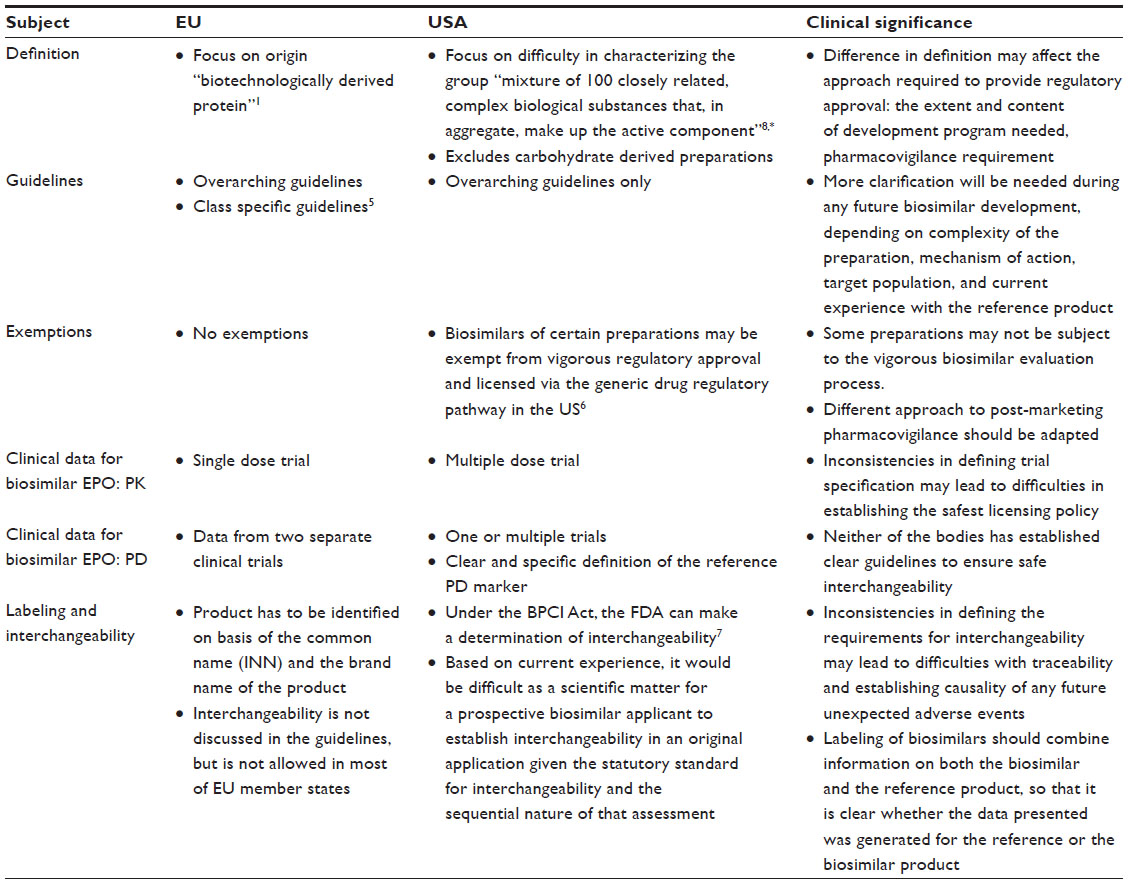 | Table 2 Comparison of general regulatory requirements for approval of biosimilars (EU vs USA) |
Epoetin zeta
Epoetin zeta, originally SB309 (Norbitec, Uetersen, Germany), was the second epoetin biosimilar to receive EMA approval. With the international nonproprietary name of epoetin zeta, it has been traded subsequently as Silapo® by Stada (Bad Vilbel, Germany) and as Retacrit® by Hospira (Lake Forest, IL, USA).
Epoetin zeta is produced in Chinese hamster ovary (CHO) cells. The molecular weight of the glycosylated protein is 30.6 kDa according to the European Pharmacopoeia monograph, 40% of which are carbohydrate structures. As a biosimilar to epoetin alfa, epoetin zeta has the same amino acid structure and relatively similar carbohydrate composition with only minor differences in the glycosylation pattern.9–12
Glycosylation patterns largely influence immunogenicity and half-life of therapeutic proteins.11 Glycosylation is dependent on the number of sialic acid residues attached to the protein molecules. The higher the number of sialic acid residues, the longer the half-life of the therapeutic protein. Both epoetin alfa and zeta have 14 sialic acid residues, both are short-acting drugs with a half-life of 6–8 hours when administered intravenously or 19–24 hours if administered subcutaneously.13 This short half-life allows epoetin zeta to be administered up to three times per week to treat anemia due to chronic kidney disease (CKD), a property that renders it ideal for administration to chronic hemodialysis patients who usually receive treatment three times weekly.14 One limitation of a short-acting erythropoiesis-stimulating agents (ESA) therapy is the need for a large storage space, and the risk of missed injections given the frequent need to administer the drug. This is one of the main motives to develop longer-acting ESAs administered fortnightly or monthly. Nevertheless, the predominant clinical practice in several renal units continues to provide short-acting ESAs to hemodialysis patients and longer-acting preparations to non-hemodialysis patients.
Pharmacokinetics
Two clinical trials comparing PK and PD of epoetin zeta and a reference product are discussed elsewhere.15–17 PK data showed that the 90% confidence interval (CI) for Cmax and AUC fell within the predefined ranges after the application of a correction factor allowing for differences in protein content.15–17
Efficacy
The efficacy of epoetin zeta has been studied in three clinical trials that evaluated the efficacy of Epoetin zeta for either correction of anemia or as maintenance ESA therapy in dialysis patients.9,18 A correction factor was used to reanalyze original data with respect to dosage, in order to adjust for the higher protein content noted in epoetin alfa compared to zeta.15 Although the correction phase study result did not meet its pre-specified criteria for equivalence of ±14 IU/kg/week (95% CI: –23.5 to 17.48 IU/kg/week), the 95% CI was within a modified post-hoc acceptance range of ±45 IU/kg/week. It is important to note that the published trial report did not state whether this correction factor was predetermined during the initial protocol or whether it was implemented during data analysis.
Similar results were noted in a crossover maintenance study.15,9,18 As in the correction phase study, a correction factor was introduced to correct for differences in protein content of the two ESAs. In the maintenance phase study, this led to a widening of the revised 95% CIs for dosage of 3.086–13.917 IU/kg/week which were within the modified acceptance range of ±45 IU/kg/week.9,15 One limitation of the methodology of this study was the comparison of the ESA dose and hemoglobin (Hb) levels between both groups over the whole 12 weeks of the study duration. To provide better analysis, it would have been ideal to define a dose titration phase followed by a dose maintenance phase to allow for accurate comparison between both groups during the dose used in the maintenance phase only.19 Reanalyzing the data after reaching a steady state of Hb level will provide a better understanding of the need to apply a correction factor and its possible value for patients well maintained on ESA therapy.19
The efficacy of epoetin zeta in correcting post-renal transplant anemia was assessed in 20 transplant recipients. Ten patients already receiving treatment with different ESA were switched to epoetin zeta (switch group) and ten ESA naïve patients were started epoetin zeta treatment (naïve group). All patients were prospectively followed up for 12 months. Iron supplements were administered during the study, as needed. In the switch group, mean plasma Hb levels >110 g/dL were maintained for the entire 1-year follow-up period, with average epoetin zeta doses 3.4% higher than the corresponding doses of previous ESA; in the naïve group, the target value was reached between the 1st and 3rd months and remained stable throughout the study. No drug-related side effect was reported.20 The authors concluded that epoetin zeta is safe and effective in correcting anemia in renal transplant recipients. The study was limited to a very small sample size (only ten patients in either arm).
The efficacy of epoetin zeta in treating chemotherapy-induced anemia was assessed in 2,333 patients with solid tumors, lymphoma, or myeloma who developed chemotherapy-induced anemia. Patients were recruited in an observational, non-interventional, longitudinal, multicenter study.21 Improvement in Hb level was achieved in 81.6% and 86.5% of patients at 3 and 6 months, respectively, and an overall mean change in Hb level was 1.52±1.61 and 1.72±1.61 g/dL at 3 and 6 months, respectively. Mean Hb response reported in the current study is similar to that reported for epoetin alfa in CIA associated with multiple myeloma.22
Safety
The long-term immunological safety of epoetin zeta was assessed by analyzing a combined outcome from patients enrolled in two earlier studies.9,18 Data from 745 chronic hemodialysis patients who completed the previous maintenance studies were evaluated. Two hundred and thirteen patients withdrew during the first 56 weeks due to adverse events, noncompliance, and other factors. A high drop-out rate is commonly observed in patients with chronic conditions who participate in long-term clinical trials. In the CREATE study the drop-out rate was 21% while in the CHOIR study it was 38%.23,24 Most patients and investigators reported tolerability as excellent or good, and most adverse events that were possibly related to study treatment were consistent with those previously reported with other ESAs. The most commonly reported adverse events were infections (34.1%) followed by injury and procedural complications (25.1%). No patient developed de novo anti-EPO antibodies during both study periods.
A further post-hoc analysis of data from 481 patients who participated in three studies (the two 24-week, randomized, double-blind correction and maintenance studies and the 56-week, open-label, follow-on study) evaluated the impact of switching hemodialysis patients with CKD between epoetin alfa and zeta on Hb concentration, epoetin dose, and safety.8,19,25,26 Mean Hb concentration was maintained at target levels throughout the drug switch. In addition, the mean differences in Hb concentration and associated 95% CIs following the switch remained within prespecified equivalence limits (±10 g/L). The 95% CIs of the mean difference in weekly epoetin dose after switch also remained within the prespecified equivalence margins (±45 IU/kg). Both treatments were similarly well tolerated. No patient developed anti-epoetin antibodies or pure red cell aplasia (PRCA) during the study.26
The safety and efficacy of epoetin zeta and epoetin alfa have also been compared in patients with renal anemia undergoing chronic hemodialysis.10 In a maintenance study, patients received epoetin zeta (n=232) or epoetin alfa (n=230) for 28 weeks after an open run-in period of 12–16 weeks, during which time the dose of epoetin was adjusted. There was no statistically significant difference in Hb levels between both groups in the last 4 weeks of the study. In addition, the 95% CI of the difference in mean ESA dose was within the 45 IU/kg/week equivalence range. There were no differences in tolerability between treatment groups, and no patient developed anti-epoetin antibodies.10
Results of post-authorization safety cohort observational study including anemic patients with CKD receiving hemodialysis and who were treated with epoetin zeta administered intravenously for up to 1 year have recently been released. The study was a part of post-approval risk-management plan as required by the EMA. The study included safety data from more than 1,600 patients in four European countries. Approximately, 94% of patients were treated previously with an ESA. The primary objective of PASCO I was to determine the incidence of adverse events of special interest in patients being treated with epoetin zeta for renal anemia. In the patients studied, treatment of renal anemia with epoetin zeta was found to have a safety profile consistent with that found in the epoetin zeta registration trials with the same route of administration and in line with other ESAs.27
Discussion
Regulatory approval designed to ensure therapeutic equivalence for biosimilars differs from that for generic drug formulations. The approval process addresses the complex formulation and manufacturing of therapeutic proteins. Because manufacturing process can affect molecular similarity, regulators face challenges in establishing efficient and appropriate review and approval pathways to ensure equivalent therapeutic efficacy and safety. Both EMA and FDA have developed robust guidelines to help biosimilar manufacturers gain marketing approval for their products. Despite the close similarity of the main structure of the guidelines produced by both bodies, some differences do exist. Such differences in approval guidelines reflect the complexity of biosimilar manufacturing process and the difficulty in establishing a robust and standardized pathway to assess their safety and efficacy.
The goal of comparability studies, needed for biosimilar approval process, is not to improve drug efficacy or patient outcome, but merely to show similar safety and efficacy to the originator product and help bring the overall cost of therapy down. Biosimilarity will be adequately assessed only if a suitable patient population and a clinical endpoint, sensitive enough to detect any difference between the products tested, are chosen for any clinical evaluation.
One important potential benefit of developing biosimilars is to allow switching of patients from the original drug. This switch could be at the prescriber (interchanging), the dispensing, or the pharmacist level (substitution). Both regulatory bodies avoided defining the trial requirements to adequately evaluate drug switching. It has been suggested that, to evaluate interchangeability, a prospective, double-blind, double crossover design trial would be ideal. Such a design is not a requirement for any biosimilar licensing based on current guidelines.28
Epoetin zeta (Retacrit) was one of the earliest biosimilar epoetins licensed in the EU.29 The application dossier submitted to the EMA contained comprehensive preclinical and toxicology data. The clinical data included several well powered studies that showed similar efficacy and safety of epoetin zeta compared with the comparator (epoetin alfa). In addition, a crossover design, Hb maintenance study was also presented. This represents an attempt by the biosimilar developer to evaluate the potential interchangeability between the biosimilar and the reference product, an issue that has not been clearly discussed in the current guideline.
In the maintenance hemodialysis study,9 switching from epoetin alfa to zeta increased the dose requirement by approximately 10%–15%. Switching from epoetin zeta to alfa reduced the dose requirement by around 10% and increased Hb levels by approximately 10%.9,18 This dose difference needs to be considered before switching from an innovator to a biosimilar for cost-saving purposes. If the switch provides a possible 10% price reduction, such a switch is unlikely to provide any effective cost saving given the potential need for dose increase. In addition, the dose conversion needs to be carefully evaluated in long-term studies involving patients requiring higher ESA dose, since it is not clear whether the 10% correction factor remains the same across the whole spectrum of anemic dialysis patients, given the wide variation in ESA requirement in this patient group. In addition, it is not clear whether the same correction factor could be used for both initiation (correction of anemia) therapy as well as maintenance treatment. A detailed pharmacoeconomic model needs to be used to provide a realistic understanding of cost implication of any drug switch.
Clinical trials comparing epoetin zeta and a reference product identified the need to introduce a correction factor to compensate for the difference in protein contents. This step is useful in helping clinician for dose conversion whenever drug conversion/substitution is considered. If it is agreed that no exact biosimilarity can be established between two products, an estimate of the correction factor required for calculating dose conversion should be a part of any drug registration application dossier.
The safety profile for epoetin zeta was shown to be similar to that of epoetin alfa, the reference product. Eight studies reported the safety of epoetin zeta in patients with renal anemia. Of those, there are four clinical trials,9,10,16,18 three observational studies,14,30,31 and one post-hoc analysis based on two clinical trials.26 Again there was no reported cases of anti-epoetin antibodies or PRCA associated with the administration of epoetin zeta in these trials. The same was also reported in a large post-authorization observational study involving more than 1,600 CKD patients. A second larger post-authorization non-interventional, multicenter, longitudinal observation study to estimate the incidence of PRCA, neutralizing antibodies, lack of efficacy, and thromboembolic events under treatment with Retacrit epoetin zeta administered subcutaneously in patients with renal anemia, expected to recruit over 6,000 patients, is currently underway.
Nowadays, it is a frequent practice to switch patients between different ESA preparations. This could be dictated by need to modify overall treatment (starting hemodialysis or switching from hemodialysis to peritoneal dialysis); or by other factors such as contractual or supply issues. With the expanding experience with different ESA preparations, it is not an infrequent practice to switch patients from epoetin to longer-acting preparations such as darbepoetin and methoxy polyethylene glycol-epoetin beta (and vice versa) and also switch patients on epoetin from the IV to the SC route (and vice versa).32,33 The conversion ratio between different innovator ESAs is hugely variable19 and dependent on previous ESA dose requirements. It is also important to know that a routine clinical practice should involve the monitoring of Hb response to administered ESA and titrating the dose of ESA accordingly. The same is required of any switch between an existing ESA and a biosimilar epoetin. Given the need to introduce a correction factor for recalculating epoetin zeta in published trials, it is important to interpret these figures cautiously since data from adequately controlled clinical trials cannot always be extrapolated to clinical practice.
Much is made of the potential risk of inducing antibody-mediated PRCA with biosimilar epoetins. It is also important to note that this is an extremely rare condition affecting 1:10,000 CKD patients treated with ESA.34 It is likely that the very stringent regulations imposed by the EMA and FDA on biosimilar production1–6 mean that the chances of inducing a more immunogenic product are probably very small. Nevertheless, it is important to note that PRCA is only one aspect of immunological response to therapeutic proteins. There are several other untoward immunological reactions that may develop in response to foreign protein (Table 3). The ill-fated experience with peginesatide (Supplementary material) is a constant reminder that, regardless of how extensive pre-registration trials could be, it is very difficult to predict the immunological response to any product; the fact that only post-marketing long-term observational studies could ensure the safety of any future product may be argued that biosimilars are potentially identical to reference product, but changes in glycosylation and secondary, tertiary, and quaternary structures with production of biosimilars are well recognized, and therefore long-term safety cannot be guaranteed. Only long-term, real-life clinical experience could provide the definitive answer. It is essential to establish fully independent registries to monitor drug-induced adverse events; such bodies may play a role in identifying and comparing relative long-term safety of different biologics. The initiative of the British Renal Association to establish a PRCA registry within the Rare Disease Groups35 represents an important step that may increase patient awareness and may stimulate research that helps evaluate relative safety of different ESAs.
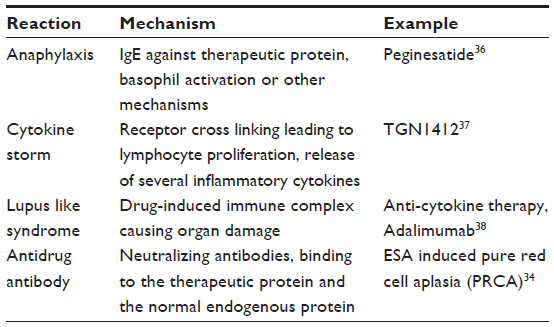 | Table 3 Potential untoward immunological responses to therapeutic proteins |
Conclusion
The last 6 years have enriched our experience on the safety and usefulness of biosimilar EPOs. Although regulatory guidelines have streamlined the marketing authorization process for biosimilars, the most important data to reassure the safety of these products will rely on post-marketing data, whether a part of well-structured pharmacovigilance plan, or from individual self-reported clinical experience. Post-marketing data of epoetin zeta provide an excellent example of how clinical experience will influence the decision-making process of choosing a biosimilar. Only long-term post-marketing data will provide the needed reassurance of long-term safety and tolerability of any future biosimilar.
Disclosure
The author reports no conflicts of interest in this work.
References
Guideline on Similar Biological Medical Products. Committee for Medicinal Products for Human Use; European Medicines Agency. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf. Accessed December 12, 2014. | |
Guidelines on Evaluaion of Similar Biotheraprutic Products. Expert Committee on biological standardization; World Health Organization. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. Accessed December 12, 2014. | |
Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Guidance for Industry. US Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologics Evaluation and Research (CBER). Available from: http://www.fda.gov/downloads/Drugs/Guidance ComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed March 12, 2014. | |
Guidance for Industry: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product. US Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologics Evaluation and Research (CBER). Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdf. Accessed March 12, 2014. | |
Guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant erythropoietins (Revision). Committee for Medicinal Products for Human Use; European Medicines Agency. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/04/WC500089474.pdf. Accessed December 9, 2014. | |
Guidance for Industry. Listed Drugs, 30-Month Stays, and Approval of ANDAs and 505(b)(2) Applications Under Hatch-Waxman, as Amended by the Medicare Prescription Drug, Improvement, and Modernization Act of 2003. Questions and Answers. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research. Available from http://www.fda.gov/OHRMS/DOCKETS/98fr/2004n-0087-gdl0001.pdf. Accessed December 9, 2014. | |
Mann J, Mahinka SP. Biosimilars: patent challenges and competitive effects. LMG Life Sci. 2014;(1):33–36. | |
Blank T, Netzer T, Hildebrandt W, Vogt-Eisele A, Kaszkin-Bettag M. Safety and toxicity of biosimilars – EU versus US regulation. GABI. 2013; 2(3):144–150. | |
Wizemann V, Rutkowski B, Baldamus C, Scigalla P, Koytchev R; Epoetin Zeta Study Group. Comparison of the therapeutic effects of epoetin zeta to epoetin alfa in the maintenance phase of renal anaemia treatment. Curr Med Res Opin. 2008;24(3):625–637. | |
Krivoshiev S, Wizemann V, Czekalsk S, et al. Therapeutic equivalence of epoetin zeta and alfa, administered subcutaneously, for maintenance treatment of renal anemia. Adv Ther. 2010;27(2):105–117. | |
Tamilvanan S, Raja NL, Sa B, Basu SK. Clinical concerns of immunogenicity produced at cellular levels by biopharmaceuticals following heir parenteral administration into human body. J Drug Target. 2010;18(17):489–498. | |
Walsh G. Post-Translational Modifications in the Context of Therapeutic Proteins: An Introductory Overview. Post-translational Modification of Protein Biopharmaceuticals. West Sussex, UK: Wiley-Blackwell. 2009;1–14. | |
Locatelli F, Del Vecchio L. Erythropoiesis-stimulating agents in renal medicine. Oncologist. 2011;16(Suppl 3):19–24. | |
Lonnemann G, Wrenger E. Biosimilar epoetin zeta in nephrology: effect of injection frequency on weekly dose. Int J Clin Med. 2012;3: 598–602. | |
Scientific discussion. European Medicines Agency. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000872/WC500054374.pdf. Accessed November 26, 2014. | |
Kirkov V, Dimitrova V, Siebert-Weigel M, et al. Evaluation of the pharmacokinetics of two recombinant human erythropoietin preparations: epoetin zeta and epoetin alfa. 1st communication: a monocentric, open, randomized, single dose, two-period crossover trial in healthy volunteers. Arzneimittelforschung. 2008;58:215–219. | |
Kirkov V, Dimitrova V, Siebert-Weigel M, et al. Evaluation of the pharmacokinetics of two recombinant human erythropoietin preparations: epoetin zeta and epoetin alfa. 2nd communication: a monocentric, double-blind, randomized, single dose, three period crossover trial in healthy volunteers. Arzneimittelforschung. 2008;58:220–224. | |
Krivoshiev S, Todorov V, Manitius J, Scigalla P, Koytchev R, Epoetin Zeta Study Group. Comparison of the therapeutic effects of epoetin zeta and epoetin alfa in the correction of renal anaemia. Curr Med Res Opin. 2008;24:1407–1415. | |
Mikhail A, Farouk M. Epoetin biosimilars in Europe: five years on. Adv Ther. 2013 ;30(1):28–40. Epub December 12, 2012. | |
Sabbatini M, Vitale S, Garofalo G, et al. Efficacy of subcutaneous epoetin-zeta on anemia in renal transplant recipients: a single-center experience. Transplant Proc. 2014;46(7):2238–2240. | |
Michallet M, Luporsi E, Soubeyran P, et al. Biosimilars in the management of anaemia secondary to chemotherapy in haematology and oncology: results of the ORHEO observational study. BMC Cancer. 2014;14:503. Erratum in BMC Cancer. 2014;14:720. | |
Dammacco F, Castoldi G, Rödjer S. Efficacy of epoetin alfa in the treatment of anaemia of multiple myeloma. Br J Haematol. 2001;113(1):172–179. Erratum in Br J Haematol. Sep 2001;114(3):738. | |
Drüeke TB, Locatelli F, Clyne N, et al. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med. 2006;355(20):2071–2084. | |
Singh AK, Szczech L, Tang KL, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355(20):2085–2098. | |
Baldamus C, Krivoshiev S, Wolf-Pflugmann M, Siebert-Wiegel M, Koytchev R, Bronn A. Long-term safety and tolerability of epoetin zeta, administered intravenously, for maintenance treatment of renal anemia. Adv Ther. 2008;25:1215–1228. | |
Wieçek A, Ahmed I, Scigalla P, Koytchev R. Switching epoetin alfa and epoetin zeta in patients with renal anemia on dialysis: post hoc analysis. Adv Ther. 2010;27:941–952. | |
Dellanna F, Fluck R, Lonnemann G, et al. Results from a safety study of patients with renal anemia receiving the biosimilar Retacrit™/Silapo™ (epoetin zeta): the PASCO I study. Poster 64. National Kidney Foundation 2014 Spring Clinical Meetings; 22–26 April, 2014; Las Vegas, USA. | |
Ebbers HC, Chamberlain P. Interchangeability: an insurmountable fifth hurdle? GaBI Journal. 2014;3(2):88–93. | |
Davis-Ajami ML, Wu J, Downton K, Ludeman E, Noxon V. Epoetin zeta in the management of anemia associated with chronic kidney disease, differential pharmacology and clinical utility. Biologics. 2014;8:155–167. | |
Bajraktar J, Lazarova B, Najdovska E, Mihajlova L. DSL-011 efficacy and safety of epoetin zeta in dialysis patients. Eur J Hosp Pharm Sci Pract. 2013;20:A91. | |
Perani L, Scolari C, Braus A, Galli E. Switch from CERA to EPO zeta in patients with anaemia and chronic kidney disease. Eur J Hos Pharm Sci Pract. 2013;20:A64. | |
Macdougall IC. Biosimilar epoetins. Nephrol Dial Transplant. 2009;24(5):1698–1699; author reply 1699–1700. | |
Besarab A, Reyes CM, Hornberger J. Meta-analysis of subcutaneous versus intravenous epoetin in maintenance treatment of anemia in hemodialysis patients. Am J Kidney Dis. 2002 ;40(3):439–446. | |
Rossert J, Casadevall N, Eckardt KU. Anti-erythropoietin antibodies and pure red cell aplasia. J Am Soc Nephrol. 2004;15(2):398–406. | |
Pure Red Cell Aplasia: Patient information. RareRenal.org; Information on rare kidney diseases. Available from: http://rarerenal.org/patient-information/pure-red-cell-aplasia-patient-infromation/. Accessed December 11, 2014. | |
Bennett CL, Jacob S, Hymes J, Usvyat LA, Maddux FW. Anaphylaxis and hypotension after administration of peginesatide. N Engl J Med. May 22, 2014;370(21):2055–2056. | |
Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–1028. | |
Piga M, Chessa E, Ibba V, et al. Biologics-induced autoimmune renal disorders in chronic inflammatory rheumatic diseases: systematic literature review and analysis of a monocentric cohort. Autoimmun Rev. 2014;13(8):873879. |
Supplementary material
The peginesatide story
Peginesatide is a synthetic, dimeric peptide that is covalently linked to polyethylene glycol. The amino acid sequence of peginesatide is unrelated to that of erythropoietin (EPO) and is not immunologically cross-reactive with EPO.1 Peginesatide binds to and activates the human EPO receptor, stimulating the proliferation and differentiation of human red cell precursors in vitro in a manner similar to other EPO-stimulating agents. In Phase II and III studies in dialysis and pre-dialysis patients, peginesatide administered once monthly was as effective as epoetin alfa given thrice weekly (dialysis patients) or darbepoetin given once weekly (non-dialysis patients), in correcting anemia of chronic kidney disease as well as maintaining hemoglobin within the desired target range.1,2 Despite published concerns regarding the safety of therapeutic pegylated proteins,3 peginesatide was licensed for treatment of anemia in dialysis patients in the US.
In July 2012, a large dialysis organization with 2,100 centers in the US started treating dialysis patients with peginesatide, with a robust risk evaluation protocol. Although registration trials revealed no new toxic effects, eight cases of anaphylaxis and hypotension among patients in the pilot initiative were reported. As a result, the manufacturer updated the product label with a warning that serious allergic reactions, including anaphylaxis reactions and hypotension, may occur in patients who receive peginesatide.4
Six months after pilot initiation, and after more than 60,000 doses of peginesatide were administered to more than 19,000 patients, five patients died from severe anaphylaxis and cardiorespiratory arrest. There were 1.4 anaphylaxis and hypotension events per 1,000 patients.5 On February 22, 2013, after the review of data from the pilot initiative, the dialysis organization discontinued administration of peginesatide. On February 23, the manufacturer voluntarily recalled the drug. The recognition of anaphylaxis and hypotension resulted in removal of peginesatide from the market. If it was not for the strict pharmacovigilance protocol initiated by the dialysis provider which implemented the pilot conversion, the immunological potential of the drug would have been missed for several months. This case clearly demonstrates the importance of post-authorization monitoring, pharmacovigilance, and risk mitigation plan.
References
Fishbane S, Schiller B, Locatelli F, et al. Peginesatide in patients with anemia undergoing hemodialysis. N Engl J Med. 2013;368(4):307–319. | |
Macdougall IC, Provenzano R, Sharma A, et al. Peginesatide for anemia in patients with chronic kidney disease not receiving dialysis. N Engl J Med. 2013;368(4):320–332. | |
Mikhail A. Profile of peginesatide and its potential for the treatment of anemia in adults with chronic kidney disease who are on dialysis. J Blood Med. 2012;3:25–31. doi: 10.2147/JBM.S23270. Epub May 23, 2012. | |
Food and Drug Administration. Revised Package Insert for Omontys (Peginesatide). Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202799s001lbl.pdf. Accessed December 4, 2012. | |
Bennett CL, Jacob S, Hymes J, Usvyat LA, Maddux FW. Anaphylaxis and hypotension after administration of peginesatide. N Engl J Med. May 22, 2014;370(21):2055–2056. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.