")
Back to Journals » Cancer Management and Research » Volume 11
Clinical Characteristics And Outcome Of Biphenotypic Acute Leukemia: 10 Case Reports And Literature Review
Authors Yu J , Li Y, Xing H, Pan Y , Sun H, Wan D, Liu Y, Xie X, Wang C, Sun L, Sun K, Jiang Z
Received 6 August 2019
Accepted for publication 5 October 2019
Published 31 October 2019 Volume 2019:11 Pages 9297—9306
DOI https://doi.org/10.2147/CMAR.S226058
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Jifeng Yu,1 Yingmei Li,1 Haizhou Xing,1 Yue Pan,1 Hui Sun,1 Dingming Wan,1 Yanfang Liu,1 Xinsheng Xie,1 Chong Wang,1 Ling Sun,1 Kai Sun,2 Zhongxing Jiang1
1Department of Hematology, First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, People’s Republic of China; 2Department of Hematology, People’s Hospital of Henan Province, Zhengzhou University, Zhengzhou, People’s Republic of China
Correspondence: Jifeng Yu; Zhongxing Jiang
Department of Hematology, First Affiliated Hospital of Zhengzhou University, 1 East Jianshe Road, Zhengzhou 450052, People’s Republic of China
Email [email protected]; [email protected]
Background: Biphenotypic acute leukemia (BAL), or mixed-phenotype acute leukemia (MPAL) represents a rare subgroup of acute leukemia which co-expresses markers for either more than one lineage in a homogenous blast population or the coexistence of two blast populations of different lineages. Proper diagnosis and classification of BAL are extremely important for patients’ outcome since BAL usually has a poor prognosis.
Purpose: The objective of this study was to identify the incidence of biphenotypic acute leukemia, their clinical characteristics and outcome of BAL patients with the chemotherapy treatment and Hematopoietic Stem Cell Transplantation (HSCT) after initial complete remission.
Patients and methods: Ten cases of biphenotypic acute leukemia were analyzed for their clinical characteristics, immunological phenotypes, chemotherapy methods for induction initial complete remission and outcome data, including induction chemotherapy, complete remission (CR) and the overall survival time, relapse and death. This study was an observational, retrospective, and descriptive study of the clinical aspects of BAL. Cytogenetics and fusion genes analysis were also done with bone marrow samples using G-banding analysis and karyotyped according to the International System for Human Cytogenetic Nomenclature. The fusion genes’ mutational status was determined by real-time PCR (RT-PCR). Gene mutation analyses were conducted with next-generation sequencing method.
Results: Among 10 BAL patients, 4 cases carried B/Myeloid phenotype, 4 cases carried T/Myeloid phenotype and 2 cases carried T/B phenotype. Cytogenetic analysis showed that 3 of the 10 cases had clonal abnormalities. Of the four cases of fusion gene aberration, two patients had RUNX1 gene mutation, one patient had BCR/ABL fusion gene mutation, and one patient had JAK1, JAK3, FBXW7 mutation. Overall, 5 of 8 (62.5%) BAL patients with chemotherapy achieved complete remission (CR) after their initial induction therapy. In the AML-directed therapy group, 1 of 2 (50%) patients achieved CR. Meanwhile, 4 of 6 (66.7%) patients achieved CR after ALL-directed induction chemotherapy. Two patients received Hematopoietic Stem Cell Transplantation (HSCT) after initial CRs, one patient died two months after transplantation due to pulmonary infection, and another patient is still alive. With an average of 14.3 (4.0–42.0) months’ follow-ups, the median survival time was 7 months. Although patients achieved CR after initial chemotherapy, the relapse rate was very high and the CR rate after relapse was very low.
Conclusion: Our results confirmed that BAL is a rare malignancy with a very poor prognosis. Patients with ALL-directed chemotherapy achieved a better CR rate compared to those with AML-directed chemotherapy. Patients should receive HSCT after initial CR whenever it is possible.
Keywords: biphenotypic acute leukemia, BAL, mixed-phenotype acute leukemia, MPAL, immunophenotyping, clinical outcome
Introduction
Acute leukemia (AL) is a malignant clonal disease originating from progenitor or multi-potential progenitor cells. Based on the cells’ antigen differentiation expression patterns, most cases of AL are classified as either myeloid or lymphoid lineage. However, there are patients with leukemic blast population who coexpresses both lymphoid and myeloid characteristics, known as biphenotypic acute leukemia (BAL) or mixed-phenotype acute leukemia (MPAL).1 These different morphologic and immunophenotypic characteristics can be present in one leukemic blast population (biphenotypic leukemia) or may be seen on two distinct leukemic blast populations from a single patient (bilineal leukemia).2 The European Group for Immunological Classification of Leukemias (EGIL) had proposed a set of diagnostic criteria for BAL based on the number and degree of specificity from certain makers for myeloid or T/B lymphoid blasts.3,4 The World Health Organization (WHO) criteria were proposed to the classification of acute leukemia of ambiguous lineage (ALAL), which included significant modifications to the diagnostic criteria for mixed phenotypic leukemia. Leukemia with blast co-expressing antigens with more than one lineage on the same cell population or with separate blast populations of different lineages is referred as MPAL. It can be further classified as B/Myeloid or T/Myeloid based on flow cytometric immunophenotyping, with either one or more than one population of blasts found. Thus, MPAL includes both biphenotypic and bilineal ALs. Leukemia expressed with non-lineage specific markers, such as CD34, HLA-DR, and/or CD38 and sometimes TdT but lack specific myeloid or lymphoid antigens, is designated as acute undifferentiated leukemia (AUL). Previously, the WHO criteria were more stringent than the criteria of the EGIL and heavily relied on positivity of myeloperoxidase (MPO).5 Recently, the WHO classification was updated in 2016, but the criteria for ALAL remained unchanged.6
A systematic review of the literature revealed 31 reports describing the prevalence of MPAL with a mean prevalence of 2.8% of acute leukemia (range 0.3–9.0%),7 while other reports on the incidence of BAL or ALAL vary based on the criteria (EGIL or 2016 WHO) used for the diagnosis. BAL is reported to account for 2–5% of all ALs based on the EGIL criteria, while ALAL reports 1–2.5% based on the WHO criteria.2,8,9 Although the incidence of AUL was not clearly defined, reports showed 0.2–1% prevalence in some studies.10,11 As for treatment, different kinds of chemotherapy for BAL or MPAL have been reported by different groups.7 ALL-therapy is associated with higher initial remission rates for MPAL and is at least equivalent to more intensive AML therapy for long-term survival. Currently, a standard chemotherapy treatment has yet to be established. This study aims to retrospectively investigate the incidence, pathological characteristics, and clinical outcome of BAL patients from the First Affiliated Hospital of Zhengzhou University (Zhengzhou, China) between January 2014 and June 2019.
Methods
Patient Cohort
From January 2014 to June 2019, the medical records of newly diagnosed BAL based on the EGIL criteria, or ALAL based on the 2008/2016 WHO criteria and who were admitted at the First Affiliated Hospital of Zhengzhou University (Zhengzhou, China) were retrospectively reviewed. Clinical data were obtained, including age, sex, blood counts, blast % in bone marrow (BM), morphology, immunophenotyping, and cytogenetic/molecular studies. Treatment methods and outcome data, including induction chemotherapy, complete remission (CR), relapse, and death, were collected and reviewed. All adult patients or child patient’s parents were given informed consent for both treatment and related laboratory examinations. This study was an observational, retrospective, and descriptive study of the clinical aspects of BAL, which was approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University. The present study was performed in accordance with the ethical standards of the 1964 Declaration of Helsinki and its later amendments.
Immunophenotyping
Immunophenotyping of BM aspirates was performed using the consensus method. The result was considered positive if the antigen was expressed on more than 20% (10% for cytoplasmic markers, anti-MPO, and TdT) of leukemic cells. The panels of monoclonal antibodies (BD Biosciences, San Jose, CA, USA) used in flow cytometric immunophenotyping to detect B-cell, T-cell, and myeloid antigens were as follows: myeloid lineage (anti-MPO, CD13, CD14, CD15, CD33, CD64, and CD117), megakaryocytes (CD41 and CD61), natural killer cells (CD56), lymphoid lineage (CD10and TdT), T-lymphoid lineage (CD2, CD3, cCD3, CD4, CD8, CD5, and CD7), B-lymphoid lineage (CD19, CD20, CD79a and cCD22), and other antigens (CD11, CD34, CD36, CD38, CD58, CD123, and HLA-DR). The flow cytometer of Beckman Coulter Gallios 4-laser and 10-color model was used for this study and the Kaluza software was used for flow cytometry data collection and analysis.
Diagnostic Criteria
The diagnostic workup of BAL was based on initial morphological, cytochemical, and immunophenotypic evaluation of the BM. BAL diagnosis was established if the score from two separate lineages was greater than 2 by using the EGIL scoring system.3 WHO criteria were used to classify the MPAL includes both biphenotypic and bilineal ALs more stringent than those of the EGIL criteria and relied heavily on positivity for myeloperoxidase (MPO).4 Different monoclonal antibodies specific to the related antigens were used in defining the pattern of lineage involvement. The myeloid lineage-defining marker was MPO as detected by flow cytometry, immunohistochemistry; monocytic differentiation was required for assigning either diffuse positivity for non-specific esterase or expression of at least two of the following: CD11c, CD14, CD36, CD64, and lysozyme. The T-lineage-defining markers were cytoplasmic CD3 or surface CD3. The B-lineage-defining marker was either a strong CD19 with at least one of the strongly expressed CD79a, cytoplasmic CD22 and CD10, or weak CD19and at least two of the strongly expressed CD79a, cytoplasmic CD22 and CD105.12–14
Cytogenetics And Fusion Genes Analysis
The bone marrow samples were studied using G-banding analysis and karyotyped according to the International System for Human Cytogenetic Nomenclature. The fusion genes’ mutational status was determined by real-time PCR (RT-PCR). We used Multiplex RTPCR Fusion Gene Kits provided by Rightongene (Shanghai, China).
Next‑generation Sequencing
Gene mutation analyses were conducted in 3 patients (UPID#4, #5, #9) with a panel of 22 genes, including FLT3-ITD, NPM1, KIT, CEBPA, DNMT3A, IDH1, IDH2, TET2, EZH2, RUNX1, ASXL1, PHF6, TP53, SF3B1, SRSF2, U2AF1, ZRSR2, NRAS, CBL, SETBP1, ETV6, and JAK2. Read pairs were aligned to Refseq hg19 (downloaded from the UCSC Genome Browser, URLs) by the Burrows–Wheeler Aligner version 0.7.13-r1126. Samtools version 1.3 was used to generate chromosomal coordinate-sorted BAM files. We used targeted next-generation sequencing with the Rightongene AML/MDS/MPN Sequencing Panel (Rightongene). The NGS libraries were paired-end sequenced (2 × 150 bp) on an Illumina MiSeq System (Illumina, San Diego, CA). The mean depth of each sample was 2500×, with an average 5% of the target sequence being covered sufficiently deeply for variant calling. SAMtools mpileup was applied for SNV/indel calling and filter workflow.
Treatment Protocols
Total of 10 BAL patients enrolled in this study. Among the 10 patients, 8 patients received induction chemotherapy, one patient (UPID#2) moved to another hospital for treatment and another patient (UPID#4) gave up the chemotherapy after the diagnosis due to personal reasons. Patients were initially treated with remission induction therapy of either ALL or AML-directed chemotherapy. ALL-directed regimen was used in 6 patients. The chemotherapy based on VDLP regimen, vincristine (VCR), prednisone (PRD), and L-asparaginase (L-asp) with daunorubicin was used in four children patients (UPID#6,7,9,10) and two adult patients (UPID#1,8). For AML-directed chemotherapy, the induction regimen MEA and IA chemotherapy were used in 2 patients (UPID#3, and #5), respectively. Patients with CR after induction chemotherapy were defined according to the criteria of the International Working Group.14 Two patients (UPID#5 and #7) received hematopoietic stem cell transplantation (HSCT) after initial CRs.
Statistical Analysis
Continuous variables were expressed as mean ± standard deviation; categorical variables were expressed as numbers and percentages. Continuous variables were compared using the Student’s t-test, while categorical variables were compared using the chi-square test. Probabilities of survival were estimated using the Kaplan–Meier (K–M) method. A P-value of <0.05 was considered significant. The software package SPSS version 21.0 (IBM Corp., Armonk, NY, USA) was used for all statistical analyses.
Results
Patient Clinical Characteristics
Among a total of 6100 patients newly diagnosed with AL, 10 (0.16%) patients satisfied the definition of BAL based on the EGIL criteria, or MPAL based on the WHO criteria, including 7 males and 3 females. The median age of these patients at diagnosis was 19 years (range 3–67 years). One patient (UPID#1) had extramedullary invasion, including neck, mediastinum (area 8), posterior septal group, left axillary, peritoneal and retroperitoneal lymph nodes. Another patient (UPID#7) had extramedullary invasion with central nervous system leukemia. The clinical characteristics of these patients were summarized as below (Table 1). Univariate analyses of clinical laboratory variables for the prediction of prognosis were performed on initial leukocyte count, hemoglobin, platelets, immunophenotype (M+B vs M+T). No significant difference had been found among these variables between different groups (Table 2).
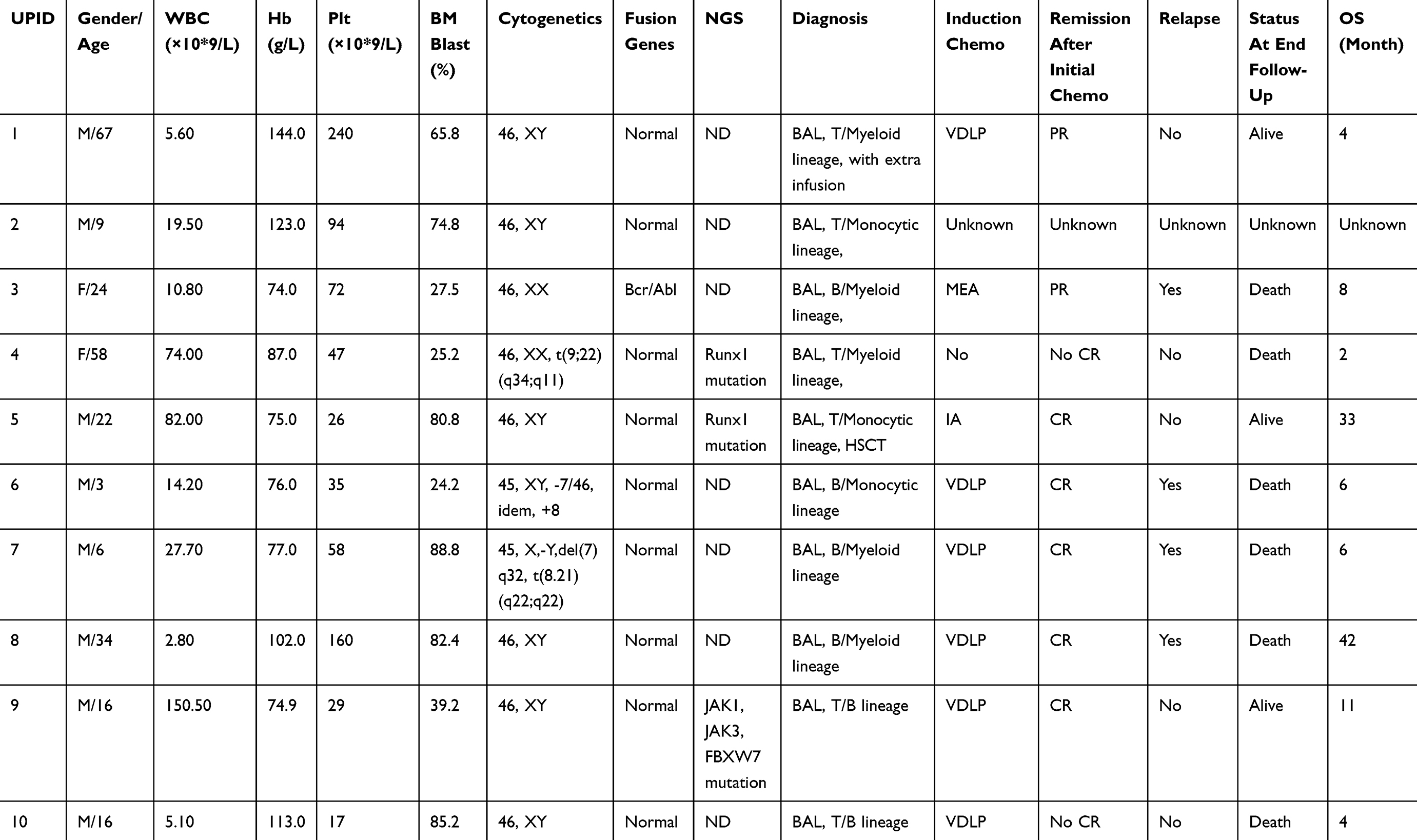 |
Table 1 Clinical Manifestations And Cytogenetic Abnormalities Of BAL Patients |
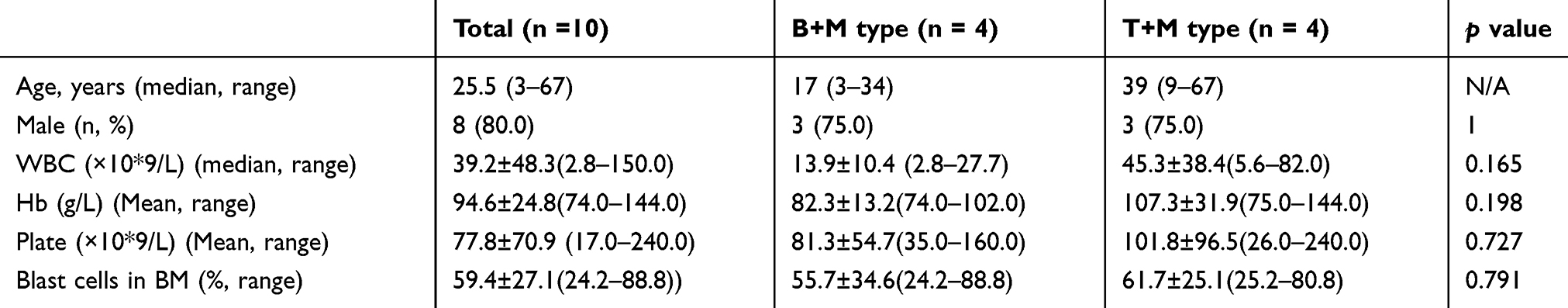 |
Table 2 Clinical Manifestations Of BAL Patients Stratified By Immunophenotypes |
Immunophenotypic Characteristics
Table 3 shows the immunophenotypic characteristics of each BAL patient. The expression of immunological and cytochemical markers on leukemic blasts according to BAL subtypes is summarized in Table 4. Among 10 BAL patients, 4 cases carried B/Myeloid phenotype, 4 cases carried T/Myeloid phenotype and 2 cases carried T/B phenotype. In 8 patients with myeloid lineage differentiation, MPO was positive in 6 (75%), CD13 in 4 (50%), CD33 in 4 (50%), CD38 in 6 (75%), CD58 in 3 (37.5%), CD117 in 4 (50%) patients. In 5 patients with B-lymphoid lineage differentiation, CD19 was positive in 4 (80%), CD79a in 5 (100%) patients. The most frequently T-lymphoid lineage positive marker was CD7 and cCD3, which were positive in 4 of 5 (80%) patients. The stem cell markers HLA-DR and CD34 were both positive in 8 (80%) patients, while CD117 was positive in 4 (40%) patients.
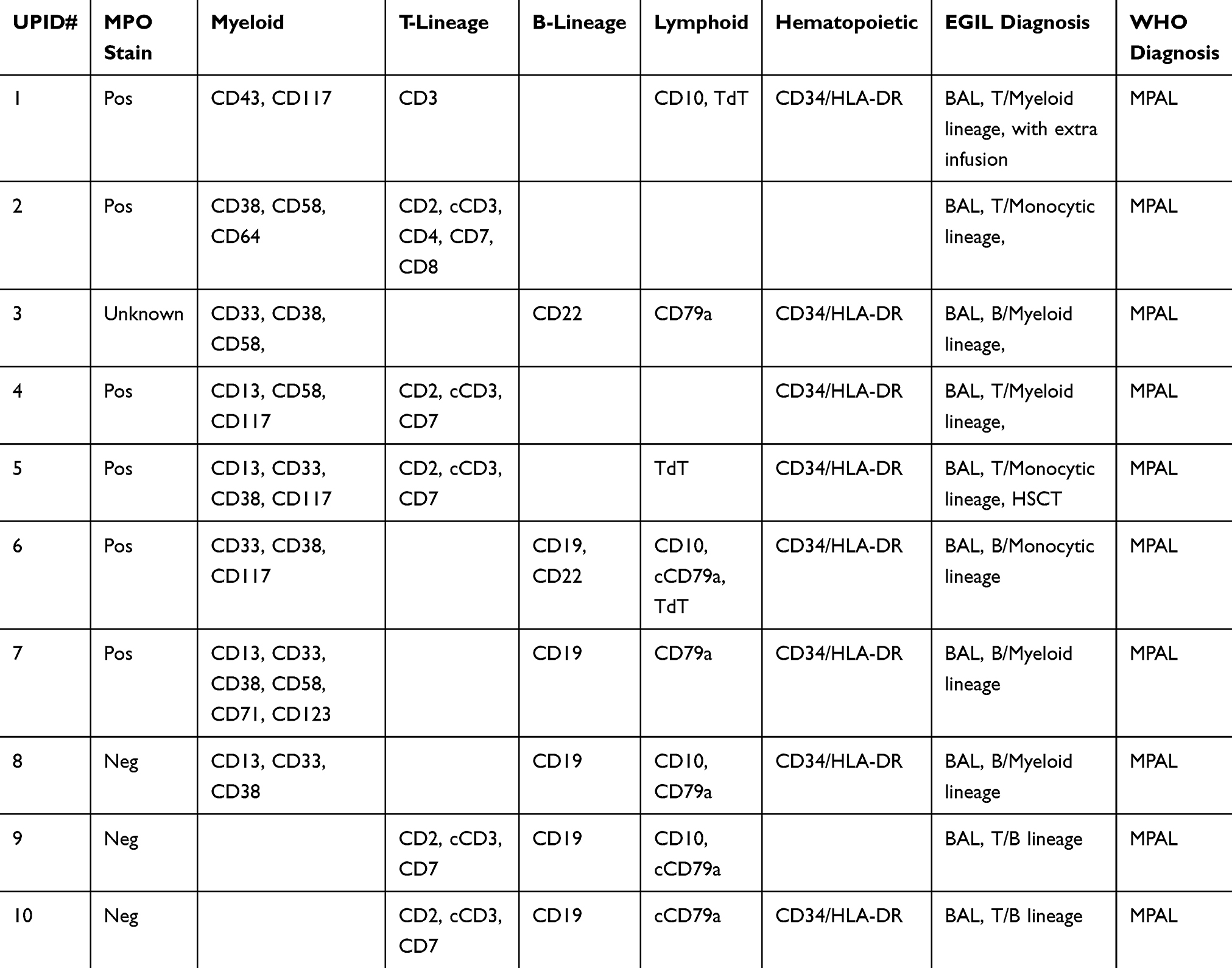 |
Table 3 EGIL Or WHO Diagnosis Based On Cytochemical And Immunophenotype Data |
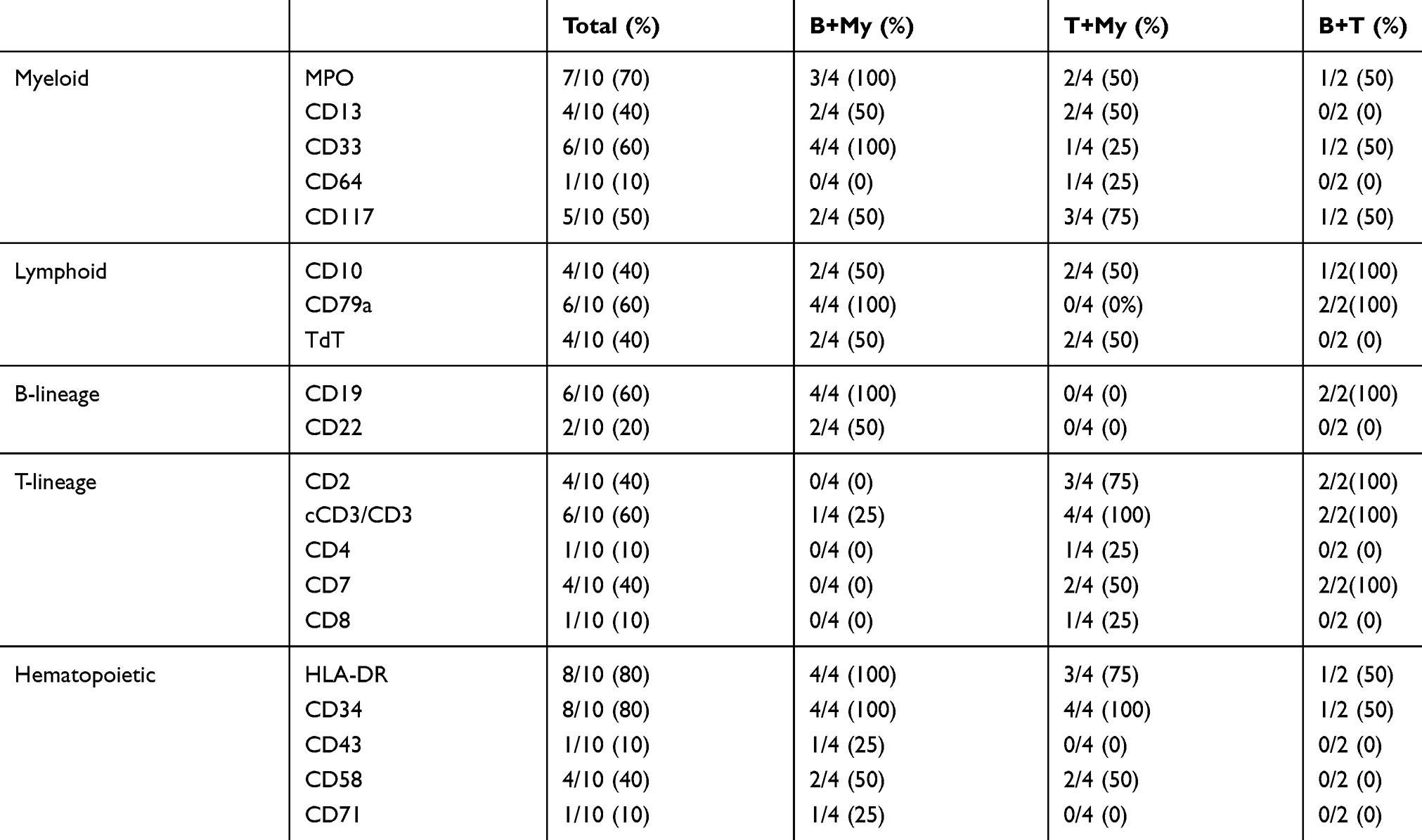 |
Table 4 Expression Of Cytochemical And Immunological Markers In BAL |
Cytogenetic Characteristics
Results of cytogenetic analysis performed on all patients were available. Details of cytogenetic analysis and molecular studies are presented in Table 1. In cytogenetic analysis, 7 of 10 patients had normal karyotypes (UPID#1, 2, 6, 7, 8, 9.10) while the other 3 patients had clonal abnormalities. UPID#4 had 46, xx, t(9;22)(q34;q11) aberration; UPID#6 had 45, XY, −719/46, idem, +8 aberration; and UPID#7 had 45, X,-Y, del(7)q32, t(8.21) (q22;q22) aberration. Molecular markers of fusion genes results showed that one patient (UPID#4) had BCR/ABL fusion gene mutation. NGS gene mutation analysis results demonstrated that two patients (UPID# 3, 5) had RUNX1 gene mutation and one patient (UPID#9) had JAK1, JAK3, FBXW7 mutation (Table 1).
Treatment Outcome
Six patients initially received ALL-directed induction therapy (VDLP) whereas two patients received standard induction therapy for AML (MEA and IA) (Table 1). Overall, 5 of 8(62.5%) patients with chemotherapy achieved complete remission (CR) after their initial induction therapy. In the AML induction group, 1 of 2 (50%) patients achieved CR. Meanwhile, 4 of 6 (66.7%) patients achieved CR after ALL-directed chemotherapy. In comparison to these two different initial induction therapy groups, there is no statistically significant difference for the CR rates (p=0.673). Among the two patients received HSCT after initial CRs, one patient (UPID#7) died two months after transplantation due to pulmonary infection, and another patient (UPID#5) is still alive. Among the 8 patients received chemotherapy, one patient only achieved partial remission and another 2 patients failed to respond to induction chemotherapy. Among the 5 patients achieved CR after initial induction chemotherapy, 3 patients had relapse (60%).
Survival Rates
Among 10 patients enrolled in this study, one patient moved to another hospital for treatment and loss of follow-up, other 6 patients died with different complications. Only three patients were still alive by end of the follow-up. With an average of 14.3 months follow-up (range, 4.0–42.0 mo), the median Overall survival (OS) time was 7.0±14.7% (Figure 1). Figure 2 demonstrates the survival probabilities and the survival comparison between ALL and AML-directed chemotherapy outcomes.
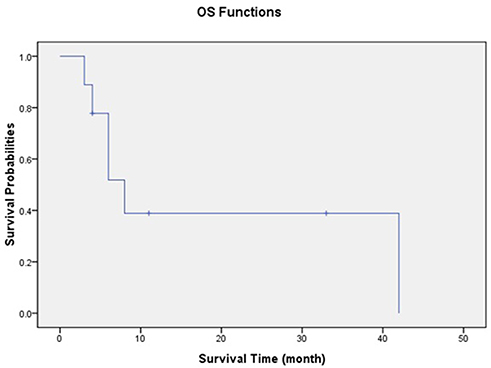 |
Figure 1 Kaplan–Meier plot of all patients’ overall survival. |
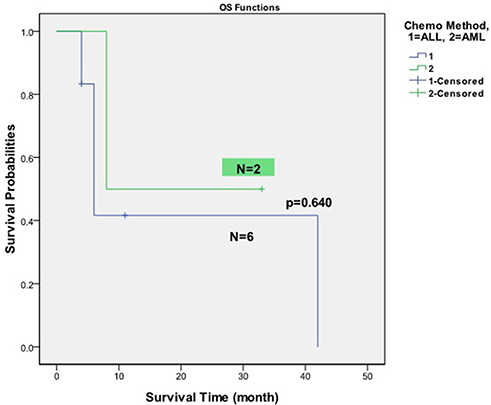 |
Figure 2 Kaplan–Meier plot of patients’ overall survival: ALL-directed vs AML-directed chemotherapy. |
Discussion
BAL is a rare subgroup of heterogeneous leukemia represented by immunophenotypic and/or karyotypic and molecular abnormalities. It has been classified as either biphenotypic acute leukemia (BAL) based on the European Group for Immunological Classification of Leukemias (EGIL) or acute leukemia of ambiguous lineage (ALAL) encompassing acute undifferentiated leukemia (AUL) and mixed-phenotype acute leukemia (MPAL) based on the World Health Organization (WHO) criteria.3–6 Due to the complexity of the disease, there is no uniform classification yet.
Based on the recent understanding of hematopoiesis, BAL pathogenesis might be due to the minute stages of differentiation blocked by deleterious molecular events. The normal immune system cannot recognize and remove leukemic cells at an early stage most likely due to the cell’s“normal” immunophenotype. Due to the different classification for AUL, MPAL and BAL, and the rarity of the disease, the morbidity of BAL or MPAL was not clearly defined. The mean prevalence of MPAL was reported as 0.2–1% in some studies.10,11 The incidence of BAL in this report was only 0.16% of AL patients, notably lower than other reports. This could possibly be due to the different patient populations. As for the rarity of this disease, some reports suggested that the MPAL incidence could be underestimated as the use of restrictive immunophenotypic panels or combinations lower the sensitivity of their detection.
Here we reported 10 cases of BAL with different immunophenotypes; 4 cases with B/Myeloid lineage, 4 cases with T/Myeloid lineage and 2 cases with T/B lineage. The diagnosis was made based on the EGIL and WHO criteria. In contrast to the EGIL scoring system based on blast immunophenotype with numerous markers, the WHO criteria emphasized key lineage-defining markers with emphasis on CD19 for B lineage, CD3 for T lineage, and MPO for myeloid lineage. Comparing to the EGIL criteria, the WHO approach was simpler but relied heavily on the sensitivity and specificity of a few markers. It did not specify positive thresholds for these key markers, leaving it up to individual laboratories to decide on the definition of significant expression. This could potentially cause some deviations between individual laboratories.
The incidence of B-lymphoid/myeloid phenotype was more than that of the T-lymphoid/myeloid phenotype according to the literature reports.2,15 In adult studies reported, more than 70% of BAL patients had the B-lymphoid/myeloid phenotype, whereas only 23–33% had the T-lymphoid/myeloid phenotype. Thus, it was found the B-lymphoid/T-lymphoid/myeloid phenotype and B-lymphoid/T-lymphoid phenotype were extremely rare.16–18 In our study, we found equal incidence of BAL with B/Myeloid phenotype and T/Myeloid phenotype. The phenotype distribution in our patients with a mixture of children and adult BAL was different from those reported in previously published studies.2,15–18
Although mutations in DNMT3A, TET2, RUNX1, WT1, and various rare chromosomal translocations had been previously identified in MPAL,19–26 recurrent molecular alterations specific to BAL have remained elusive. Our study demonstrated recurrent RUNX1 gene mutations in 2 cases of BAL patients, BCR/ABL gene mutation in 1 case, and 2 alternative mutations in 2 other cases. Meta review results showed that the top 3 common gene mutations in BAL patients were BCR/ABL,7 ETV6/RUNX1and KMT2Ar. Recent report showed that majority of the children BAL patients with clonal abnormalities involving the MLL gene, additional to the BCR/ABL fusion gene.27 The relatively small number of patients with this rare disease made an estimation of the precise frequency of gene mutation in BAL difficult.
A systematic review and meta-analysis of quantitative synthesis for MPAL therapy in children and adults with mixed phenotype acute leukemia showed that the use of an ALL induction regimen is more likely to achieve an initial remission than more toxic AML regimens.7 Meta-analyses supported the benefit of starting with ALL therapy for OS, but this finding was not replicated in multivariable analysis of the smaller compiled case series. It is unclear if this discrepancy is due to differences in post-induction therapy, variable use of SCT, or other differences not minimized by the large number of patients in the aggregate meta-analyses. Most recent reports showed that Ph+ MPAL was more common in male and inclined to high WBC counts at diagnosis.28 The prognosis of patients with Ph+ MPAL was poor. Although many patients achieved CR with the initial induction chemotherapy, but the relapse rate was very high and the CR rate after relapse was very low. Imatinib and allogeneic HSCT may improve survival of patients with Ph+ MPAL.28
In most of the publications, the selection of an induction chemotherapy regimen for AL is largely based on whether a case is classified as myeloid or lymphoid. So far, there are no agreed chemotherapy protocols for patients with BAL yet. In our study, the induction regimen was selected based on the morphology of the blasts, cytochemical stains and immunophenotyping results. We chose to use the ALL induction regimen for 6 of the 8 patients who received induction chemotherapy based on the systematic review and meta-analysis data supporting improved CR rates with ALL therapy. The other two patients received AML induction regimen based on the morphology and immune phenotyping was more toward to the AML types.
The survival rate of BAL from different studies in pediatric and adult patients had a broad variation ranged from 8.1% to 60%.2,15,29 In the recent two studies on pediatric patients, the 5-year OS of the total cohort was 51.1±15.8% from Korean patients and the survival rate of MPAL based on the WHO classification was about 80%.11,27 One study demonstrated a better OS for the younger patients than older counterparts (75% vs 17% at 2 yrs; P=0.01).30 A comparison study results demonstrated that the 5-year EFS probability of ALAL patients (62±5%) was lower than those of ALL patients (80±1%, P<0.001), but better than those of AML patients (49±2%, P=0.027).15 In a meta-review study the results showed that although the survival of MPAL had been improved in recent years, but the prognosis of MPAL is still poor especially evidenced by the multivariate analysis.29
Our results showed that 5 of 8 BAL patients (62.5%) achieved CR after their initial induction chemotherapy. Patients with ALL-directed chemotherapy achieved a different CR rate (4/6, 66.7%) compared to patients with AML-directed chemotherapy (1/2, 50%), but no statistically significant difference. The CR rates were lower than the meta-analysis results and other reports.27,28 Although the CR rates after initial induction chemotherapy were satisfactory, our results showed a low OS duration, indicating that the prognosis of the BAL is poor. Many patients in the different studies with a OS benefit received either HSCT after the initial CR or a new chemotherapy regimen such as Imatinib.7,11,28,29 In our study, only two patients received HSCT after initial CR, this could possibly be one of the reasons that our results for the OS duration were much lower than other studies, additional to the small number of cases. While the meta-analysis data supporting improved CR rates with ALL therapy is consistent across all aspects of this quantitative analysis, the impact of therapy type on OS is not as clear. Due to the rarity of BAL among patients, larger sample sizes prove difficult to organize. Although ALL-directed chemotherapy is more favorable than the alternative AML-directed regimen,7,11,28 but the relapsing and refractory issue had been the clinical challenges for many BAL patients. Other challenges including the obscurity of pathogenesis and related molecular mechanisms in BAL make the selection of the chemotherapy regimen quite difficult. Our results demonstrated that there was no difference between ALL and AML-direction induction regimen for BAL chemotherapy. However, due to the rarity of this disease, more thorough studies on pathogenesis and targeted chemotherapy with multicenter prospective studies incorporating large number of cases are urgently needed to improve the clinical outcomes for the BAL patients.
Conclusion
BAL is a rare subgroup of acute leukemia with a poor prognosis. We reported 10 cases of BAL, including 4 cases with B/Myeloid phenotype, 4 cases with T/Myeloid phenotype and 2 cases with T/B phenotype. Five of 8 patients with chemotherapy achieved CR (62.5%) after their initial induction therapy. Immunophenotypic characteristics and cytogenetic characteristics are critical for the diagnosis and chemotherapy outcomes. Although many patients achieved CR after initial chemotherapy, but the relapse rate was very high and the CR rate after relapse was very low. Patients should receive HSCT after initial CR whenever it is possible. Our results re-confirmed that BAL is a rare malignancy with a very poor prognosis. Due to the rarity of the disease, further studies of pathogenesis on the cellular and molecular anomalies with some new therapeutic targets need to be explored to improve patients’ prognosis.
Acknowledgments
This work was supported by the Talent Research Fund of the First Affiliated Hospital of Zhengzhou University, and Research Fund of the Key Scientific and Technological Research Project of Henan Education Department (project number 20A320062) and the Special Talents Project of the First Affiliated Hospital of Zhengzhou University.
Author Contributions
J.Y. and Z.J. designed and directed the study and wrote the manuscript. Y.L., H.X., H.S., D.W., Y.L., C.W., X.X., L.S., K.S. and Z.J. contributed to the clinical data and patients’ treatment. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Béné MC, Porwit A. Acute leukemias of ambiguous lineage. Semin Diagn Pathol. 2012;29:12–18.
2. Rubnitz JE, Onciu M, Pounds S, et al. Acute mixed lineage leukemia in children: the experience of St Jude Children’s Research Hospital. Blood. 2009;113:5083–5089. doi:10.1182/blood-2008-10-187351
3. Bene MC, Castoldi G, Knapp W, et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia. 1995;9:1783–1786.
4. Matutes E, Morilla R, Farahat N, et al. Definition of acute biphenotypic leukemia. Haematologica. 1997;82:64–66.
5. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–951. doi:10.1182/blood-2009-03-209262
6. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi:10.1182/blood-2016-03-643544
7. Maruffi M, Sposto R, Oberley MJ, Kysh L, Orgel E. Therapy for children and adults with mixed phenotype acute leukemia: a systematic review and meta-analysis. Leukemia. 2018;32(7):1515–1528. Epub 2018 Feb 27. doi:10.1038/s41375-018-0058-4
8. Lee MY, Tan TD, Feng AC. Clinicopathologic analysis of acute myeloid leukemia in a single institution: biphenotypic acute myeloid leukemia may not be an aggressive subtype. J Chin Med Assoc. 2007;70:269–273. doi:10.1016/S1726-4901(07)70003-5
9. Matutes E, Pickl WF, Van’t Veer M, et al. Mixed-phenotype acute leukemia: clinical and laboratory features and outcome in 100 patients defined according to the WHO 2008 classification. Blood. 2011;117:3163–3171. doi:10.1182/blood-2010-10-314682
10. Gerr H, Zimmermann M, Schrappe M, et al. Acute leukaemias of ambiguous lineage in children: characterization, prognosis and therapy recommendations. Br J Haematol. 2010;149:84–92. doi:10.1111/j.1365-2141.2009.08058.x
11. Hrusak O, de Haas V, Stancikova J, et al. International cooperative study identifies treatment strategy in childhood ambiguous lineage leukemia. Blood. 2018;132:264–276. doi:10.1182/blood-2017-12-821363
12. Weinberg OK, Seetharam M, Ren L, Alizadeh A, Arber DA. Mixed phenotype acute leukemia: a study of 61 cases using World Health Organization and European Group for the immunological classification of leukaemias criteria. Am J Clin Pathol. 2014;142:803–808. doi:10.1309/AJCPPVUPOTUVOIB5
13. Wolach O, Stone RM. How I treat mixed-phenotype acute leukemia. Blood. 2015;125:2477–2485. doi:10.1182/blood-2014-10-551465
14. Charles NJ, Boyer DF. Mixed-phenotype acute leukemia: diagnostic criteria and pitfalls. Arch Pathol Lab Med. 2017;141:1462–1468. doi:10.5858/arpa.2017-0218-RA
15. Al-Seraihy AS, Owaidah TM, Ayas M, et al. Clinical characteristics and outcome of children with biphenotypic acute leukemia. Haematologica. 2009;94:1682–1690. doi:10.3324/haematol.2009.009282
16. Carbonell F, Swansbury J, Min T, et al. Cytogenetic findings in acute biphenotypic leukaemia. Leukemia. 1996;10:1283–1287.
17. Owaidah TM, Al Beihany A, Iqbal MA, Elkum N, Roberts GT. Cytogenetics, molecular and ultrastructural characteristics of biphenotypic acute leukemia identified by the EGIL scoring system. Leukemia. 2006;20:620–626. doi:10.1038/sj.leu.2404128
18. Xu XQ, Wang JM, Lü SQ, et al. Clinical and biological characteristics of adult biphenotypic acute leukemia in comparison with that of acute myeloid leukemia and acute lymphoblastic leukemia: a case series of a Chinese population. Haematologica. 2009;94:919–927. doi:10.3324/haematol.2008.003202
19. Kern W, Grossmann V, Roller A, et al. Mixed phenotype acute leukemia, T/ myeloid,NOS (MPAL-TM) has a high DNMT3A mutation frequency and carries further genetic features of both AML and T-ALL: results of a comprehensive next-generation sequencing study analyzing 32 genes. Blood. 2012;120(21):403. doi:10.1182/blood.V120.21.403.403
20. Yan L, Ping N, Zhu M, et al. Clinical, immunophenotypic, cytogenetic, and molecular genetic features in 117 adult patients with mixed-phenotype acute leukemia defined by WHO-2008 classification. Haematologica. 2012;97(11):1708–1712. doi:10.3324/haematol.2012.064485
21. Ma Q, Tong Y, Jin J, Lou Y. Mixed T/myeloid phenotype acute leukemia with rare variants of IDH1 and CEBPA. Leuk Lymphoma. 2013;54(11):2568–2570. doi:10.3109/10428194.2013.783212
22. Manola KN. Cytogenetic abnormalities in acute leukaemia of ambiguous lineage: an overview. Br J Haematol. 2013;163(1):24–39. doi:10.1111/bjh.12484
23. Eckstein OS, Wang L, Punia JN, et al. Mixed-phenotype acute leukemia (MPAL) exhibits frequent mutations in DNMT3A and activated signaling genes. Exp Hematol. 2016;44(8):740–744. doi:10.1016/j.exphem.2016.05.003
24. Yamamoto K, Kawamoto S, Mizutani Y, et al. Mixed phenotype acute leukemia with t(12;17)(p13;q21)/TAF15-ZNF384 and other chromosome abnormalities. Cytogenet Genome Res. 2016;149(3):165–170. doi:10.1159/000448447
25. Pallavajjala A, Kim D, Li T, et al. Genomic characterization of chromosome translocations in patients with T/myeloid mixed-phenotype acute leukemia. Leuk Lymphoma. 2017;59:1231–1238. doi:10.1080/10428194.2017.1372577
26. Yao L, Cen J, Pan J, et al. TAF15-ZNF384 fusion gene in childhood mixed phenotype acute leukemia. Cancer Genet. 2017;211:1–4. doi:10.1016/j.cancergen.2016.12.002
27. Lee HG, Baek HJ, Kim HS, et al. Biphenotypic acute leukemia or acute leukemia of ambiguous lineage in childhood: clinical characteristics and outcome. Blood Res. 2019;54:63–73. doi:10.5045/br.2019.54.1.63
28. Jia Y, Li Y, Gong XY, et al. Clinical characteristics of patients with Ph+ mixed phenotype acute leukemia. J Exp Hematol. 2019;27(2):354–359.
29. Shi R, Munker R. Survival of patients with mixed phenotype acute leukemias: a large population-based study. Leuk Res. 2015;39:606–616. doi:10.1016/j.leukres.2015.03.012
30. Killick S, Matutes E, Powles RL, et al. Outcome of biphenotypic acute leukemia. Haematologica. 1999;84:699–706.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.