Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
Clinical Characteristics and Novel ZEB2 Gene Mutation Analysis of Three Chinese Patients with Mowat-Wilson Syndrome
Authors Han X, Zhang Q, Wang C, Han B ![]()
Received 25 April 2023
Accepted for publication 11 August 2023
Published 23 August 2023 Volume 2023:16 Pages 777—783
DOI https://doi.org/10.2147/PGPM.S414161
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Xiao Han,1 Qianjuan Zhang,2 Chengcheng Wang,3 Bingjuan Han4
1Department of Pediatrics, Jining First People’s Hospital, Jining, Shandong, 272011, People’s Republic of China; 2Department of Children’s Medical Rehabilitation Center, Jinan Maternity and Child Care Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, 250001, People’s Republic of China; 3Department of Pediatric Surgery, Jining First People’s Hospital, Jining, Shandong, 272011, People’s Republic of China; 4Department of Children’s Health Prevention, The Second Children & Women’s Healthcare of Jinan City, Jinan, Shandong, 271100, People’s Republic of China
Correspondence: Bingjuan Han, Email [email protected]
Purpose: Mowat-Wilson syndrome (MWS) is an autosomal dominant disease caused by a pathogenic variant of the ZEB2 gene. The main clinical manifestations include special facial features, Hirschsprung disease (HSCR), global developmental delay and other congenital malformations. Here, we summarize the clinical characteristics and genetic mutation analysis of three Chinese patients with MWS.
Patients and Methods: The clinical characteristics of the patients were monitored and the treatment effect was followed up. DNA was extracted from peripheral blood and analyzed by sequencing. Whole exome sequencing was then performed.
Results: Three novel ZEB2 gene mutations were identified in 3 patients (c.1147_1150dupGAAC, p.Q384Rfs*7, c.1137_1146del TAGTATGTCT, p.S380Nfs *13 and c.2718delT, p.A907Lfs*23). They all had special facial features, intellectual disability, developmental delay, microcephaly, structural brain abnormalities and other symptoms. After long-term regular rehabilitation treatment, the development quotient of each functional area of the patient was slightly improved.
Conclusion: Our study expanded the mutation spectrum of ZEB2 and enriched our understanding of the clinical features of MWS. It also shows that long-term standardized treatment is of great significance for the prognosis of patients.
Keywords: Mowat-Wilson syndrome, ZEB2, gene mutation, rehabilitation treatment, long-term follow-up
Introduction
Mowat-Wilson syndrome (MWS) is an autosomal dominant disease caused by a pathogenic variant of the ZEB2 gene. The main clinical manifestations include special facial features, Hirschsprung disease (HSCR), moderate to severe intellectual disability, global developmental delay, epilepsy and other congenital malformations. Special face is mainly manifested in forehead protrusion, eye distance is increased, eyebrows are widened and have horn like protrusion inside, nose tip protrusion is round, open mouth with M-shaped upper lip, small jaw, etc. In addition, other congenital malformations may include congenital heart disease, agenesis of the corpus callosum, genitourinary abnormalities (especially in men with hypospadias), and ocular defects.1–3 The syndrome is caused by de novo mutations in one allele of zinc-finger E-box-binding homeobox 2 gene (ZEB2). ZEB2 also known as ZFHX1B (zinc finger homeobox 1B) or SIP1 (Smad Interacting protein 1).4,5 The ZEB2 protein contains a number of functional domains, including a nucleosome remodeling and deacetylase‑interaction motif, one zinc‑finger (ZF) cluster in the amino terminal region (N‑ZF), a SMAD binding domain, a homeodomain, a C‑terminal binding protein interacting domain, and one ZF cluster in the carboxyl terminal region (C‑ZF). ZEB2 is a member of the two-handed zinc-finger/homeodomain transcription factor family, it consists of nine coding exons (exons 2–10) and one non-coding exon (exon 1).6,7 ZEB2, like its family members, interacts with Smad1 protein and functions as a transcriptional repressor of TGF-β signaling pathway.8 ZEB2 plays important roles in development, such as neural crest formation, gastrula formation, cardiac morphogenesis, musculoskeletal system formation, and craniofacial structure establishment.6,7
MWS was first described in 1998 by Mowat et al, and the genetic locus was identified on chromosome 2q22-q23.9 It has been reported that the incidence of the disease is about 1:50, 000 to 1:70, 000.10 By reviewing the literature, it can be found that about 300 MWS patients have been reported, and about 280 ZEB2 variants have been found (Human Gene Mutation Database).11–14 It has mainly been reported from countries such as Europe, Australia and the United States, with Japan also reporting a large number of cases.14 35 cases of MWS and 25 pathogenic ZEB2 variants have been reported in China.11,15–18 Here, we report three novel ZEB2 variants in Chinese MWS patients and summarize their clinical manifestations, genetic mutations, and follow-up.
Patients and Methods
All three patients were from The Second Children & Women’s Healthcare of Jinan City. All tests were performed as routine clinical investigations in accordance with the ethical principles of the Declaration of Helsinki. Written informed consent was obtained from the patient’s guardian. This study was approved by the Medical Ethics Committee of The Second Children & Women’s Healthcare of Jinan City.
Genomic DNA was extracted from peripheral blood leukocytes using a commercial kit (Qiagen). Whole exome sequencing was then performed. The genetic analysis was approved by the Medical Ethics Committee of The Second Children & Women’s Healthcare of Jinan City.
Results
Patient One
The patient was the first child of a healthy unrelated couple. She was born by vaginal delivery at 40 weeks of gestation. Her birth weight was 3.74 kg. She had tears in her eyes and a lot of secretions after her birth. When she was 7 months old, she came to the hospital because she could not turn over and sit down. At this time, she has unique facial features, including wide eyebrows, a slightly wider distance between the eyes, and a prominent rounded nose tip.
After examination, the echocardiography of the patient showed that the intracardiac structure was generally normal, and the liver, gallbladder, pancreas, spleen and kidney were not significantly abnormal. The results of tandem mass spectrometry and gas-chromatography mass spectrometry were normal. Gastrointestinal ultrasound showed no obvious abnormalities, and craniocerebral MRI showed suspicious white matter retardation. SNP array did not detect clinically significant chromosome copy number abnormalities. A novel heterozygous variant (NM_014794.3:c.1147_1150dupGAAC, p.Q384Rfs*7) was found in this patient by whole exome detection, which was not detected in the peripheral blood of the patient’s parents after Sanger verification, so it may be a de novo variant, as shown in Supplementary Figure 1. This variant has not been previously reported and is defined as a pathogenic variant based on the available evidence.
Clinical follow-up showed that when the patient was diagnosed with MWS (8 months old), he was tested by neuropsychological examination. The results showed that the development quotient of the patient was 55, which was mild intellectual disability. The development quotient of each functional region was shown in Table 1. At this time, the height of the patient was 72cm, her weight was 9.6kg, and head circumference was 42.5cm. At 11 months, the patient showed more severe growth retardation and intellectual disability, and all developmental milestones were delayed. At this time, she could not sit alone very well. The neuropsychological examination of children showed that the development quotient of the patient was 44.1, which was moderate intellectual disability. The development quotient of each functional area was shown in Table 1. At this time, in addition to her previous special appearance, she also developed uplifted earlobes, as shown in Figure 1. We recommended long-term regular rehabilitation training, but the patient did not comply. At the age of 1 year, the patient showed nodding motion. We performed video electroencephalography (EEG) monitoring on the patient, and the results showed abnormal infant EEG. During the follow-up, we monitored whether the child had epileptic symptoms. Fortunately, it did not occur, but the child had high fever and convulsion. At the age of 1 year and 2 months, the intellectual disability of the patient was worse than before, and the developmental quotient was 38.4, which was severe intellectual disability. The developmental quotient of each functional area is shown in Table 1. She was able to stand alone at 2 years of age and walk alone at 2 years and 2 months of age, but she is still nonverbal and has a poor prognosis. At the age of 2 years and 2 months, her weight was 12.2kg, in the 50th percentile of the average weight of infants of the same age.
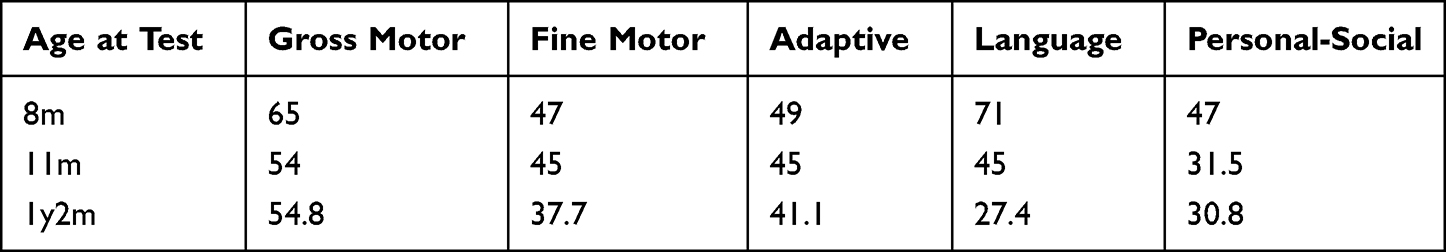 |
Table 1 Results of DQ for Patient 1 |
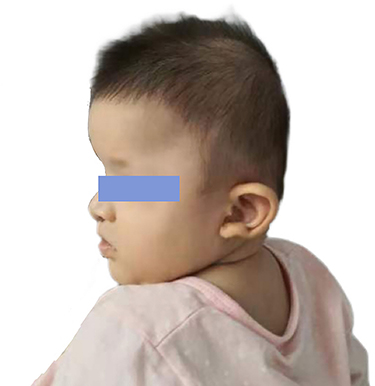 |
Figure 1 Distinctive facial characteristics of Patient 1 with Mowat-Wilson syndrome. The patient shows characteristic facial features of Mowat-Wilson syndrome, including wide eyebrows, a slightly wider distance between the eyes, and a prominent rounded nose tip and uplifted ear lobes with a central depression. |
Patient Two
The patient is the second child of a healthy unrelated couple, and his older brother is healthy. He was born by caesarean section at 37 weeks gestation. The birth weight was 2.65 kg and the birth length was 48 cm. When he was 3 months old, he came to our hospital because he could not lift his head and was prone to crying. At this time, his length was 59.5 cm, weight was 5.8 kg and head circumference was 37.2 cm. As we can see, he has microcephaly (<1%). In terms of facial features, there was no other special manifestation except earlobe bulge with central depression. In addition, his cardiac ultrasound revealed a ventricular septal defect and patent ductus arteriosus. Cryptorchidism was detected. Brain magnetic resonance imaging (MRI) showed agenesis of the corpus callosum. Since he did not pass neonatal hearing screening, transient evoked otoacoustic emission and automatic auditory brainstem response (ABR) tests were performed. However, the results showed bilateral sensorineural deafness.
Through the whole-exome testing, a novel heterozygous variant was identified in this patient (NM_014795: c.1137_1146del TAGTA TGTCT, p. S380Nfs*13), which was not detected in the peripheral blood of the patient’s parents after Sanger verification, so it may be a de novo variant, as shown in Supplementary Figure 2. And this variant has only been previously reported by our group.
Clinical follow-up showed that the patient was initially diagnosed with MWS at 6 months of age. The results of the neuropsychological examination showed that the development quotient of the patient was 60.9, indicating mild mental deficiency. The development quotient of each functional area is shown in Table 2. At this time, the child was just able to lift his head, and he could sit alone at 12 months. At 17 months, a neuropsychological examination was conducted, and the results showed that the development quotient was 46.5, which was a moderate mental deficiency. The development quotient of each area is shown in Table 2. At 18 months, he stood without help, and at 22 months, he walked on a wide base supported by support. At 30 months, his developmental quotient was 27.6, which developed into severe intellectual disability, and the developmental quotient of each energy area decreased greatly, as shown in Table 2. During this period, children still adhere to long-term regular rehabilitation training. The child could walk alone at 3 years old. The developmental quotient was 28.4 at 3 years and 3 months, At the age of 4, the face is shown in Figure 2 and his weight was 16kg, in the 45th percentile of the average weight of children of the same age, and the developmental quotient was 28.6. The development quotient of each energy zone is shown in Table 2. We can see that although the overall development quotient of the child is not obvious, the development quotient of each functional area is improved. Surprisingly, he never had a seizure during follow-up. But his hearing loss did not improve as he grew.
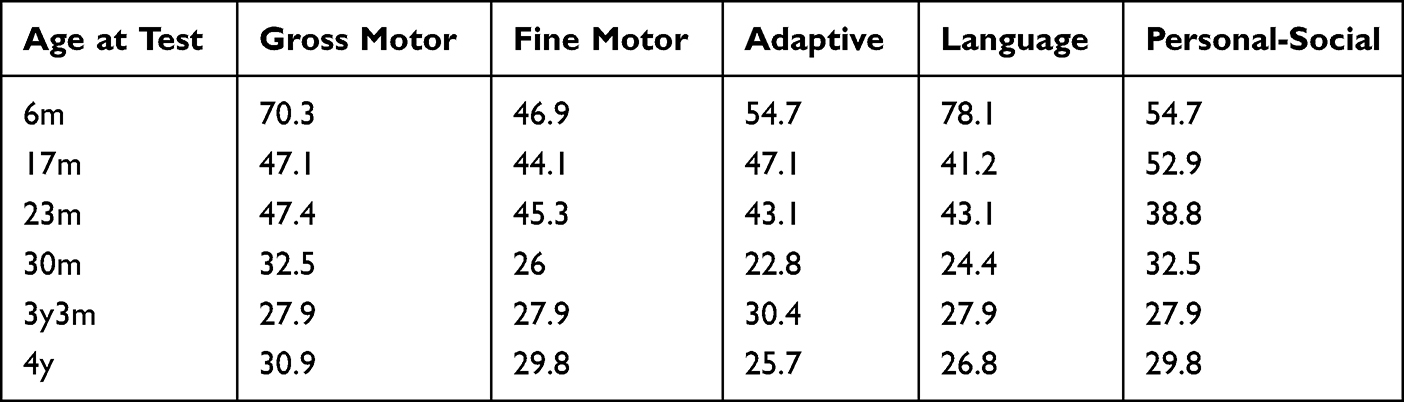 |
Table 2 Results of DQ for Patient 2 |
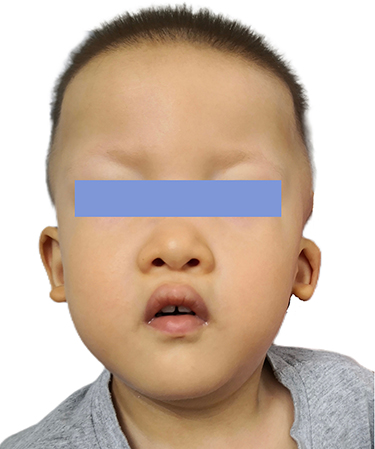 |
Figure 2 Distinctive facial characteristics of Patient 2 with Mowat-Wilson syndrome. The patient shows characteristic facial features of Mowat-Wilson syndrome, including frontal bossing, rounded nasal tip, sparse eyebrows, prominent chin, widely spaced teeth, and uplifted ear lobes with a central depression. |
Patient Three
The patient is the first child of a healthy unrelated couple, she was born by vaginal delivery at 40 weeks of gestation. The birth weight was 3.16 kg and the head circumference was 31cm, slightly smaller than that of normal neonates. The patient was admitted to the hospital for repeated vomiting for 14 hours after birth, but the patient still vomited repeatedly after admission, and the gastric lavage effect was not good. Barium meal angiography combined with clinical consideration of sigmoid colon, descending colon, transverse colon and partial ascending colon megacolon is highly likely, and the diagnosis of congenital megacolon was considered after pediatric surgical consultation. In addition, echocardiography showed patent ductus arteriosus, ventricular septal defect (perimembranous), and atrial septal defect (patent foramen ovale), which indicated congenital heart disease. Brain magnetic resonance imaging (MRI) is consistent with corpus callosum dysplasia. During hospitalization, the child had a lot of eye discharge, and after ophthalmic consultation, it was suggested that the right eye retinal hemorrhage and conjunctivitis were present. Due to the wide eye distance, small head circumference, congenital heart disease and HSCR, the patient underwent genetic testing. The heterozygous variation of ZEB2 gene c.2718delT (p. A907Lfs*23) was found. The variant was not detected in the peripheral blood of the patient’s parents as verified by Sanger, so it may be a de novo variant and was not reported in our reference population gene database.
During the follow-up, we can see that when the child was 9 months old, a neuropsychological examination was performed, and the developmental quotient was 32, indicating severe intellectual disability, including 33 for gross motor development, 33 for fine motor development, 33 for adaptive ability, 22 for language, and 39 for social behavior. The developmental quotient of each functional area is shown in Table 3. We recommended long-term standardized rehabilitation, but the patient did not comply. At present, the child is 3 years old, her weight was 13.6kg, in the 40th percentile of the average weight of infants of the same age. She still could not stand alone and walk alone, only know the simple two-word address. She still displayed persistent growth delay, and the prognosis is not good.
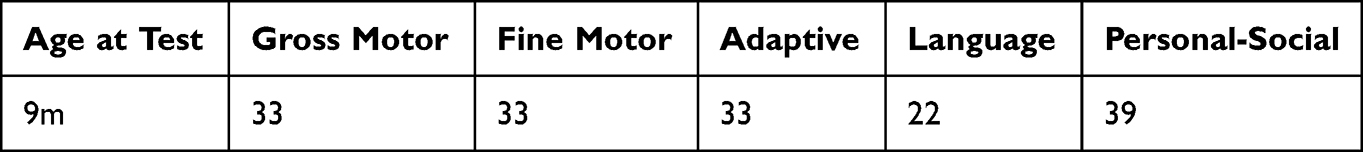 |
Table 3 Results of DQ for Patient 3 |
Discussion
Mowat-Wilson syndrome is a rare congenital malformation syndrome. Due to the diversity of clinical phenotypes, MWS is difficult to be diagnosed clinically. Currently, more than 300 MWS patients have been reported,11–13 and more than 280 ZEB2 variants have been found. However, only 35 cases of MWS and 25 pathogenic ZEB2 variants were reported in China.11,15–18 In this study, we found three new ZBE2 variants in three Chinese patients with MWS, all of whom showed special facial features, intellectual disability, developmental delay, microcephaly, structural brain abnormalities and other abnormalities consistent with MWS.
Hirschsprung disease is one of the most characteristic manifestations of MWS. Hirschacolon incidence has been reported to be 44%,1 However, the rate of HSCR is decreasing as more patients with MWS are diagnosed.19 Among the three patients in this study, only one patient (patient 3) was observed and surgically treated with HSCR, and the other two patients had no history of constipation. Previous studies have shown that the incidence of Hirschsprung’s disease in Chinese MWS patients is low,11 and this study is consistent with previous reports.
Brain structural abnormalities are also common in MWS and can be detected by cranial MRI. Garavelli et al studied the neuropathology of 54 MWS patients and showed that 96% of the patients had brain MRI abnormalities, of which corpus callosum dysplasia was the most common (79.6%). Other common abnormalities included hippocampal abnormalities (77.8%), lateral ventricle enlargement (68.5%), and white matter abnormalities (40.7% thickness reduction). Local signal changes of 22.2%).6,7 In our study, three patients showed abnormal brain MRI, including two with agenesis of corpus callosum (Patients 2 and 3) and one with white matter retardation. This is consistent with previous reports. Because agenesis of the corpus callosum is the only feature of MWS that can be detected antenatal, special attention to facial features during ultrasound or fetal pathology may be helpful in diagnosing MWS in fetuses with agenesis of the corpus callosum.2
Epilepsy is one of the most common neurological problems in MWS patients, accounting for about 80%.20,21 Seizures usually occur in the second year of life, and the average age of seizure onset in 87 MWS patients studied by Ivanovski et al was 27.5 months. It can occur as early as 1 month old and as late as 11 years old.21 Fortunately, only one of the three patients we followed (patient 1) had febrile convulsions, and the other two patients did not have seizure-related symptoms.
According to statistics, all children with MWS present with intellectual disability and overall developmental delay of varying severity.21 At least moderate to severe intellectual disability. Developmental milestones such as sitting and walking are very delayed, with literature reporting that the average age of sitting without support is 20 months and the average age of walking is 4 years and 3 months.2 Our three patients were about 1 year old sitting alone. And all fine motor skill milestones are delayed in MWS patients. Most patients over the age of 20 still need help with dressing and other daily activities. Speech is rarely more than a few words and starts at the average age of four.21 Just like our patient, all three patients were basically speechless, or only known by simple names such as mom and dad. In addition, few previous studies have reported formal intelligence tests and have mostly been limited to imaging changes and rough clinical findings. This study is the first to describe mental development and rehabilitation treatment, which fills the gap of follow-up treatment effect research. At the same time, by comparing the intellectual development level of patient 2 with patients 1 and 3, it can be seen that the prognosis of patients with standardized rehabilitation treatment is better. Although patient 2 was also severely mentally retarded, the development quotient of each functional area of the child was improved through regular rehabilitation treatment. This shows the importance of regular follow-up treatment in the prognosis of children, and is more conducive to promote the healthy growth of children. It also illustrates the difficulty of MWS treatment.
In addition, another common feature of MWS is congenital heart disease,1 Two patients in this study had ventricular septal defects and patent ductus arteriosus. Eye abnormalities can also be detected in WMS.1,2 In our study, one patient developed retinal hemorrhage. About 60% of men with genital abnormalities have hypospadias, and about 40% of men have cryptorchidism.1,22 Only one of our 3 patients was male, and he had cryptorchidism. In addition, our team has reported that MWS patients have symptoms of hearing loss.
In our study, we report three different novel ZEB2 mutations that have not been previously mentioned in the literature. It greatly enriches the mutation spectrum and helps to understand the relationship between clinical phenotypes and genotypes.
Conclusion
Our study expanded the mutation spectrum of ZEB2 and enriched our understanding of the clinical features of MWS. It also shows that long-term standardized treatment is of great significance for the prognosis of patients.
Data Sharing Statement
All data generated or analyzed in this study are included in this published article.
Ethical Approval
All three patients were from The Second Children & Women’s Healthcare of Jinan City. All tests were performed as routine clinical investigations in accordance with the ethical principles of the Declaration of Helsinki. All patients’ guardians provided written informed consent for the publication of images as well as personal and medical information for data analysis and publication. This study was approved by the Medical Ethics Committee of The Second Children & Women’s Healthcare of Jinan City. It agreed to publish.
Acknowledgments
We would like to express our sincere gratitude to our patients and their families for their cooperation.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ayyildiz Emecen D, Isik E, Utine GE, et al. Clinical and molecular spectrum of four patients diagnosed with Mowat-Wilson syndrome. Mol Syndromol. 2020;11(5–6):296–301. doi:10.1159/000511609
2. Garavelli L, Mainardi PC. Mowat-Wilson syndrome. Orphanet J Rare Dis. 2007;2(1):42. doi:10.1186/1750-1172-2-42
3. Ho S, Luk HM, Chung BH, et al. Mowat-Wilson syndrome in a Chinese population: a case series. Am J Med Genet A. 2020;182(6):1336–1341. doi:10.1002/ajmg.a.61557
4. Liu F, Wu Y, Li Z, et al. Identification of MMACHC and ZEB2 mutations causing coexistent cobalamin C disease and Mowat-Wilson syndrome in a 2-year-old girl. Clin Chim Acta. 2022;533:31–39. doi:10.1016/j.cca.2022.06.004
5. Birkhoff JC, Huylebroeck D, Conidi A. ZEB2, the Mowat-Wilson syndrome transcription factor: confirmations, novel functions, and continuing surprises. Genes. 2021;12(7):1037. doi:10.3390/genes12071037
6. Garavelli L, Ivanovski I, Caraffi SG, et al. Neuroimaging findings in Mowat–Wilson syndrome: a study of 54 patients. Genet Med. 2017;19(6):691–700. doi:10.1038/gim.2016.176
7. Wakamatsu N, Yamada Y, Yamada K, et al. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nat Genet. 2001;27(4):369–370. doi:10.1038/86860
8. Cacheux V, Dastot-le MF, Kaariainen H, et al. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum Mol Genet. 2001;10(14):1503–1510. doi:10.1093/hmg/10.14.1503
9. Mowat DR, Croaker GD, Cass DT, et al. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J Med Genet. 1998;35(8):617–623. doi:10.1136/jmg.35.8.617
10. Ghoumid J, Drevillon L, Alavi-Naini SM, et al. ZEB2 zinc-finger missense mutations lead to hypomorphic alleles and a mild Mowat-Wilson syndrome. Hum Mol Genet. 2013;22(13):2652–2661. doi:10.1093/hmg/ddt114
11. Fu Y, Xu W, Wang Q, et al. Three Novel De Novo ZEB2 variants identified in three unrelated Chinese patients with Mowat-Wilson syndrome and a systematic review. Front Genet. 2022;13:853183.
12. Wei L, Han X, Li X, et al. A Chinese Boy with Mowat-Wilson Syndrome Caused by a 10 bp Deletion in the ZEB2 gene. Pharmgenomics Pers Med. 2021;14:1041–1045. doi:10.2147/PGPM.S320128
13. Zou D, Wang L, Wen F, et al. Genotype-phenotype analysis in Mowat-Wilson syndrome associated with two novel and two recurrent ZEB2 variants. Exp Ther Med. 2020;20(6):263. doi:10.3892/etm.2020.9393
14. Moore SW, Fieggen K, Honey E, et al. Novel Zeb2 gene variation in the Mowat Wilson syndrome (MWS). J Pediatr Surg. 2016;51(2):268–271. doi:10.1016/j.jpedsurg.2015.10.070
15. Ju Y, Ji TY. Clinical characteristics of epilepsy in 5 patients with Mowat-Wilson syndrome. Chin J Pediatr. 2022;60(6):5.
16. She MC, Zhao ZH, Shi PL, et al. Genetic analysis of a patient with Mowat-Wilson syndrome caused by nonsense mutation of ZEB2 gene. Chin J Med Genet. 2022;39(8):889–892. doi:10.3760/cma.j.cn511374-20210510-00393
17. Zhou TC, Wang YC, Liang D, et al. Clinical characteristics and gene analysis of three children with Mowat-Wilson syndrome. Chin J Med Genet. 2022;39(9):944–948. doi:10.3760/cma.j.cn511374-20210829-00703
18. Wang H, Yan YC, Li X, et al. Clinical characteristics and genetic analysis of 3 children with Mowat-Wilson syndrome. Chin J Contemp Pediatr. 2019;21(05):468–473. doi:10.7499/j.issn.1008-8830.2019.05.014
19. Coyle D, Puri P. Hirschsprung’s disease in children with Mowat-Wilson syndrome. Pediatr Surg Int. 2015;31(8):711–717. doi:10.1007/s00383-015-3732-x
20. Cordelli DM, Garavelli L, Savasta S, et al. Epilepsy in Mowat-Wilson syndrome: delineation of the electroclinical phenotype. Am J Med Genet A. 2013;161A(2):273–284. doi:10.1002/ajmg.a.35717
21. Ivanovski I, Djuric O, Caraffi SG, et al. Phenotype and genotype of 87 patients with Mowat–Wilson syndrome and recommendations for care. Genet Med. 2018;20(9):965–975. doi:10.1038/gim.2017.221
22. Adam MP, Conta J, Bean L. Mowat-Wilson syndrome; 1993.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.