")
Back to Journals » The Application of Clinical Genetics » Volume 11
Clinical and molecular genetic features of Hb H and AE Bart’s diseases in central Thai children
Authors Traivaree C , Boonyawat B , Monsereenusorn C , Rujkijyanont P , Photia A
Received 30 December 2017
Accepted for publication 10 February 2018
Published 3 April 2018 Volume 2018:11 Pages 23—30
DOI https://doi.org/10.2147/TACG.S161152
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Chanchai Traivaree,1,* Boonchai Boonyawat,2,* Chalinee Monsereenusorn,1 Piya Rujkijyanont,1 Apichat Photia1
1Division of Hematology/Oncology, Department of Pediatrics, Phramongkutklao Hospital and College of Medicine, Bangkok, Thailand; 2Division of Genetics, Department of Pediatrics, Phramongkutklao Hospital and College of Medicine, Bangkok, Thailand
*These authors contributed equally to this work
Background: α-Thalassemia, one of the major thalassemia types in Thailand, is caused by either deletion or non-deletional mutation of one or both α-globin genes. Inactivation of three α-globin genes causes hemoglobin H (Hb H) disease, and the combination of Hb H disease with heterozygous hemoglobin E (Hb E) results in AE Bart’s disease.
Objective: This study aimed to characterize the clinical and hematological manifestations of 76 pediatric patients with Hb H and AE Bart’s diseases treated at Phramongkutklao Hospital, a tertiary care center for thalassemia patients in central Thailand.
Patients and methods: Seventy-six unrelated pediatric patients, 58 patients with Hb H disease and 18 patients with AE Bart’s disease, were enrolled in this study. Their clinical presentations, transfusion requirement, laboratory findings, and mutation analysis were retrospectively reviewed and analyzed.
Results: A total of 76 pediatric patients with Hb H and AE Bart’s diseases who mainly lived in central Thailand were included in this study. The clinical severities of patients with non-deletional mutations were more severe than those with deletional mutations. Eighty-six percent of patients with non-deletional AE Bart’s disease required more blood transfusion compared to 12.5% of patients with deletional AE Bart’s disease. Non-deletional AE Bart’s disease also had a history of urgent blood transfusion with the average of 6±0.9 times compared to 1±0.3 times in patients with deletional Hb H disease. The difference was statistically significant.
Conclusion: This study revealed the differences in clinical spectrum between patients with Hb H disease and those with AE Bart’s disease in central Thailand. The differentiation of α-thalassemia is essential for appropriate management of patients. The molecular diagnosis is useful for diagnostic confirmation and genotype–phenotype correlation.
Keywords: genotype, phenotype, Hb H disease, AE Bart’s disease, Thai children
Introduction
Thalassemia is the most common inherited blood disorder in Southeast Asia, which is caused by reduced or absent synthesis of the globin chains of hemoglobin leading to imbalance of the globin chains. α-Thalassemia is one of the major thalassemia types and is caused by mutation in either α1-globin gene (HBA1) or α2-globin gene (HBA2) on chromosome 16.1 The genetic mutation results in clinical variable depending on the decreased or absent α-globin chain. In Thailand, the most common type of α-globin gene mutation is the deletion of two α-globin genes (α-thalassemia 1) specifically named as Southeast Asian (SEA) type deletion (--SEA), followed by the deletion of one α-globin (α-thalassemia 2) including 3.7 kb or rightward deletion (-α3.7) and 4.2 kb or leftward deletion (-α4.2), respectively. The most common non-deletional α-globin gene mutation in Thailand is hemoglobin Constant Spring (Hb CS, ααCS). Hemoglobin E (Hb E) which is one of the most common HBB gene mutations in Southeast Asia can present with either hemoglobinopathy or thalassemic phenotype associated with diverse clinical manifestations.
The clinical and hematological manifestation of α-thalassemia is variable ranging from silent carrier to fatal Hb Bart’s hydrops fetalis syndrome. Interaction of α-thalassemia 1 and α-thalassemia 2 causes hemoglobin H (Hb H) disease and interaction of Hb H disease with heterozygous Hb E results in AE Bart’s disease.2,3 Hb H is characterized into two main forms including deletional and non-deletional Hb H diseases. Deletional Hb H disease is caused by a combination of deletion removing both α-globin genes on one chromosome 16 and deletion removing only single α-globin gene on the other chromosome 16. Non-deletional Hb H disease results from a combination of deletion removing both α-globin genes on one chromosome 16 and point mutation or small insertion/deletion involving either the HBA1 or HBA2 gene on the other chromosome 16. The correlation between clinical phenotypes, hematological parameters, α-globin genotypes, and laboratory biomarkers in different populations had been studied.4–6
The aim of this study was to investigate phenotypic manifestations of these α-globin gene mutations by studying their hematologic parameters, hemoglobin typing, age at presentation, transfusion requirement, age and height in both Hb H disease and AE Bart’s disease forms of α-thalassemia. The genotype–phenotype correlation of each mutation including laboratory biomarkers and genetic profiles in children with Hb H disease and AE Bart’s disease treated at Phramongkutklao Hospital, a tertiary care center for thalassemia patients in central Thailand, was presented.
Patients and methods
Patient selection
Seventy-six α-thalassemia patients, who were treated at the Hematology Clinic, Department of Pediatrics, Phramongkutklao Hospital, Bangkok, Thailand, were enrolled in this study. Written informed consent and assent form were obtained from all participants as well as parents or guardians of the children prior to the enrollment in the study. The study protocol was approved by the Institutional Review Board of Phramongkutklao Hospital and College of Medicine, Bangkok, Thailand, following the ethical principles of the Declaration of Helsinki of 1975 and its revision. The inclusion criteria include patients who were diagnosed with Hb H disease and AE Bart’s disease at ≤18 years of age and written informed consent and assent to participate in our study. The patients who had incomplete data including hematological data, hemoglobin typing, and α-globin genes mutation analysis from medical records were excluded from the study.
A total of 58 patients with Hb H disease and 18 with AE Bart’s disease were included. The majority of patients came from the central part of Thailand. Patients with α-thalassemia were clinically classified into transfusion dependent and non-transfusion dependent. Patients were also examined for growth parameters. Patients with α-thalassemia were diagnosed on the basis of age at first transfusion, transfusion requirement, physical examination, and also on the basis of tests including hematological data, laboratory biomarkers, hemoglobin typing, and α-globin genes mutation analysis. A complete clinical history was recorded along with growth parameters. Hematological data, laboratory biomarkers, and hemoglobin typing were obtained retrospectively from medical records.
Hematological and biochemical parameters
Hematological analyses were carried out using Coulter HMX Automated Hematology Analyzer (Beckman Coulter Corporation, Miami, FL, USA). Hemoglobin profiles and fetal hemoglobin (HbF) concentrations were determined using capillary electrophoresis (CE; Minicap system, Sebia, Parc Technologique Leonard de Vinci, France).
Genetic analysis
A total of 76 peripheral blood EDTA samples from all individuals were collected. Genomic DNA was extracted from peripheral blood lymphocytes using commercially available kits according to manufacturer’s protocol. Molecular analysis for HBA1 and HBA2 mutation was performed.7,8 In brief, multiplex gap-polymerase chain reaction (PCR) was first used to characterize common α-globin gene deletions in Southeast Asian populations including α-thalassemia 1 [SEA (--SEA) and THAI (--THAI) deletion] and α-thalassemia 2 [3.7 kb (-α3.7) and 4.2 kb (-α4.2) deletion]; multiplex-amplification refractory mutation system was performed to detect common non-deletional α-globin gene mutations including Hb CS and hemoglobin Paksé (Hb PS);9 and direct DNA sequencing of all coding regions and exon–intron boundaries of both genes was finally used to detect unknown non-deletional α-globin gene mutations.10
Statistical analysis
The estimated number of participants was calculated by setting an α error of 0.05, prevalence 0.0611 and set at 80 patients. Baseline values of the selected variables were calculated as mean, median, and average according to percentile. Distribution of the quantitative variables was analyzed using the Shapiro–Wilk test. Continuous variables were compared between two groups using the unpaired t-test for data with a parametric distribution and the Mann–Whitney test for non-parametric distribution. The Chi-square test and Fisher’s exact test were used to analyze the categorical variables for data with a parametric distribution and non-parametric distribution, respectively. Statistical Package for the Social Science (SPSS) version 23 software (IBM Corporation, Armonk, NY, USA) was used for statistical analysis, and p-value <0.05 was considered to be statistically significant.
Results
Characteristics of patients
Seventy-six α-thalassemia patients treated at Phramongkutklao Hospital were retrospectively reviewed and analyzed. Characteristics of patients are listed in Table 1. Fifty-eight patients were diagnosed with Hb H disease (31 males [53.4%] and 27 females [46.6%]). The remaining 18 patients (7 males [38.9%] and 11 females [61.1%]) were diagnosed with AE Bart’s disease. The mean age at diagnosis of patients with Hb H disease and those with AE Bart’s disease was 12.5±5.3 years and 10.7±5.6 years, respectively. The mean weight and height were 36.4±16.7 kg and 140±30 cm for patients with Hb H disease and 30.2±15.2 kg and 130±21 cm for patients with AE Bart’s disease. The mean body mass indexes (BMIs) of patients with Hb H disease and those with AE Bart’s disease were 18.2±3.6 kg/m2 and 17.6±3.8 kg/m2, respectively. There were no statistical differences in gender, mean age at diagnosis, weight, height, and BMIs between patients with Hb H disease and those with AE Bart’s disease. Most of the patients with Hb H disease (55 patients; 94.8%) resided in central Thailand. Only patients with AE Bart’s disease (5 patients; 27.8%) resided in northeastern Thailand.
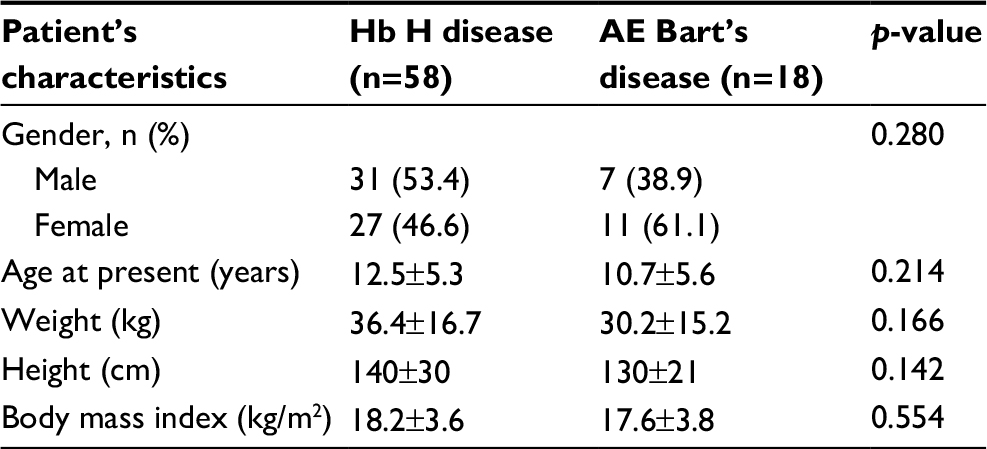 | Table 1 Characteristics of patients (n=76) Notes: Data are shown as mean±SD or number (%). p-value was obtained from the unpaired t-test or chi-square test for data with a parametric distribution and the Mann–Whitney test or Fisher’s exact test for non-parametric distribution. p<0.05 is statistically significant. Abbreviation: Hb H, hemoglobin H. |
Hematologic and hemoglobin typing data
Clinical characteristics and laboratory findings of the patients with Hb H disease were analyzed and compared with those of patients with AE Bart’s disease, as described in Table 2. Among various hematologic data, only mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) showed statistical difference in patients with Hb H disease (60.1±9.1 fL and 17.8±2.7 pg) compared to those with AE Bart’s disease (51.6±6.3 fL and 15.9±1.9 pg) (p<0.001 and 0.009, respectively).
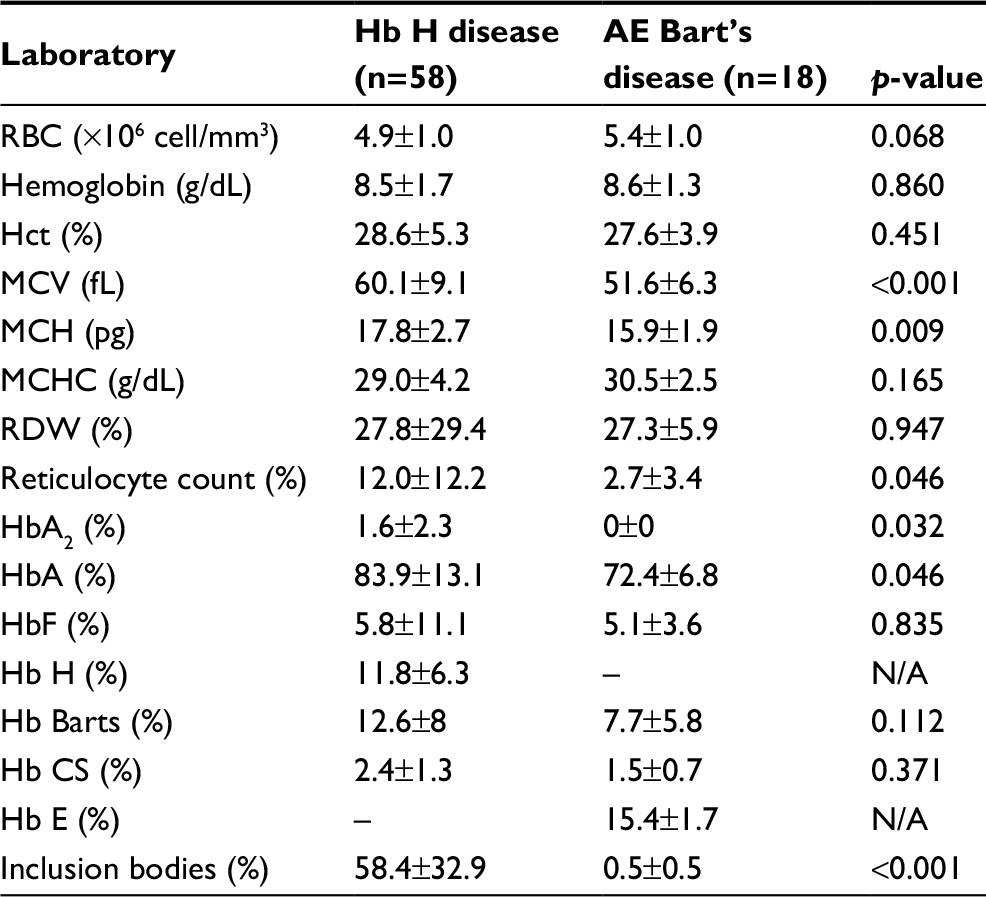 | Table 2 Hematologic findings and hemoglobin typing of Hb H and AE Bart’s disease (n=76) Notes: Data are shown as mean±SD or number (%). p-value was obtained from the t-test or chi-square test for data with a parametric distribution and the Mann–Whitney test or Fisher’s exact test for non-parametric distribution. p<0.05 is statistically significant. Abbreviations: CS, Constant Spring; Hb E, hemoglobin E; Hb H, hemoglobin H; Hct, hematocrit; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; N/A, not available; RBC, red blood cells; RDW, red cell distribution width.. |
Reticulocyte count, HbA2, and HbA were significantly higher in patients with Hb H disease compared to those with AE Bart’s disease (p <0.05), as described in Table 2. In addition, inclusion bodies that are dense blue-stained particles in red blood cells containing protein, ribosomal components, or DNA/RNA fragments were found to be significantly higher in patients with Hb H disease (58.4±32.9%) than in patients with AE Bart’s disease (0.5±0.5%) (p<0.001).
Genotypic data
α-Globin gene mutation analysis was performed in all patients and revealed seven different mutations (Table 3). Deletional and non-deletional mutations were found to be equal in patients with Hb H disease. Nevertheless, non-deletional mutations were frequently found in patients with AE Bart’s disease (55.6% vs 44.4%).
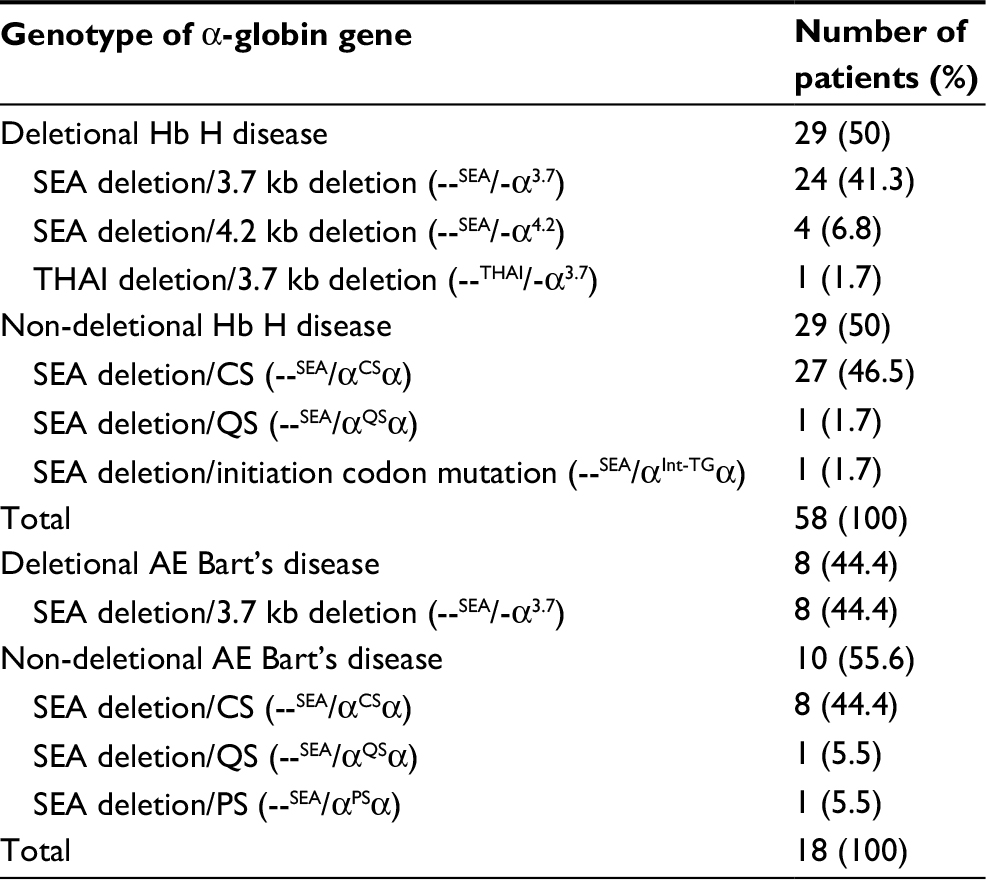 | Table 3 Distribution of detected α-globin gene mutations in patients (n=76) Note: 3.7 kb deletion, rightward deletion of 3.7 kb of the α2 gene heterozygous; 4.2 kb deletion, leftward deletion of 4.2 kb of the α2 gene heterozygous. Abbreviations: CS, Constant Spring; Hb H, hemoglobin H; PS, Paksé; QS, Quong Sze; SEA, Southeast Asian type. |
Among 29 patients with deletional Hb H disease, SEA deletion/3.7 kb deletion (--SEA/-α3.7) mutation was most commonly found in 24 patients (41.3%) followed by SEA deletion/4.2 kb deletion (--SEA/-α4.2) mutation (6.8%) and THAI deletion/3.7 kb deletion (--THAI/-α3.7) mutation (1.7%). SEA deletion/CS (--SEA/αCSα) was the most common mutation among non-deletional Hb H disease found in 27 patients (46.5%) followed by SEA deletion/Quong Sze (QS) (--SEA/αQSα) mutation (1.7%) equally with SEA deletion/initiation codon (--SEA/αInt-TGα) mutation.
For AE Bart’s disease, SEA deletion/3.7 kb deletion (--SEA/-α3.7) mutation was found in all patients with deletional AE Bart’s disease, whereas for non-deletional AE Bart’s disease, SEA deletion/CS (--SEA/αCSα) mutation was most commonly found (44.4%) followed by SEA deletion/QS (--SEA/αQSα) mutation equally with SEA deletion/PS (--SEA/αPSα) mutation (5.5%).
SEA (--SEA) mutation was the most common deletional α-globin mutation in α-thalassemia 1 (98.6%) followed by THAI (--THAI) mutation (1.4%). In α-thalassemia 2, 3.7 kb deletion (-α3.7) mutation was most commonly found (89.2%) followed by 4.2 kb deletion (-α4.2) mutation (10.8%). Hb CS was the most common non-deletional α-globin mutation in α-thalassemia 2 (89.7%). The less common non-deletional α-globin mutations included hemoglobin QS (Hb QS), Hb PS, and initiation codon mutation, respectively.
Genotypic and phenotypic correlation
Clinical and baseline laboratory parameters in patients with Hb H disease were reviewed (Table 4). Baseline complete blood count (CBC) in patients with Hb H disease was reviewed, and hemoglobin level was found to be significantly lower in patients with non-deletional Hb H disease (7.9±1.4 g/dL) compared to hemoglobin level in patients with deletional Hb H disease (9.0±1.8 g/dL) (p=0.011). In addition, patients with non-deletional Hb H disease was noted to have significantly higher MCV (64.3±7.3 fL) compared to patients with deletional Hb H disease (55.9±8.8 fL) (p<0.001).
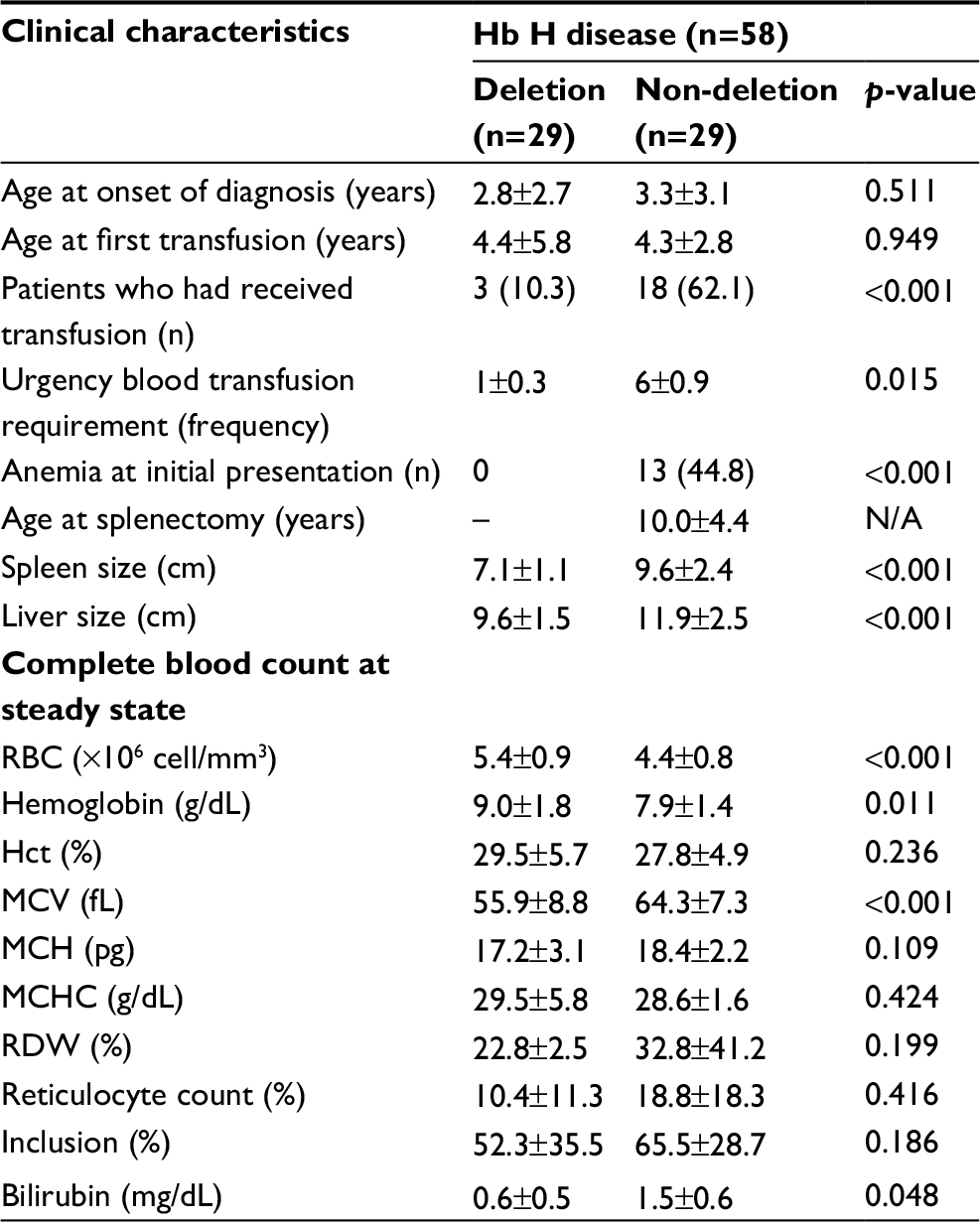 | Table 4 Clinical characteristics of Hb H (n=58) Notes: Data shown as mean±SD or number (%). p-value was obtained from the unpaired t-test or chi-square test for data with a parametric distribution and the Mann–Whitney test or Fisher’s exact test for non-parametric distribution. p<0.05 is statistically significant. Abbreviations: Hb H, hemoglobin H; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; N/A, not available. |
Clinical and baseline laboratory parameters in patients with AE Bart’s disease were reviewed as described in Table 5. Characteristics of patients with AE Bart’s disease showed that 7 out of 10 patients with non-deletional AE Bart’s disease (70%) required blood transfusion compared to 1 out of 8 patients with deletional AE Bart’s disease (12.5%) (p=0.025).
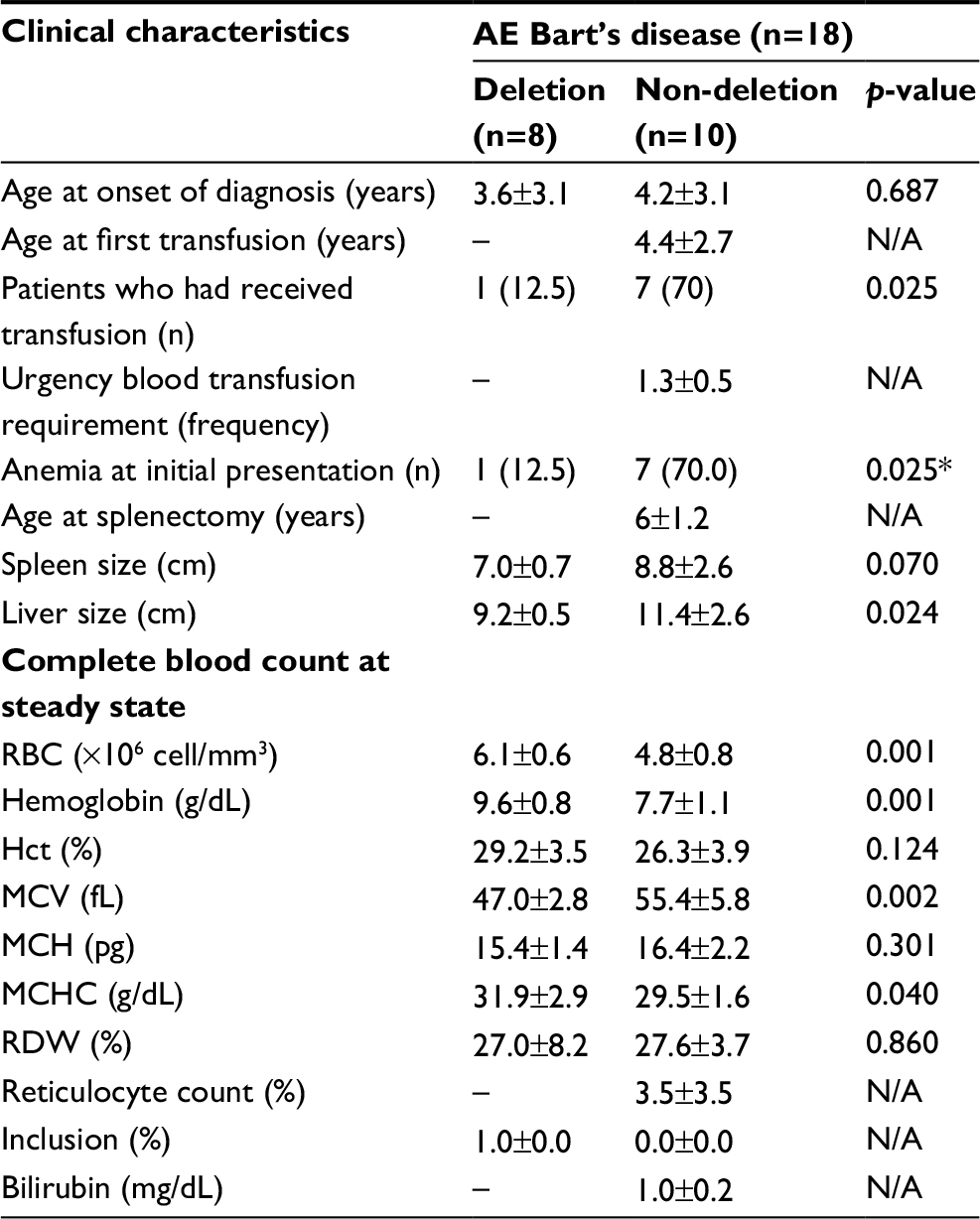 | Table 5 Clinical characteristics of AE Bart’s disease (n=18) Notes: Data are shown as mean±SD or number (%). p-value was obtained from the unpaired t-test or chi-square test for data with a parametric distribution and the Mann–Whitney test or Fisher’s exact test for non-parametric distribution. p<0.05 is statistically significant. Abbreviations: Hb H, hemoglobin H; Hct, hematocrit; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; N/A, not available; RBC, red blood cells; RDW, red cell distribution width. |
Baseline CBC and hemoglobin level were found to be significantly lower in patients with non-deletional AE Bart’s disease (7.7±1.1 g/dL) compared to patients with deletional AE Bart’s disease (9.6±0.8 g/dL) (p=0.001). In addition, patients with non-deletional AE Bart’s disease were noted to have significantly higher MCV (55.4±5.8 fL) compared to patients with deletional AE Bart’s disease (47.0±2.8 fL) (p=0.002).
Among all 58 patients with Hb H disease and AE Bart’s disease enrolled in this study, 19 patients (25%) were categorized into the transfusion-dependent thalassemia (TDT) group in which they required regular blood transfusion to keep the hemoglobin level between 9.5 and 10.5 g/dL along with iron chelation therapy in those with iron overload. Fifty-seven out of 76 patients (75%) were categorized into the non-transfusion-dependent thalassemia (NTDT) group in which they usually had mild anemia and had average hemoglobin level between 7 and 10 g/dL and might require intermittent blood transfusion when their hemoglobin decreased from some specific reasons such as infections. Growth parameters were also reviewed and analyzed between TDT and NTDT patients. There were no statistical differences in height (p=0.41), average weight (p=0.37), and BMI (p=0.49) between the two groups.
Discussion
α-Thalassemia is one of the most common genetic disorders in Thailand. Molecular pathology of disease can be due to the deletion of one α-globin gene called α-thalassemia 2 or -α, the deletion of two α-globin genes called α-thalassemia 1, or the specific mutation of α-globin gene (αTα or ααT). Lal et al12 evaluated 86 patients with Hb H disease and reported that 60 of those patients (70%) had deletional α-globin mutation, 23 patients (27%) had non-deletional Hb CS (ααCS), and the remaining three patients (3%) had other specific non-deletional α-globin mutations.
The occurrence of α-thalassemia 1 and α-thalassemia 2 contributes to Hb H disease, which is common in Southeast Asia where there is a high prevalence of SEA deletion α-thalassemia 1 and 3.7 kb deletion α-thalassemia 2. In addition, b-globin gene mutation, especially βE, is also common in Southeast Asia and accounted for 20%–50% in Thai population.13 Therefore, it is not uncommon to witness AE Bart’s disease patients having both Hb H disease and heterozygous Hb E. Genetic analysis was performed to identify α-globin gene mutation in patients with Hb H disease, and the results showed that the most common α-globin gene mutation in patients with Hb H disease was SEA deletion α-thalassemia 1 with Hb CS (ααCS) (--SEA/αCSα) that accounted for 50% of patients, followed by SEA deletion α-thalassemia 1 with 3.7 kb or rightward deletion α-thalassemia 2 (--SEA/-α3.7) that accounted for 41.3% of patients.14
In our study, genetic analysis was also performed in 18 patients with AE Bart’s disease. Among those 18 AE Bart’s disease patients, non-deletional α-globin mutation (55.6%) was found to be more common than deletional α-globin mutation (44.4%). The most common type of non-deletional AE Bart’s disease was SEA deletion α-thalassemia 1 with Hb CS [(--SEA/αCSα), βE/β] (44.4%). The most common type of deletional AE Bart’s disease was SEA deletion α-thalassemia 1 with 3.7 kb or rightward deletion α-thalassemia 2 [(--SEA/-α3.7), βE/β] accounting for 44.4% of AE Bart’s disease patients, as previously published by Cai et al.15
The diversity in α-globin gene mutations in patients with Hb H disease and AE Bart’s disease was largely resulting from geographical differences in the population in Thailand. In this study, most of our patients treated at Phramongkutklao Hospital resided in central Thailand. Although Thai type α0-thalassemia was initially found in Thai patients with Hb H disease, the mutation was only reported in 1 out of 58 Hb H patients (1.7%) in this study. This mutation is ATG>_TG (HBA2:c.1delA), which is the initiation codon mutation that possibly affects the downstream α-globin gene expression. Recently, it has been reported in Thai population.16 Our patient may be one of those cases who carries this rare mutation. In our study, comparison of clinical spectrum and severity between patients with Hb H disease and AE Bart’s disease was evaluated. Patients with Hb H disease had higher MCV (60.1±9.1 fL) than patients with AE Bart’s disease (51.6±6.3 fL) as shown in the study by Boonsa et al,9 who reported average MCV of patients with Hb H disease (59.6 fL) being higher than those with AE Bart’s disease (52.6 fL). The pathophysiology of low MCV is from oxidative damage secondary to free globin chains from abnormality of cell membrane in patients with thalassemia.
Hemoglobin typing was also performed on those patients with Hb H disease and AE Bart’s disease. Hb H was found in patients with Hb H disease, but not in patients with AE Bart’s disease. The pathogenesis of Hb H disease was from reduced or absent α-globin mRNA and α-globin chain leading to the α/β globin mRNA ratio being <0.5 and the α/β globin chain synthetic ratio in the range of 0.2–0.7. During the newborn period, overproduced g-globin chains will form g4 tetramers (hemoglobin Bart). In adults, overproduced b-globin chains will form β4 tetramers (Hb H), which can be detected by hemoglobin typing assay in very small numbers. Therefore, in the case of patients with AE Bart’s disease who had additional βE heterozygous, the chance of detecting Hb H from hemoglobin typing assay will be very low and may be undetectable, especially using less sensitivity assay such as low-pressure liquid chromatography (LPLC). In addition, Hb H (β4) is unstable and has high affinity to oxygen causing less oxygen being transported to tissues. Therefore, erythrocytes produced in patients with Hb H disease will be unstable, broken easily and have short life-span. Furthermore, erythrocyte membrane in patients with Hb H disease and Hb CS is rigid and less flexible when passing through microcirculation causing the cell to be damaged. Accordingly, patients with Hb H disease and Hb CS will have clinical severity more than patients with Hb H disease alone.17
The differences in clinical spectrum between patients with Hb H disease and patients with AE Bart’s disease were observed in this study. The clinical severity of patients with non-deletional disease was generally more severe than clinical severity of patients with deletional disease as reported in the study by Bowden et al.18 Due to less clinical severity of the patients with deletional α-thalassemia disease including Hb H disease and AE Bart’s disease, some patients might be asymptomatic and therefore will not be diagnosed until adulthood when having infections causing severe hemolysis. The variation in clinical spectrum in those patients generally resulted from the different amounts of α-globin production in patients with deletional and non-deletional Hb H disease. α2-Globin gene can function and produce α-globin chain almost 3/4 of total α-globin production. This is more than α-globin production by α1-globin gene, which can produce α-globin chain only 1/4 of total α-globin production.19
Most of the genotype abnormalities of deletional Hb H disease in this study, including SEA deletion α-thalassemia 1 with 3.7 kb or rightward deletion α-thalassemia 2 (--SEA/-α3.7), SEA deletion α-thalassemia 1 with 4.2 kb or leftward deletion α-thalassemia 2 (--SEA/-α4.2), and THAI deletion α-thalassemia 1 with 3.7 kb or rightward deletion α-thalassemia 2 (--THAI/-α3.7), had remaining functional α-globin gene (α1-globin gene or α2-globin gene) which can produce normal α-globin chain; therefore, the patients with deletional Hb H disease had no significant differences in their clinical severity. When patients with deletional Hb H disease are compared with patients with non-deletional Hb H disease, clinical severity of patients with non-deletional Hb H disease specifically Hb CS was more prominent than patients with deletional Hb H disease. The remaining α-globin gene (α2-globin gene) in patients with deletional Hb H disease had the capability to produce more α-globin chain than α1-globin gene, thereby causing imbalance between α-globin chain and accordingly b-globin chain being less severe than those from patients with non-deletional Hb H disease. This could be the reason of less clinical severity in patients with deletional Hb H disease compared to patients with non-deletional Hb H disease as described in the study by Bowden et al.18
In our study, four patients underwent splenectomy. Three patients had non-deletional Hb H disease and one patient had non-deletional AE Bart’s disease. All patients also had Hb CS. Sripichai et al20 also reported that patients with non-deletional Hb CS had more clinical severity due to the unstable α-globin chain produced from mutated α-globin gene, which causes increased breakage of red blood cells leading to increased spleen size and requirement for blood transfusion.
In general, thalassemia can be divided into two groups: TDT and NTDT. TDT patients usually are anemic and do require regular blood transfusion to maintain hemoglobin level between 9.5 and 10.5 g/dL, with iron chelation administration in the case of developing iron overload. In our study, 19 patients were classified as TDT, which accounted for 25% of total patients. NTDT patients usually are less anemic with an average hemoglobin level of 7–10 g/dL and do not require regular blood transfusion. Fucharoen et al21 and Galanello et al22 reported that adult patients with a deletion Hb H disease also had less transfusion requirements than those with a non-deletional Hb H disease which higher serum ferritin levels than those with the deletional type of disease. Our study also showed increased serum ferritin level though the patients did not receive blood transfusion. These mechanisms may be explained due to the increased absorption of dietary iron, ineffective erythropoiesis, and higher proportions of patients with transfusion therapy.23 These patients are recommended to be closely monitored for tissue damage due to iron overload and iron chelation therapy.
Conclusion
α-Thalassemia is a very heterogeneous disease in terms of presentation, and the genotype–phenotype correlation is not clear. The molecular characterization as performed in this study is useful not only for diagnostic confirmation but also for carrier detection and genotype–phenotype correlation for both α-thalassemia and complex αβ thalassemia syndrome.
The differentiation of α-thalassemia is essential for appropriate management of patients. The accurate diagnosis of patient with mild clinical characteristics would avoid unnecessary transfusions and their complications.
Acknowledgment
This study was approved and funded by the Phramongkutklao College of Medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155–167. | ||
Fucharoen S, Viprakasit V. Hb H disease: clinical course and disease modifiers. Hematology. 2009;2009(1):26–34. | ||
Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood. 2003;101(3):791–800. | ||
Origa R, Barella S, Paglietti ME, et al. Hematological phenotypes in children according to the alpha-globin genotypes. Blood Cells Mol Dis. 2017;69:102–106. | ||
Nuinoon M, Jeenduang N, Kesornsit A, Horpet D, Plyduang T. Hematological and molecular characterization of a novel Hb A2 variant with homozygous alpha-thalassemia-2 in a Southern Thai individual. Hemoglobin. 2017;41(3):213–215. | ||
Charoenkwan P, Wanapirak C, Thanarattanakorn P, et al. Hemoglobin E levels in double heterozygotes of hemoglobin E and SEA-type alpha-thalassemia. Southeast Asian J Trop Med Public Health. 2005;36(2):467–470. | ||
Chong SS, Boehm CD, Higgs DR, Cutting GR. Single-tube multiplex-PCR screen for common deletional determinants of alpha-thalassemia. Blood. 2000;95(1):360–362. | ||
Boonyawat B, Photia A, Monsereenusorn C, Rujkijyanont P, Traivaree C. Molecular characterization of Hb H and AEBart’s diseases in Thai children: Phramongkutklao Hospital experiences. J Med Assoc Thai. 2017;100(2):167–174. | ||
Boonsa S, Sanchaisuriya K, Fucharoen G, Wiangnon S, Jetsrisuparb A, Fucharoen S. The diverse molecular basis and hematological features of Hb H and AE Bart’s diseases in Northeast Thailand. Acta Haematol. 2004;111(3):149–154. | ||
Chan V, Chan VW, Tang M, Lau K, Todd D, Chan TK. Molecular defects in Hb H hydrops fetalis. Br J Haematol. 1997;96(2):224–228. | ||
Wasi P, Na-Nakorn S, Pootrakul P, Panich V. Incidence of haemoglobin Thai: a re-examination of the genetics of alpha-thalassaemic diseases. Ann Hum Genet. 1972;35(4):467–470. | ||
Lal A, Goldrich ML, Haines DA, Azimi M, Singer ST, Vichinsky EP. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med. 2011;364(8):710–718. | ||
Fucharoen S, Winichagoon P, Pootrakul P, Piankijagum A, Wasi P. Differences between two types of Hb H disease, alpha-thalassemia 1/alpha-thalassemia 2 and alpha-thalassemia 1/Hb constant spring. Birth Defects Orig Artic Ser. 1987;23(5A):309–315. | ||
Nguyen VH, Sanchaisuriya K, Wongprachum K, et al. Hemoglobin Constant Spring is markedly high in women of an ethnic minority group in Vietnam: a community-based survey and hematologic features. Blood Cells Mol Dis. 2014;52(4):161–165. | ||
Cai R, Li L, Liang X, et al. [Prevalence survey and molecular characterization of alpha and beta thalassemia in Liuzhou city of Guangxi]. Zhonghua Liu Xing Bing Xue Za Zhi. 2002;23(4):281–285. | ||
Viprakasit V, Ekwattanakit S, Chalaow N, et al. Clinical presentation and molecular identification of four uncommon alpha globin variants in Thailand. Initiation codon mutation of alpha2-globin Gene (HBA2:c.1delA), donor splice site mutation of alpha1-globin gene (IVSI-1, HBA1:c.95 + 1G>A), hemoglobin Queens Park/Chao Pra Ya (HBA1:c.98T>A) and hemoglobin Westmead (HBA2:c.369C>G). Acta Haematol. 2014;131(2):88–94. | ||
Viprakasit V, Tyan P, Rodmai S, Taher AT. Identification and key management of non-transfusion-dependent thalassaemia patients: not a rare but potentially under-recognised condition. Orphanet J Rare Dis. 2014;9:131. | ||
Bowden DK, Hill AV, Higgs DR, Oppenheimer SJ, Weatherall DJ, Clegg JB. Different hematologic phenotypes are associated with the leftward (-alpha 4.2) and rightward (-alpha 3.7) alpha+-thalassemia deletions. J Clin Invest. 1987;79(1):39–43. | ||
Laosombat V, Viprakasit V, Chotsampancharoen T, et al. Clinical features and molecular analysis in Thai patients with HbH disease. Ann Hematol. 2009;88(12):1185–1192. | ||
Sripichai O, Makarasara W, Munkongdee T, et al. A scoring system for the classification of beta-thalassemia/Hb E disease severity. Am J Hematol. 2008;83(6):482–484. | ||
Fucharoen S, Winichagoon P, Prayoonwiwat W, Pootrakul P, Piankijagum A, Wasi P. Clinical and hematologic manifestations of AE Bart disease. Birth Defects Orig Artic Ser. 1987;23(5A):327–332. | ||
Galanello R, Melis MA, Paglietti E, Cornacchia G, de Virgiliis S, Cao A. Serum ferritin levels in hemoglobin H disease. Acta Haematol. 1983;69(1):56–58. | ||
Chen FE, Ooi C, Ha SY, et al. Genetic and clinical features of hemoglobin H disease in Chinese patients. N Engl J Med. 2000;343(8):544–550. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.