")
Back to Journals » International Journal of General Medicine » Volume 16
Clinical and Laboratory Characteristics in Children with Alagille Syndrome: Experience of a Single Center
Authors Li D, Mao K, Sun J, Liu J, Zhang C
Received 19 July 2022
Accepted for publication 9 November 2022
Published 6 January 2023 Volume 2023:16 Pages 77—83
DOI https://doi.org/10.2147/IJGM.S382430
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Dalei Li, Kangwei Mao, Jun Sun, Jiangyang Liu, Chenxi Zhang
Department of Pediatric Internal Medicine, The First People’s Hospital of Lianyungang & Xuzhou Medical University Affiliated Hospital of Lianyungang & The First Affiliated Hospital of Kangda College of Nanjing Medical University, Lianyungang, 222061, People’s Republic of China
Correspondence: Kangwei Mao, Department of Pediatric Internal Medicine, The First People’s Hospital of Lianyungang & Xuzhou Medical University Affiliated Hospital of Lianyungang & The First Affiliated Hospital of Kangda College of Nanjing Medical University, No. 6 Zhenhua East Road, Haizhou District, Lianyungang, Jiangsu, 222061, People’s Republic of China, Email [email protected]
Background: This study aimed to explore the clinical predictors of Alagille syndrome (ALGS) in children and to provide a basis for early diagnosis.
Methods: We retrospectively analyzed the clinical data of 14 children diagnosed with ALGS at the First People’s Hospital of Lianyungang City from March 2016 to March 2021 and followed up the children.
Results: Among the 14 patients, 9 (64.28%) had cholestasis, 12 (85.71%) had heart malformations, 13 (92.85%) had characteristic facial features, 2 (14.28%) had pruritus, and 2 (14.28%) had a positive family history. Among the 13 patients who were examined by pediatric ophthalmologists, 3 patients had ocular lesions. Among the 13 patients who underwent spine radiography, 2 had typical butterfly vertebrae. Among the 6 patients with hepatic pathology, 2 had intracellular cholestasis, 2 had reduced or no small bile duct in the portal area, 2 had small bile duct hyperplasia with massive fibrous hyperplasia and extensive inflammatory cell infiltration, and 2 underwent biliary tract exploration. Genetic testing of 12 children with ALGS revealed JAG1 gene mutations in 7 cases and NOTCH2 gene mutations in 2 cases. The abovementioned two mutant genes were not detected in any of the 3 cases. Among the 12 followed-up patients, 7 were in stable condition, 5 underwent liver transplantation, and 1 died of severe pneumonia.
Conclusion: Cholestatic liver disease, cardiac malformations, and abnormal facial development are predictors of ALGS in children and can be definitively diagnosed by genetic testing.
Keywords: Alagille syndrome, cholestasis, genetic testing
Introduction
Alagille syndrome (ALGS) is a rare autosomal dominant disease manifested as a multi-organ, multi-directional developmental disorder.1 Heterogeneity of clinical manifestations is also common in families, generally characterized by liver biopsy for cholestasis, lack of bile ducts, and variable involvement of other organs such as the vascular system, heart, kidneys, eyes, face, and bones.2 Changes in ALGS range from subclinical to life-threatening, with a high mortality rate of 10%.1–3 Owing to the variable participating organs of this genetic disease and the different clinical manifestations, calculating the prevalence and incidence of ALGS in clinical research and treatment is difficult. The estimated prevalence of ALGS is 1:30,000–1:100,000.4
Current molecular genetic studies have suggested that the pathogenesis of ALGS is mainly caused by heterozygous mutations in genes of the two Notch signal transduction pathways, JAGGED1 (JAG1) and NOTCH2.5 The Notch signaling pathway, which includes five transmembrane ligand families, is a highly conserved system comprising five transmembrane ligand families: JAG1, DLL1, JAG2, DLL3, and DLL4. Notch receptors are recognized by the binding of the NOTCH1, NOTCH2, NOTCH3, and NOTCH4 ligands. In response to this effect, Notch receptors are cleaved by proteolysis, and intracellular peptides are released and transported to the nucleus to co-regulate gene transcription.6,7 These signaling pathways indirectly suggest the diversity of clinical manifestations of ALGS and play a key role in multi-phylogeny. ALGS caused by NOTCH2 gene mutation accounts for approximately 1.5%, and 94% of morbidity is caused by mutation or deletion of the JAG1 gene encoding Jagged1; however, 4.5% of cases remain undetected.8
The clinical diagnosis of ALGS is difficult because of the diversity of clinical difficulties. This study included 14 children with ALGS, and their clinical data and characteristics, laboratory test results, liver histopathological characteristics, and genetic test results in the past period were analyzed and summarized. This study aimed to explore the clinical predictors of ALGS in children and provide a basis for the early diagnosis of ALGS.
Materials and Methods
Study Population
From March 2016 to March 2021, data from 14 children diagnosed with ALGS in the Department of Pediatrics, Lianyungang First People’s Hospital, were collected and retrospectively analyzed in this study. All the selected children met the revised ALGS diagnostic criteria proposed by Kamath9 and Guru Murthy.10 As all patients were aged <18 years, informed consent was obtained from their guardians during the hospital stay. The study was approved by the Ethics Committee of the First People’s Hospital of Lianyungang (no. 2,015,000,358). The study was conducted in accordance with the Declaration of Helsinki. The samples for liver biopsy histology were donated voluntarily with written informed consent, and that this was conducted in accordance with the Declaration of Istanbul. The clinical characteristics of the patients were shown in Table S1.
Inclusion and Exclusion Criteria
The inclusion criteria were as follows: age ≤ 13 years and patients who met the revised ALGS diagnostic criteria proposed by Kamath and Guru Murthy.
Meanwhile, patients who met the following criteria were excluded: received outpatient pretreatment, patients’ guardians disagreed to sign the informed consent form, and lost to follow-up.
Collection of Clinical Data
The clinical data of the children were all estimated from related records, including sex, age of onset, age at diagnosis, clinical symptoms, signs, cardiac ultrasound, abdominal (hepatobiliary–pancreatic–splenic) ultrasound, biochemical indicators such as liver function and lipid metabolism, X-ray of the spine, liver histopathological results, ophthalmic examination, and genetic test results (sent to Guangzhou Jiajian Medical Laboratory or the Precision Medication Laboratory of The First People’s Hospital of Lianyungang).
Laboratory Examination
The patients fasted for at least 12 h before the venous blood was drawn in the morning. Indicators included total bilirubin, conjugated bilirubin, unconjugated bilirubin, alanine transaminase, aspartate transaminase, alkaline phosphatase, γ-glutamyl transpeptidase, total bile acid, total cholesterol, triacylglycerol, high-density lipoprotein, and low-density lipoprotein.
Genetic Analysis
The venous blood of the children was drawn, genomic DNA was extracted, and the whole genome library containing the target genes associated with metabolic liver disease (JAG1, NOTCH2, etc.) was established. The target gene was captured with a liquid phase capture kit (MyGenostics, China), and the sequencer HiSeq2000 (Illumina, USA) was used for high-throughput sequencing to analyze the gene mutation. Sequencing analysis of the target exome capture was performed by Beijing Mackinaw Gene Technology Co., Ltd.
Statistical Analysis
The SPSS software (version 22.0; IBM Corp., Armonk, NY, USA) was used for the statistical analysis. Normally distributed data were compared using the t-test and are described as mean ± standard deviation (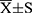 ). The abnormally distributed data were compared using the Mann–Whitney U-test and are expressed as the median and quartile [M(Q1, Q3)]. Statistical significance was set at P < 0.05.
). The abnormally distributed data were compared using the Mann–Whitney U-test and are expressed as the median and quartile [M(Q1, Q3)]. Statistical significance was set at P < 0.05.
Results
Clinical Manifestations of the Study Population
In this study, 14 children with ALGS were included (six girls and eight boys; male-to-female ratio, 1.33 to 1.0). The age distribution ranged from 1 month to 27 days, 12 years, and 10 months, with a median of 20 months. The onset of disease occurred within 2 months of birth in 10 cases, and the time of onset in the other four older children was unknown. Among them, nine patients exhibited yellow staining of the skin and sclera, three patients had abnormal liver function, and one had abnormal blood coagulation. Among the 14 children with ALGS in this study, 10 (71.4%) had congenital cardiac malformations of varying degrees, including two cases of atrial septal defect with pulmonary artery stenosis, five cases of the atrial septal defect, two cases of pulmonary artery stenosis, and one case of the ventricular septal defect. In this study, 13 children underwent spinal X-ray imaging, which revealed 2 cases of typical butterfly vertebrae (Figure 1) and 1 case of scoliosis. In the ophthalmology of 14 children, 3 patients had eye lesions, including 1 case of retrokeratoemic embryonic ring, 1 case of conjunctivitis, 1 case of strabismus in both eyes, and 13 cases had characteristic facial features (protruding forehead, deep depression in orbit, and hypertrophy of the tip of the mandibular and front of the nose). Of these, three cases were accompanied by growth retardation, and the main symptoms were abnormal mental development and short stature. Two patients (14.28%) had skin itching symptoms. Two cases (14.28%) had a positive family history (one case with a mother and one case with a father). Among the 14 children, 2 cases had biliary tract exploration, and 6 cases had a liver biopsy, of which 2 cases had intrahepatic stasis of the liver pathology, 2 cases had decreased or disappeared small bile ducts in the sink area (Figure 2), and 2 cases of micro-bile duct hyperplasia with polyfibrosis were observed in 2 cases, and polyflammatitis cell invasion was detected.
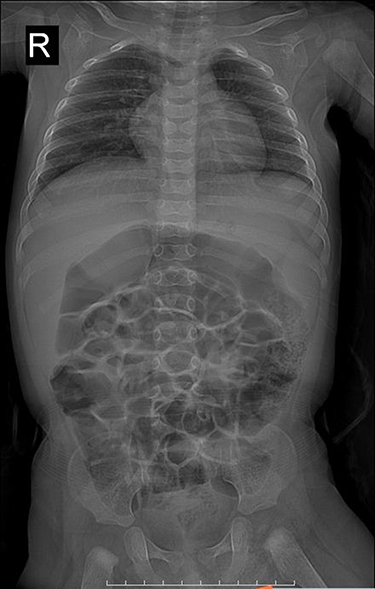 |
Figure 1 Butterfly vertebrae seen in the thoracic region in children with Alagille syndrome (arrow showed). |
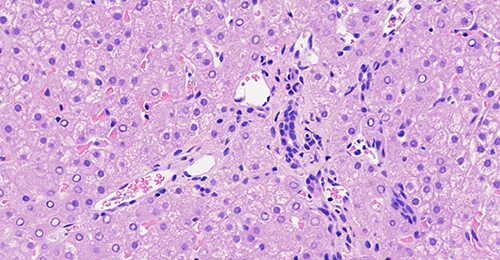 |
Figure 2 Liver biopsy histology showed the absence of bile ducts in the portal area in children with Alagille syndrome. |
Biochemical Results
In this study, 11 cases had different degrees of increase in total bilirubin, and the combination of elevated bilirubin levels was the main clinical realization. These 11 children were also accompanied by the clinical realization of varying degrees of unbound elevation of bilirubin levels. Twelve children had different degrees of elevation of aminotransferase levels, and the degree of elevation was several times higher. The highest alanine aminotransferase and aspartate aminotransferase levels were 521 U/L and 654 U/L, respectively. Nine cases demonstrated significantly elevated glutamyltransferase levels, and four cases had varying degrees of elevation of alkaline phosphatase levels. Of all children who underwent lipid metabolism testing, six cases had hypercholesterolemia, and four cases had hypertriglyceridemia (Table 1).
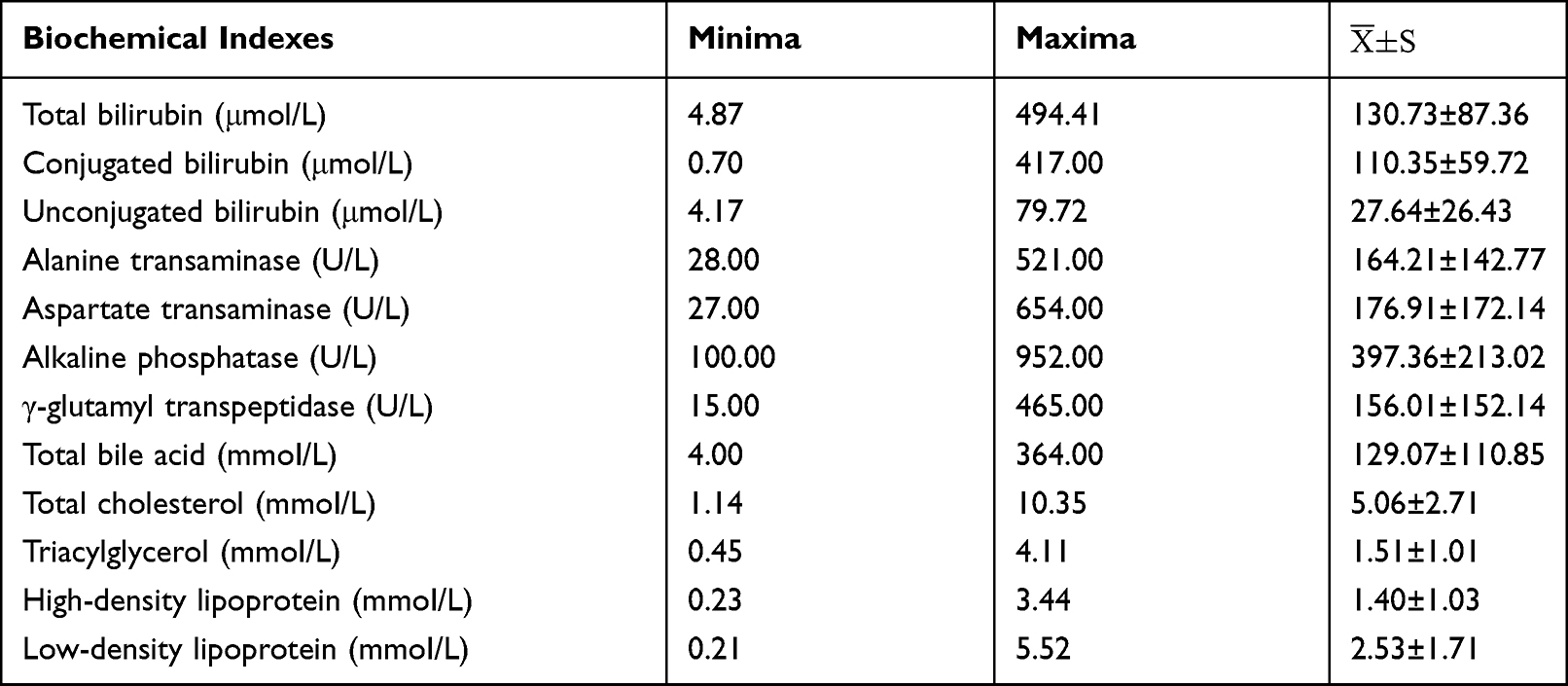 |
Table 1 Blood Biochemical Examination Results of 14 Children with Alagille Syndrome |
Genetic Test Results
As presented in Table 2, among the genetic tests in 12 cases, seven cases (accounting for 58.33%) had JAG1 gene mutations, and two cases (accounting for 16.67%) had NOTCH2 gene mutations, while such mutant genes were not detected in three cases.
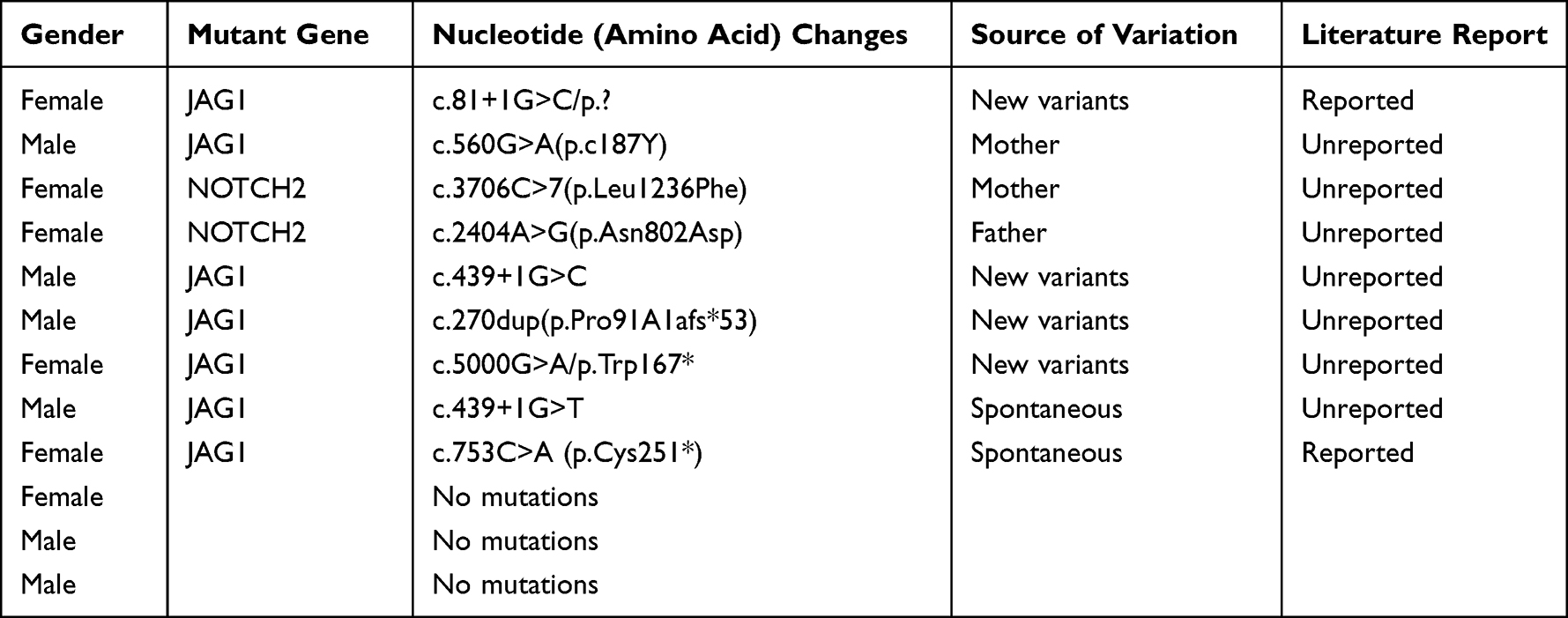 |
Table 2 Genetic Test Results of 12 Children with Alagille Syndrome |
Follow-Up
Fourteen children with ALGS were followed up for 1 to 3 years, of which two were lost to follow-up. Of the 12 remaining children, 7 were in stable condition, and 5 patients underwent liver transplantation (2 had congenital biliary atresia or severe cholestasis, 1 had recurrent thrombocytopenia, 1 had liver failure, and 1 died of severe pneumonia).
Discussion
This study confirmed that ALGS causes abnormal development of the pancreas, kidneys, and neurovasculature, as well as abnormalities in the development of the heart, eyes, liver, spine, and face, with various clinical features. The incidence of chronic cholestasis in ALGS cases is high at 95%, and its onset is often within 3 months after birth.11 With elevated serum total bile acid and transaminase levels in these children, yellowing of the skin and mucous membranes caused by hyperconjugated bilirubinemia may lead to pruritus and delayed growth and development. Liver pathology typically presents with a lack of bile ducts between the lobules of the liver.12 Some patients with ALGS may have no interlobular bile ducts decreasing or disappearing in early infancy, and the lack of bile ducts is progressive, which is also a clinical manifestation proposed in recent studies.13 In the present study, 6 patients underwent liver biopsy, of which 2 cases (1 case at 2 months and 1 case at 6 months) had intrahepatic cholestasis in liver pathology, and 2 cases (1 case at 2 months and 1 case at 3 years and 10 months) had a decrease or disappearance of the small bile ducts in the sink area. Two cases had interlobular cholangiopathy with polyfibrosis (one was 2 months old and the other was 8 months old), and numerous inflammatory cells were infiltrated. No significant progressive changes in the reduction of the small bile ducts were observed in this study. On the one hand, it may be related to compensatory hyperplasia of the liver, and on the other hand, it may be due to the few samples in this study. Approximately 15% of children with ALGS develop progressive liver disease due to cholestasis, which may lead to cirrhosis and liver failure and eventually require liver transplantation.14
Because ALGS affects the development of children, the height and weight of children with ALGS are lower than those of children of the same age, and more than half of them are lower than the 5th percentile of height and weight of their normal peers.15 Among the children with ALGS in this study, three cases (21.4%) of growth retardation with severe malnutrition were caused by multiple factors. Rovner et al16 indicated that only 20% of the children with ALGS received sufficient calories and other nutrients. The main factor is an inadequate intake, while fat malabsorption due to chronic diseases and cholestasis are additional contributing factors. Studies have demonstrated that growth delay is more significant in children with ALGS than in those with other chronic liver diseases, and some children with ALGS have pyramidal dysplasia, suggesting that the JAG1 gene may play a role in the process of body growth and development, and the specific mechanism needs to be further studied.17
A foreign study has reported that >90% of children with ALGS experience congenital cardiovascular structural malformations.18 The six-year survival rate of children without cardiac involvement and malformations is high at 90%. In cases of congenital heart malformations, the six-year survival rate of children can be reduced to 40%.14 The presence of complex congenital heart disease may be the most important indicator of early mortality, whereas liver complications account for a larger proportion of later deaths. Of the 14 children with ALGS, 71.4% of the children with congenital cardiovascular structural malformations accounted for 10 cases, which is consistent with the results of previous studies. Most children with ALGS have congenital cardiovascular structural abnormalities; however, the proportion of cardiac malformation types varies. Despite the small sample size of this study, the key role of JAG1 in cardiovascular system development has been confirmed in both mouse and human embryo studies. However, whether definite pathogenesis exists between JAG1 gene mutations and cardiovascular disease is unclear,3 and further research is needed.
Turnpenny et al have reported that children with ALGS often have mild yet recognizable deformities, including a protruding forehead, deep depression in orbit, and hypertrophy at the tip of the mandible and front of the nose.19 Since the characteristic facial features have great diagnostic value for the disease, the facial features of children with cholestasis should be observed during the physical examination; however, the characteristic facial features in early infancy may not be significant. The use of facial features in diagnostic criteria is controversial because of subjectivity and differences between observers.20 In our study, 13 cases had characteristic facial features, including two 2-month-old infants who were treated for repeated skin yellowing. No relevant characteristic manifestations were observed in the disease onset, and the changes in facial appearance gradually became evident over time. Although the mechanism of facial feature formation is unclear, the role of JAG1 in the NOTCH signaling pathway of mouse cranial crest cells has been established, and further research is needed to determine whether ALGS affects the formation of facial features.
Vertebral abnormalities are often associated with ALGS, and symptoms mostly manifest as butterfly vertebrae, in which the anterior arch of the vertebral body does not fuse, and the vertebral body is sagittally divided into semi-vertebral bodies.21 Although patients are asymptomatic, this can help diagnose 33–66% of patients. Similarly, ocular abnormalities generally do not affect vision, although they are helpful in diagnosis.22 Posterior embryotoxon are the most common, and ophthalmic ultrasound of the optic disc is a more specific method of diagnosing ALGS, with a detection rate of 95%.3 Among the 14 children with ALGS, 13 underwent spine radiography, which revealed that two had typical butterfly vertebrae and one had scoliosis. Among the 13 children with ophthalmic consultation, three had ocular lesions, including one case with the posterior embryonic ring, one with binocular strabismus, and one with conjunctivitis.
With the continuous development of science and medical technology, remarkable progress has been made in the field of molecular genetics. Genetic diagnosis technology has become an important auxiliary diagnostic method for ALGS. For suspicious cases, the JAG1 gene or NOTCH2 gene can be detected by genome sequencing for further clarification.23 Genome sequencing provides not only a gold standard for the diagnosis of the disease but also much information on prognosis and genetic counseling. Additionally, it also protects children from traumatic examinations.24 In our study, the whole genomes of 12 children with ALGS were sequenced, of which seven cases had JAG1 gene mutations, two cases had NOTCH2 gene mutations, and three cases had none of the two mutant genes. With the continuous accumulation of cases and the continuous improvement of diagnostic technology, the molecular mechanism of the disease will be gradually elucidated, and more susceptible genes will be discovered.
Conclusions
Cholestatic liver disease, cardiac malformations, and abnormal facial development are predictors of ALGS in children and can be definitively diagnosed by genetic testing.
Abbreviations
ALGS, Alagille syndrome.
Funding
This study was supported by funds from Lianyungang Health Science and Technology Project (Grant No.: ZD202101).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Benabed Y, Chaillou E, Denis MC, et al. Alagille syndrome: a case report. Ann Biol Clin. 2018;76(6):675–680.
2. Chen HL, Wu SH, Hsu SH, et al. Jaundice revisited: recent advances in the diagnosis and treatment of inherited cholestatic liver diseases. J Biomed Sci. 2018;25(1):75. doi:10.1186/s12929-018-0475-8
3. Mitchell E, Gilbert M, Loomes KM. Alagille syndrome. Clin Liver Dis. 2018;22(4):625–641. doi:10.1016/j.cld.2018.06.001
4. Diaz-Frias J, Kondamudi NP. Alagille syndrome. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2021.
5. Kamath BM, Baker A, Houwen R, et al. Systematic review: the epidemiology, natural history, and burden of alagille syndrome. J Pediatr Gastroenterol Nutr. 2018;67(2):148–156. doi:10.1097/MPG.0000000000001958
6. Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. 2016;17(11):722–735. doi:10.1038/nrm.2016.94
7. Grochowski CM, Loomes KM, Spinner NB. Jagged1 (JAG1): structure, expression, and disease associations. Gene. 2016;576(1 Pt 3):381–384. doi:10.1016/j.gene.2015.10.065
8. Leonard LD, Chao G, Baker A, et al. Clinical utility gene card for: alagille Syndrome (ALGS). Eur J Hum Genet. 2014;22(3):435. doi:10.1038/ejhg.2013.140
9. Kamath BM, Suchy FJ, Sokal RJ, et al. Liver Disease in Children.
10. Guru Murthy GS, Rana BS, Das A, et al. Alagille syndrome: a rare disease in an adolescent. Dig Dis Sci. 2012;57(11):3035–3037. doi:10.1007/s10620-012-2226-0
11. Kohut TJ, Gilbert MA, Loomes KM. Alagille syndrome: a focused review on clinical features, genetics, and treatment. Semin Liver Dis. 2021;41(4):525–537. doi:10.1055/s-0041-1730951
12. Gilbert MA, Loomes KM. Alagille syndrome and non-syndromic paucity of the intrahepatic bile ducts. Transl Gastroenterol Hepatol. 2021;6:22. doi:10.21037/tgh-2020-03
13. Kamath BM, Munoz PS, Bab N, et al. A longitudinal study to identify laboratory predictors of liver disease outcome in Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;50(5):526–530. doi:10.1097/MPG.0b013e3181cea48d
14. Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet. 2016;9:75–82. doi:10.2147/TACG.S86420
15. Jesina D. Alagille Syndrome: an Overview. Neonatal Netw. 2017;36(6):343–347. doi:10.1891/0730-0832.36.6.343
16. Rovner AJ, Schall JI, Jawad AF, et al. Rethinking growth failure in Alagille syndrome: the role of dietary intake and steatorrhea. J Pediatr Gastroenterol Nutr. 2002;35(4):495–502. doi:10.1097/00005176-200210000-00007
17. Zhang W, Zhao X, Huang J, et al. Alagille syndrome: an uncommon cause of intrahepatic cholestasis in adults. Rev Esp Enferm Dig. 2019;111(4):323–326. doi:10.17235/reed.2019.5679/2018
18. Ayoub MD, Kamath BM. Alagille syndrome: diagnostic challenges and advances in management. Diagnostics. 2020;10(11):907. doi:10.3390/diagnostics10110907
19. Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012;20(3):251–257. doi:10.1038/ejhg.2011.181
20. Yang WH, Zhang L, Xue FS, Riaz A, Zhu ZJ. Pediatric liver transplantation for alagille syndrome: anesthetic evaluation and perioperative management. Ann Transplant. 2020;25:e924282. doi:10.12659/AOT.924282
21. Berniczei-Royko A, Chałas R, Mitura I, Nagy K, Prussak E. Medical and dental management of Alagille syndrome: a review. Med Sci Monit. 2014;20:476–480. doi:10.12659/MSM.890577
22. Sanderson E, Newman V, Haigh SF, et al. Vertebral anomalies in children with Alagille syndrome: an analysis of 50 consecutive patients. Pediatr Radiol. 2002;32(2):114–119. doi:10.1007/s00247-001-0599-x
23. Fischetto R, Palmieri VV, Tripaldi ME, et al. Alagille syndrome: a novel mutation in JAG1 gene. Front Pediatr. 2019;7:199. doi:10.3389/fped.2019.00199
24. Andersson ER, Chivukula IV, Hankeova S, et al. Mouse model of alagille syndrome and mechanisms of jagged1 missense mutations. Gastroenterology. 2018;154(4):1080–1095. doi:10.1053/j.gastro.2017.11.002
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.