Back to Journals » Journal of Inflammation Research » Volume 15
Clenbuterol, a Selective β2-Adrenergic Receptor Agonist, Inhibits or Limits Post-Stroke Pneumonia, but Increases Infarct Volume in MCAO Mice
Authors Xu Y, Ge Y, Zhou M, Zhang Z
Received 26 October 2021
Accepted for publication 24 December 2021
Published 14 January 2022 Volume 2022:15 Pages 295—309
DOI https://doi.org/10.2147/JIR.S344521
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Ning Quan
Younian Xu,1 Yangyang Ge,1 Miaomiao Zhou,2 Zongze Zhang2
1Anesthesiology Department, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 2Anesthesiology Department, Zhongnan Hospital of Wuhan University, Wuhan, People’s Republic of China
Correspondence: Miaomiao Zhou
Anesthesiology Department, Zhongnan Hospital of Wuhan University, No. 169 Donghu Road, Wuchang District, Wuhan, 430071, Hubei Province, People’s Republic of China
Tel/Fax +86 027-67812903
Email [email protected]
Background: Large ischemic stroke provokes an inflammatory response, promoting the release of norepinephrine (NE) by intensifying the sympathetic nervous system. This augmented sympathetic outflow was deemed to act on β 2-adrenergic receptors (β 2-ARs) expressed by immune cells, rendering organisms to post-stroke infections, like pneumonia. To clarify this issue, we introduced selective β 2-ARs agonist clenbuterol (CLEN) to stroke mice to investigate how β 2-adrenergic signaling augmentation after stroke affects immune response and post-stroke outcomes, including central and peripheral.
Methods: We developed a middle cerebral artery occlusion (MCAO) stroke model in mice to induce large ischemic stroke and administered CLEN 24 h after the onset of MCAO stroke. First, we assessed infarct volume and NE levels in plasma and spleen 3ds later. Next, the immune state was identified by analyzing the spleen index, immune cell populations, and immune cytokines. Finally, peripheral outcomes were assessed by measuring signs of pneumonia, such as pathology, bacterial burden, and lung cytokines.
Results: We report that CLEN treatment 24 h after MCAO stroke causes an enlarged infarct volume and a decrease in NE levels at 3ds after stroke. Consistent with a reduction of total T cells, T helper cells, and increase of cytotoxic T cells, the immune milieu after CLEN treatment presents an anti-inflammatory landscape, showing raised expression of anti-inflammatory cytokines: IL-4, IL-10, and TGF-β 1, and decreased expression of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IFN-γ, with a dramatically reduced percentage of Gr-1+ neutrophils and B cells but an increased percentage of NK cells. In our study, CLEN treatment results in no higher risk of pneumonia but relieves bacterial burden, inhibits or limits pneumonia, and diminishes TNF-α expression in lung tissues after MCAO.
Conclusion: We identified increased β 2-adrenergic signaling after MCAO stroke, inhibits or limits post-stroke pneumonia but enlarges stroke volume in MCAO mice. Thus, careful consideration must be taken to improve post-stroke outcomes by manipulation over β 2-adrenergic receptors.
Keywords: ischemic stroke, β 2-adrenergic receptor, infarct, pneumonia, immunity, sympathetic
Introduction
It is well documented that the sympathetic nervous system (SNS) plays a dominant role in post-stroke immune modulation by giving rise to an intense activation of the SNS and emission of norepinephrine (NE), which thus activates β2-adrenergic receptors (β2-ARs).1 The β2-ARs, highly expressed on all significant immune cell subtypes, then transmitted the signaling pathway and regulated the peripheral immune system.2 It has been previously demonstrated that β2-ARs stimulation decreases the synthesis and release of inflammatory mediators from activated macrophages and lymphocytes,3,4 and induces the release of anti-inflammatory cytokines.5,6 Such anti-inflammatory (immunosuppressive) response was regarded to be beneficial by attenuating autoimmune response in the central nervous system.
However, previous studies investigating how β2-ARs signaling affects stroke-induced neuronal outcome in the brain by manipulating the receptor precede or post-stroke have yielded conflicting results. Pretreatment with a selective β2 agonist has been proved to reduce infarct size in mice and rats,7,8 while post-stroke treatment with a selective β2 agonist enlarged infarct size in mice.9 Global knockout of β2-ARs or pretreatment with β2-ARs selective antagonist ICI 118,551 has been shown to reduce infarct size and improve behavioral outcome.10,11 β2-ARs stimulation although may inhibit immune response against ischemic injury. It might as well potentiate individuals to immunosuppressive-induced high susceptibility to infections.
Among the post-stroke infections, pneumonia was the most common complication. The pneumonia-related inflammatory state was thought to release a bystander autoimmune response against central nervous system antigens, initiating a vicious circle.12 The largest clinical trial implemented to investigate the effect of pneumonia on 30-day mortality in hospitalized patients with acute stroke reported a 27% mortality rate in 635 patients due to pneumonia compared with a 4% death rate among patients free from respiratory infections.13 Sympathetic nerves locally innervate lung tissue.14 The use of β-adrenergic receptor antagonists (β-blockers) in post-stroke mice was demonstrated to reduce pneumonia and mortality rates.15 The underlined mechanism of β-adrenergic receptor blockade for decreasing bacterial burden was considered by promoting the production of pro-inflammatory cytokines.16 On the contrary, in LPS-elicited acute septic injury, activation of β2-ARs by NE or selective agonist could intrinsically inhibit the immune response in primary alveolar macrophages, reducing inflammatory cytokines, bronchial inflammation, atelectasis, and airway damage, and protect the lung from injury induced by systemic inflammatory responses.4,14,17 To the best of our knowledge, there has been no research concerning the actual effects of β2-ARs activation on post-stroke pneumonia.
The present study was undertaken to estimate SNS activity after ischemic stroke and evaluate whether facilitating response of β2-ARs is beneficial or detrimental to MCAO mice considering both neuronal and peripheral outcomes.
Materials and Methods
Eight to ten weeks old male C57BL/6 mice (24–26 g) were used for all studies. Animals have been housed in individually ventilated cages (temperature 21 ± 2 °C; humidity 55% ± 5%; 12-h light/12-h dark cycle) and given access to food and water ad libitum. Animals were allocated treatments and surgical fates in a randomized order across cages. All procedures were approved by the Institutional Animal Care and Use Committee at Tongji Medical College, Huazhong University of Science and Technology (IACUC Number: 2419). All experiments were performed according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Efforts were made to minimize the number of animals used.
Stroke Model and Drug Treatment
The middle cerebral artery occlusion (MCAO) was performed mainly according to the study of Chiang.18 Briefly, mice were anesthetized with Chloral hydrate (300 mg/kg, i.p.). After ventral cervical incision, silicone rubber-coated monofilament diameter of 0.24 ± 0.02 mm (Yushun Biotech, Pingdingshan, Henan, China) was introduced into the common carotid artery, advanced along the internal carotid artery to the origin of the right MCA, and left there for 60 min until reperfusion. After surgeries, local analgesia (bupivacaine 1 mg kg,−1 subcutaneously (s.c.)) was applied. Sham operation was performed identically, except that the sham group only briefly introduced the filament into the external carotid artery. A feedback-controlled heating pad was used to maintain body temperature during the operations and until recovery from anesthesia to control a percentage of infarct area/total brain area between 20% and 40%. β2-ARs agonist clenbuterol (CLEN) (Adooq Bioscience A13407, USA) dissolved in 0.9% sodium chloride at 0.1 mM (1 mg/kg) or vehicle (saline) was administered intraperitoneally at 24 h and 48 h after MCAO (groups defined as M-CLEN or M-saline respectively).
Blood and Tissue Sample Collection
Mice were rapidly euthanized by cervical dislocation 72 h after MCAO. Blood was obtained by cardiac puncture from mice, tissues of spleen, lung, and brain were collected. Spleens were weighed immediately after sacrifice. The spleen index was evaluated as the weight of the spleen (mg)/body weight (g)×10.
Assessment of Infarct Volume
The infarct volume was assessed by TTC staining as previously described.19 Briefly, the mice were decapitated, and the brain was rapidly removed and sliced into 6 coronal sections, 1 mm thick each. Sections were immersed in pre-warmed 2% TTC in saline for 30 min at 37 °C and then fixed in 4% paraformaldehyde (PFA) for 24 h. The infarct areas and total area on each slice were calculated using Image-Pro Plus 6.0 software and then expressed as the percentage of infarction in the total area.
Evaluation of Sympathetic Activity
NE levels could serve as an indirect index of sympathetic activity.20–22 For determination of the baseline NE level, mice were treated to obtain sedation (chloral hydrate 300 mg/kg, i.p.), then blood was immediately collected by eyeball removal, and only the first two drops of blood were collected to avoid NE spillover. For measurement of the spleen NE level, homogenized tissues first. After that, blood and tissue samples were then centrifuged, and NE levels were measured according to the manufacturer’s protocol. The OD value at 450 nm was detected using a microplate reader (AMR-100, Aosheng, China).
Flow Cytometry
Cell populations were identified by flow cytometry. Briefly, red blood cells were lysed by ACK lysis buffer (Beyotime, China), isolated leukocytes were centrifugated by 37–70% Percoll density gradient centrifugation. Cells were washed with buffer (PBS with 0.5% bovine serum albumin and 0.02% sodium azide) three times and stained with antibodies listed in Supplementary Table 1 for 20 min at 4 °C. CD3 expressed on T cells is a part of the T cell receptor complex. CD4 and CD8 are transmembrane glycoproteins on T helper cells and cytotoxic T cells. CD45R, a cell surface molecule on B cells that assists with antigen-based B cell activation. Gr-1’s expression is restricted to monocytes within the bone and on neutrophils in peripheral organs. The F4/80 molecule was established as a unique marker of murine macrophages. CD49b is a Pan NK cell marker. CD11c is a transmembrane protein found at high levels on most dendritic cells. We used NovoCyte 452161026119 (Agilent, USA) with NovoExpress 1.5.0 software (Agilent, USA) to determine the phenotypes of leukocytes.
Quantification of Plasma Cytokines
Pro-inflammatory and anti-inflammatory cytokines (TNF-α, INF-γ, IL-1β, IL-4, IL-10, TGF-β1) were determined by commercially available immunoenzymatic commercial assays dedicated to equine species (Cloud-Clone Corp, Wuhan, China). For analysis of all cytokines, plasma was prepared by centrifugation of whole blood at 1500 × g for 10 min and stored in aliquots at −80 °C. Then, samples were thawed and filtered for analysis using mouse-specific ELISAs (Bioswamp, China) following the manufacturer’s instructions. A Luminex 200 plate reader equipped with xPOnent software (University of Rochester Flow Cytometry Core) was used to determine median fluorescence intensity for each analyte. The concentration of each analyte was calculated using the corresponding standard curve fit to a 5-parameter logistic equation. For all ELISAs, absorption was measured at 450 nm using a multi-well plate reader (AMR-100, Aosheng, China). Curve fitting and sample concentration calculations were conducted with Gen5 software (Biotek).
Histopathological Analysis of Lung Tissues
Lungs were removed, immersed in 10% formalin fixative, and processed for histological examination. The lung tissue was dehydrated, transparent, soaked, and embedded in paraffin wax, and cut into 12-µm thick sections by microtome dissection. After this, the sections were stained with hematoxylin/eosin (HE) for assessment. Twenty representative sections of lungs were chosen per animal (n = 4 in each group) and evaluated by two investigators, blinded to the treatment groups. The sections were graded according to the following criteria: (1) normal, (2) focal (<50% lung section) interstitial congestion and inflammatory cell infiltration, (3) diffuse (>50% lung section) interstitial congestion and inflammatory cell infiltration, (4) focal (<50% lung section) consolidation and inflammatory cell infiltration, (5) diffuse (>50% lung section) consolidation and inflammatory cell infiltration, as previously described.23
Bacteriological Analysis
To determine colony-forming units (CFU), bronchoalveolar lavage fluid (BALF) was collected. After the mice were euthanized, the trachea was exposed and subsequently cannulated with a polyethylene catheter. The lungs were washed with three flushes containing 500 μL of 0.9% NaCl solution. Then, BALF was plated onto blood agar plates, incubated at 37 °C for 18 h. Positive colonies were recorded, and CFU was calculated per milliliter of BALF.
Statistical Analysis
Bacteriological data are presented as violin plots. Other data are expressed as mean ± SEM. GraphPad Prism software (version 8.0) was used for statistical analyses. The ROUT method identified the outliers (Q = 1.000%) and excluded them from the analysis. After validating the normal distribution of these datasets (Kolmogorov–Smirnov test), significant differences were assessed by two-tailed Student’s t-test in the comparison between two groups, the one-way ANOVA with Holm–Sidak correction in pairwise comparison among three groups, or Kruskal–Wallis test for analysis of the histopathological score. A p-value <0.05 was regarded as significant, whereas p-value <0.01, p-value <0.001 and p-value <0.0001 were highly significant.
Results
β2-ARs Agonist CLEN Increased Infarction Area After Ischemic Stroke, and Partly Reversed Elevated NE Levels Induced by Ischemic Stroke
We induced stroke by transient (60 min) proximal MCA filament occlusion, and the procedures are conducted as Figure 1A. The locations of ischemic lesions 3ds following MCAO stroke are shown in Figure 1B. The mean size of ischemic lesions in the MCAO stroke was 35.0% of the whole brain (Figure 1C). We first investigated the plasma levels of sympathetic neurotransmitters (NE) in mice with infarcts compared with sham-operated mice. At d3 after MCAO, there was a more than a two-fold increase in the plasma NE concentrations, compared with the SHAM group (348.8 ± 40.2 pg/mL vs 156.5 ± 12.5 pg/mL, p < 0.0001; Figure 1D). However, the elevated plasma levels of NE in the M-saline group hardly impacted the immunity because NE exerts anti-inflammatory effects only in micromolar concentrations.24 The norepinephrine nerve terminals which govern the spleen and provide a high concentration of NE in the direct vicinity of antigen-activated immune cells were suggested to play a dominant role in the modulatory effect.25 Hence, spleen NE level was supplementary measured. We detected a similar increase of spleen NE level in the M-saline group as seen in plasma level 3 d after stroke (p < 0.0001; Figure 1E). We next sought to define whether activation of β2-ARs protects mice from MCAO stroke and introduced β2-ARs agonist CLEN once daily to MCAO mice for 24 h after stroke until mice were sacrificed. Unlike we expected, though, the infarct volume was enlarged after CLEN treatment from 35.0 ± 7.1% to 45.8 ± 6.4% (p < 0.05; Figure 1B and C). In contrast, the NE levels in the M-CLEN group compared to the M-saline group were slightly decreased in both plasma and spleen. However, compared to the SHAM group, the NE levels in the M-CLEN group are still highly increased in both plasma and spleen (p < 0.0001; Figure 1D and E).
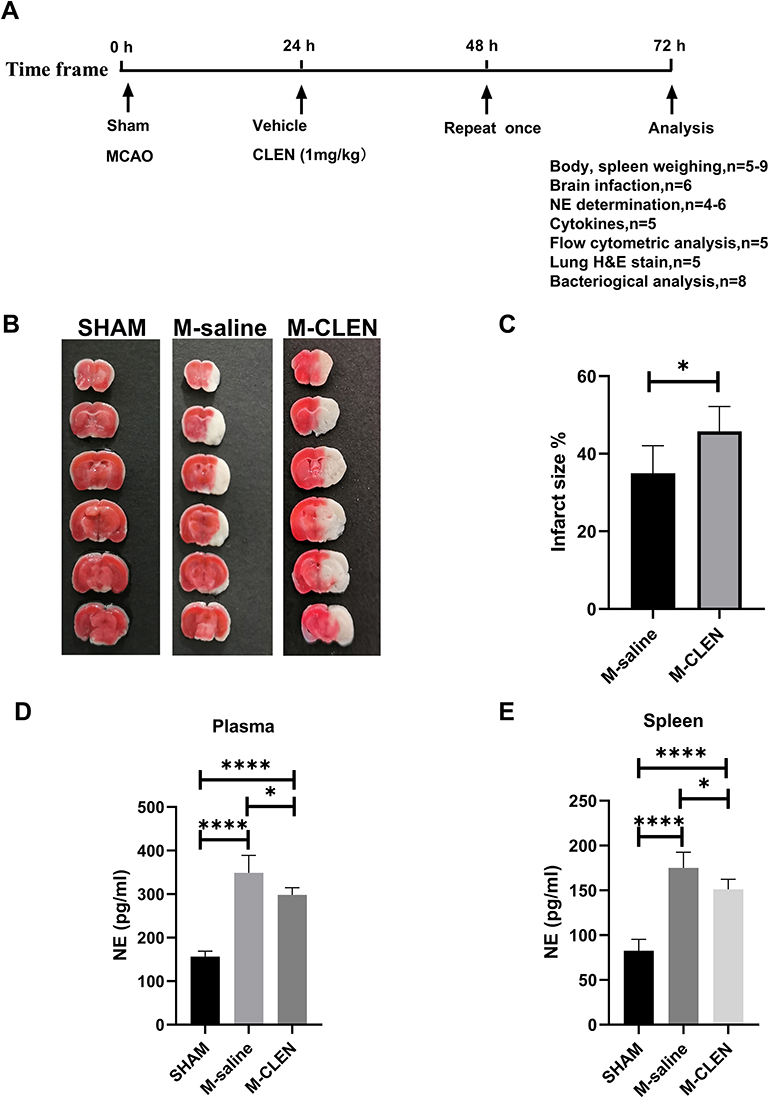 |
Figure 1 The schematic diagram of animal study design and infarct volumes and NE levels 3ds after MCAO stroke. (A) 8–10 weeks old male C57BL/6 mice were allocated randomly to the SHAM, M-saline, and M-CLEN groups. Mice were given intraperitoneally vehicle or CLEN respectively at 24 h and 48 h after stroke. 72 h following the stroke, mice were allocated into the indicated analyses. (B) Representative TTC-stained coronal serial sections of cortical infarcts in saline and CLEN treated MCAO mice 3 d after stroke. (C) Infarct volumes of MCAO in the saline and CLEN treated MCAO mice 3 d following the stroke (n = 6 per group). (D) Plasma NE levels were determined 3 d following the stroke in the sham-operated and saline, CLEN treated MCAO mice (SHAM n = 4, M-saline n = 5, M-CLEN n = 6). (E) Spleen levels of NE in the sham-operated and saline, CLEN treated MCAO mice 3 d following stroke (SHAM n = 5, M-saline n = 9, M-CLEN n = 6). *p < 0.05, ****p < 0.0001, one-way ANOVA with the post-hoc. |
The Effects of β2-ARs Agonist CLEN Treatment on Body Weight Lost, Spleen Weight and Spleen Index
Body weight loss was used as general parameters reflecting physical activity, food intake, and potential infectious illness after an operation. There was no statistical difference in body weight noted in saline and CLEN treated MCAO mice measured 3ds after stroke (Figure 2A). A profound reduction was observed in the spleen weight in the saline-treated M-saline group at 3 d after MCAO (Figure 2B), which is conversely consistent with the above NE analysis, which showed a dramatic increase in the M-saline group. Administration of CLEN countered the reduction in the spleen weight after MCAO, consistent with the above NE results that CLEN partly counteracts the NE elevation induced by MCAO (Figure 2B). When the spleen weight was normalized to spleen index as the spleen to body weight ratio, there was a more considerable statistical difference between the M-saline and M-CLEN groups (Figure 2C). The pictures of the spleens in the three groups are shown in Figure 2D.
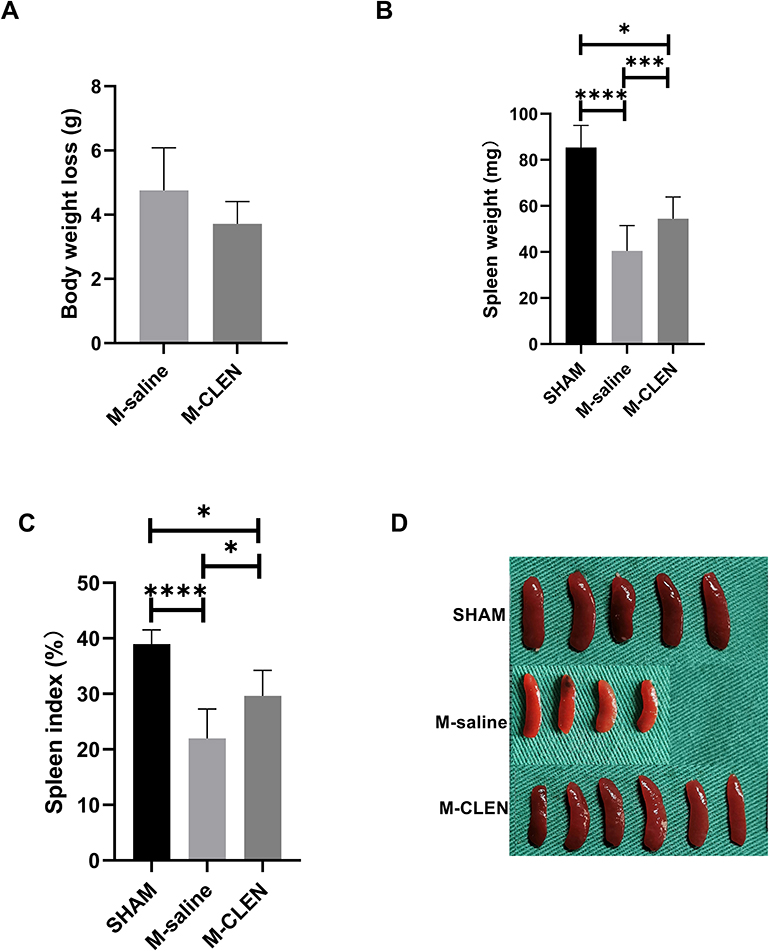 |
Figure 2 Body weight loss and spleen index in the sham-operated and saline, β2-ARs agonist CLEN treated MCAO mice. (A) Body weight loss was measured as an index of general stress elicited by cerebral ischemia (M-saline n = 5, M-CLEN n = 6). (B) Spleen weight and (C) Spleen index 72 h following MCAO measured as an immunosuppression index in the sham-operated and CLEN treated MCAO mice (SHAM n = 5, M-saline n = 9, M-CLEN n = 6). (D) Spleens in the above three groups were shown (SHAM n = 5, M-saline n = 4, M-CLEN n = 6). *p < 0.05, ***Indicates p < 0.001, and ****Indicates p < 0.0001 by one-way ANOVA with Holm–Sidak correction. |
The Effects of β2-ARs Agonist CLEN Treatment on Adaptive Immune State
Previous work indicated that adrenergic nerve endings in the spleen terminate in the white pulp, which contains rich T and B –lymphocytes, and enhances NE release from the spleen by adrenergic stimulation.26 We, therefore, investigated the effects of MCAO stroke on lymphocyte subpopulations, including T cells and B cells, in blood 3 d after MCAO stroke. The results showed that the percentage of CD3+ total T cells was not changed, but the percentage of B cells was decreased in the MCAO groups compared with the SHAM group (Figure 3A and B). We next compared the two main T-lymphocyte subpopulations, including percentages of T helper cells (CD3+CD4+) and cytotoxic T cells (CD3+CD8+) within the total T cell populations in blood. We found that experiments with mice undergoing MCAO stroke revealed no differences in the percentages of T helper cells and cytotoxic T cells between the SHAM group and M-saline group (Figure 3C and D). The above results were contrary to our expectation because a decrease in the percentages of CD3+ total T cell, T helper cells (CD3+CD4+), and cytotoxic T cells (CD3+CD8+), which indicates a state of immune deficiency, is likely to appear in post-stroke organisms, especially in individuals with large infarction volume.27 Treatment of CLEN induced a drop in the percentage of total T cells and T helper cells, but an increase in the percentages of cytotoxic T cells, all of which indicated immune deficiency (Figure 3A, C and D). Besides, CLEN resulted in a further drop in the percentage of B cells compared with the M-saline group (Figure 3B).
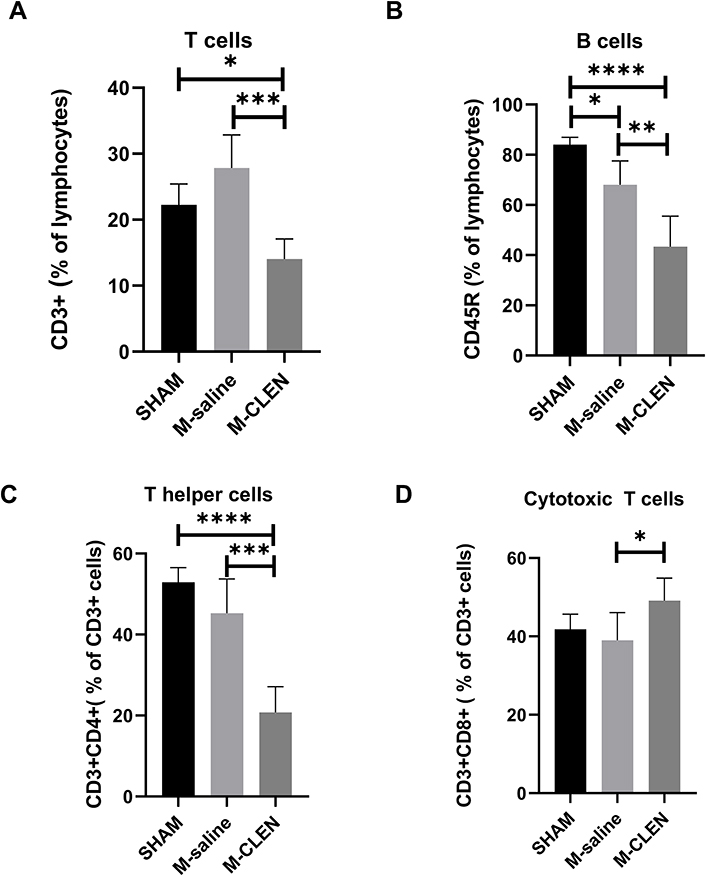 |
Figure 3 Adaptive cell subpopulations in the sham-operated and saline, β2-ARs agonist CLEN treated MCAO mice. (A) Percentages of CD3+ total T cells (n = 5 per group), (B) B cells (CD45R+) in the blood 72 h after stroke in the SHAM, M-saline, and M-CLEN groups (n = 5 per group). (C) Percentages of T helper cells (CD3+CD4+) and (D) Cytotoxic T cells (CD3+CD8+) in CD3+ total T cells in the blood 72 h after stroke in the SHAM, M-saline and M-CLEN groups (n = 5 per group). *p < 0.05, **Indicates p < 0.01, ***Indicates p < 0.001 and ****Indicates p < 0.0001, one-way ANOVA with Holm–Sidak correction. |
The Effects of CLEN Administration on the Innate Immune State
We further analyzed innate immune cells, including neutrophil (Gr-1+), macrophage (F4-80+), NK cell (CD49b+), and dendritic cell (CD11c+) subpopulations in blood by flow cytometry at 72 h after MCAO stroke. Neutrophil cell populations were decreased in the M-saline group compared with the SHAM group, treatment of CLEN results in a further decrease of neutrophil cell populations (Figure 4A). Figure 4B shows that blood macrophages did not differ among the SHAM, M-saline, and M-CLEN groups. Dendritic cell populations were decreased in the M-saline group compared with the SHAM group. CLEN treatment also resulted in a further decrease, although no statistical differences were seen among the three groups (Figure 4C). In contrast, NK cell counts were increased in the M-saline group compared with the SHAM group, while CLEN treatment resulted in a further increase of NK cell counts (Figure 4D).
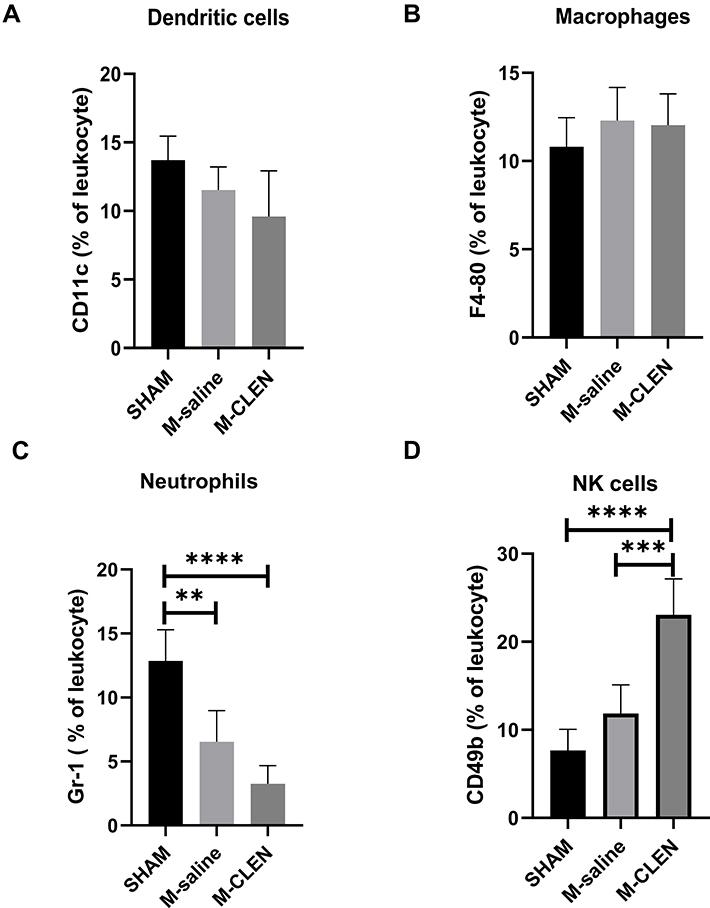 |
Figure 4 Innate cell subpopulations in the sham-operated and saline, β2-ARs agonist CLEN treated MCAO mice. Here shows the percentage of neutrophil, macrophage, NK cell, and dendritic cell populations in peripheral blood of sham-operated mice and mice treated with vehicle or CLEN following MCAO. (A) Neutrophils (Gr-1+) cells, (B) Macrophages (F4-80+), (C) NK cells (CD49b+), (D) Dendritic cells (CD11c+) in the blood 72 h after stroke in the SHAM, M-saline and M-CLEN groups. The sample size was n = 5 for the analysis of antigens. **p < 0.01, ***Indicates p < 0.001 and ****Indicates p < 0.0001, one-way ANOVA with Holm–Sidak correction. |
CLEN Modulation of Plasma Pro- and Anti-Inflammatory Cytokine Levels After MCAO Stroke
We analyzed serum cytokine concentrations to determine the impact of the MCAO stroke models on lymphocyte function and systemic inflammatory activation (Figure 5). The pro-inflammatory cytokines TNF-α, IFN-γ, and IL-1β, have been proved to be elevated in the stroke mice 3ds after MCAO, especially the cytokines of TNF-α and IL-1β, which showed a substantial increase (Figure 5A–C). There were no statistical differences between SHAM and M-saline on the expression of IL-4 and IL-10 (Figure 5D and E). However, a pronounced difference between the M-saline and SHAM groups was found for concentrations of anti-inflammatory cytokine TGF-β1 (Figure 5F). The consecutive administration of β2-ARs agonist CLEN since 24h after MCAO until being sacrificed at 3 d after MCAO has reversed the expression of pro-inflammatory cytokines TNF-α, IFN-γ, and IL-1β. After CLEN treatment, all anti-inflammatory cytokines of IL-4, IL-10, and TGF-β1 have been up-regulated. Overall, the immune milieu in the M-saline group exhibited a marked inflammatory state characterized by increased TNF-α, IFN-γ, and IL-1β, while after CLEN treatment, the immune milieu presented an anti-inflammatory state characterized by increased IL-4, IL-10, and TGF-β1.
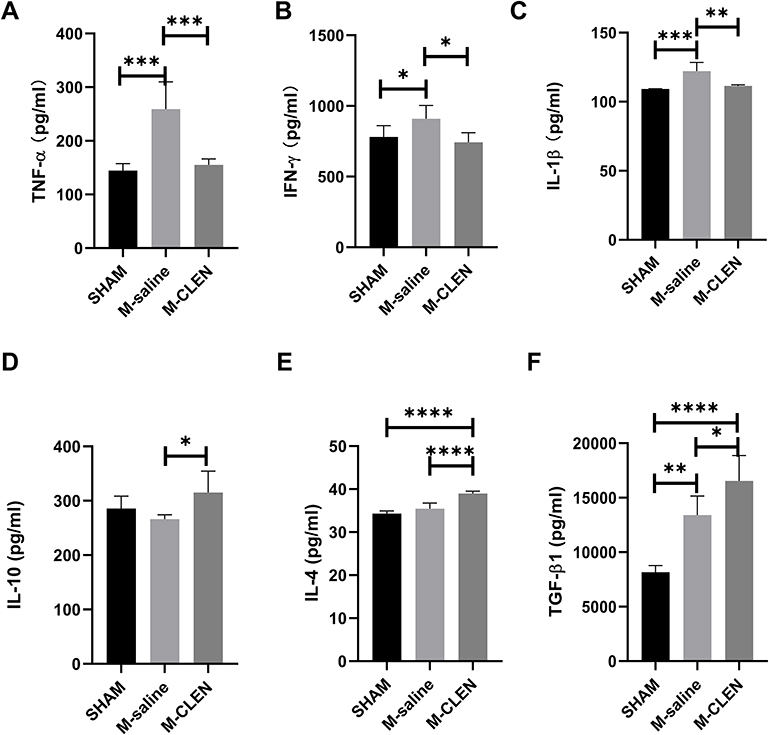 |
Figure 5 Effects of stroke and β2-ARs agonist CLEN application on the expression of blood cytokines in mice. (A) Pro-inflammatory cytokines TNF-α, (B) IFN-γ levels, and (C) IL-1β in sham-operated and saline, CLEN treated MCAO mice 72 h after occlusion. (D) Anti-inflammatory cytokines IL-10, (E) IL-4, and (F) TGF-β1 levels were assessed in sham-operated and saline, CLEN treated MCAO mice 72 h after occlusion. *p < 0.05, **p < 0.01, ***p < 0.001 and ****Indicates p < 0.0001 (one-way ANOVA with Holm–Sidak correction, n = 5 per group in all the analysis of cytokines). |
β2-ARs Agonist CLEN Inhibits or Limits Bacterial Pneumonia
To test whether β2-ARs agonist CLEN treatment in MCAO animals will get a higher risk of bacterial pneumonia, we conducted a histological examination of lungs 72 h after MCAO. Histological examination revealed more or less typical signs of bacterial pneumonia, showing thickened alveolar septa, intra-alveolar inflammatory infiltration, and interstitial congestion in all the mice subjected to the M-saline group (n = 5 per group, Figure 6A), while only one mouse in five had signs of pneumonia in the M-CLEN group. The remaining four HE-stained lungs in the M-CLEN group showed no signs of pneumonia, typically seen in the representative 12-µm section of HE-stained lungs (n = 5, Figure 6A). Histopathological scores of the three groups (SHAM, M-saline, M-CLEN groups) are shown in Figure 6B. All histopathological scores in the M-saline group were 2, while only one in five of the histopathological scores in the M-CLEN group was 2. These data are statistically shown as an outlier by the ROUT method and excluded from the group. Subsequently, we concluded that CLEN treatment provided inhibition or limitation of pneumonia.
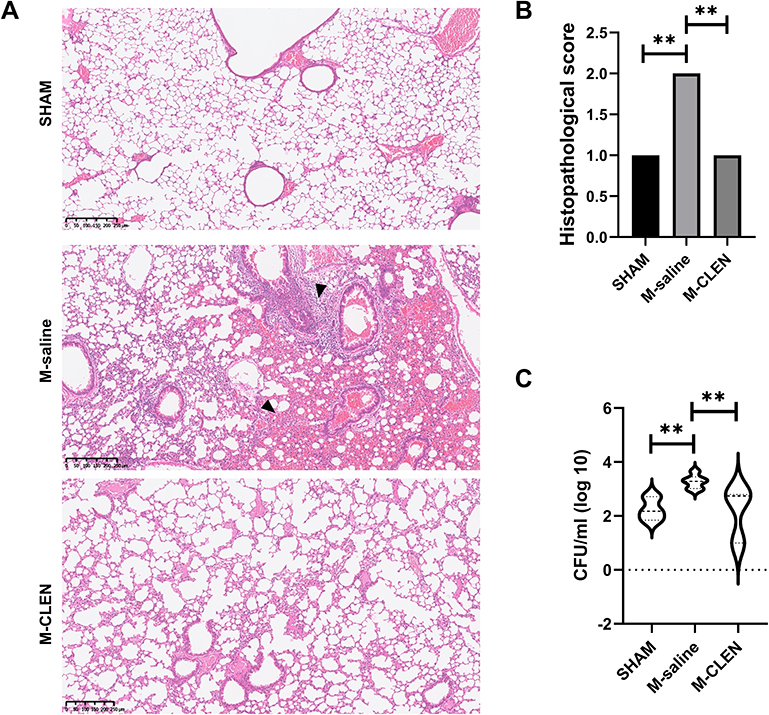 |
Figure 6 β2-ARs agonist CLEN inhibits or limits pneumonia in post-stroke MCAO mice. MCAO mice received the CLEN or only diluent after stroke, as described in Materials and Methods. BALF and lung samples were collected 72 h after stroke. (A) Lungs from sham-operated and CLEN or diluent-treated MCAO mice (each group n = 5) were collected after 72 h for histological examination. A representative 12-µm section of HE-stained lung from CLEN-treated MCAO revealed signs of pneumonia (thickening of alveolar walls and neutrophilic infiltrates) ×40, arrows. (B) Quantitative analysis of histopathological lung injury scores (n = 5 animals per group). Kruskal–Wallis test, **p < 0.01. (C) BALF samples from sham-operated and CLEN or diluents-treated MCAO mice (n = 8 per group) were collected for bacteriological analysis 72 h after stroke. Data are given in CFU/mL (log 10) BALF. One-way ANOVA with Holm–Sidak correction. **p < 0.01. |
Bacterial counts in BALF were simultaneously determined 72 h after MCAO. Microbiological analysis revealed significant bacterial loads in BALF cultures of saline-treated stroke mice at 72 h after MCAO (Figure 6C). In addition, little or no bacterial growth was observed in the BALF cultures of either CLEN-treated MCAO mice or sham-operated, consistent with histological examination.
β2-ARs Agonist CLEN Affects Cytokine Release in Lung Tissues
Pro-inflammatory cytokines such as TNF-α, IL-1β, and IFN-γ are critical for promoting bacterial clearance during lung infections. We found that level of TNF-α, an important pro-inflammatory cytokine that regulates the early phase of inflammation reaction, was elevated 72 h following MCAO operation, and the enhancement was inhibited by CLEN treatment (Figure 7A). However, levels of IL-1β, IFN-γ remained unaltered (Figure 7B and C). Interestingly, IL-4, IL-10, and TGF-β1 were unchanged, decreased, and sharply increased, respectively, in the MCAO mice compared with the SHAM mice. The levels of IL-4 appeared no statistical significance between the M-CLEN and M-saline groups, while the levels of IL-10 and TGF-β1 were elevated in mice subjected to CELN treatment (Figure 7D–F). Overall, these data suggested that mice would suffer from pneumonia 72 h after MCAO stroke, and CLEN treatment could alleviate pneumonia 72 h following MCAO stroke by decreasing production of pro-inflammatory cytokines TNF-α and increasing expression of anti-inflammatory mediator IL-10.
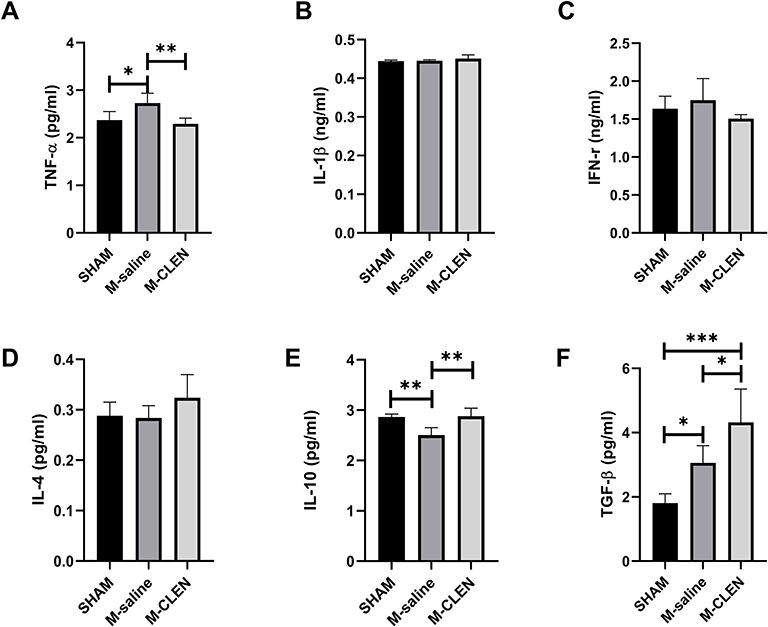 |
Figure 7 Effects of β2-ARs agonist CLEN on the expression of lung cytokines in MCAO mice. (A) Proinflammatory cytokines TNF-α, (B) IL-1β, and (C) IFN-γ levels in the sham-operated and saline, CLEN treated MCAO mice 72 h after occlusion. (D) Anti-inflammatory cytokines IL-4, (E) IL-10, and (F) TGF-β1 levels were assessed in the sham-operated and saline, CLEN treated MCAO mice 72 h after occlusion. *p < 0.05, **Indicates p < 0.01, ***Told p < 0.001 by one-way ANOVA with Holm–Sidak correction, n = 5 per group in all the analysis of cytokines. |
Discussion
This paper demonstrated that the SNS activity was raised after cerebral infarction. Considering that β2-ARs were involved in the immune modulation after MCAO stroke, we evaluated the impact of selective β2-ARs agonist CLEN on the outcomes of MCAO stroke and found that it inhibited or limited pneumonia but expanded infarct area of MCAO stroke.
The results in the literature on the effects of CLEN (1 mg/kg) application on cerebral outcome after stroke were controversial.9,28 Considering that early immune responses are beneficial, we treated MCAO mice with CLEN (1 mg/kg) 24 h after MCAO stroke, and the results showed undesired enlargement of infarct size. Kendra J, in his study, partly attributed the enlargement of infarct volume to the reduction in blood pressure and, therefore, cerebral perfusion by application of CLEN at the dose of 1 mg/kg. The stroke model in Kendra J’s experiment was a permanent thrombotic stroke model, with lesions limited to the cortical, which did not impact the release of NE transmitter. However, a large stroke was induced in our experimental stroke model, and the NE transmitter was drastically elevated. Though NE level after MCAO insult was slightly decreased by CLEN treatment, it remained mainly above the sham level. As a vasodilator, CLEN may have limited effects on the post-stroke blood pressure due to a burst of NE. In another research on focal cerebral ischemia, 1 mg/kg CLEN was not found to affect the blood pressure of the stroke mice.8 Kendra J, in his study, mainly attributed the enlargement of infarct volume to the disruption of early inflammation after focal ischemic stroke, considering that early inflammation is beneficial for stroke outcomes. In our experiment, we introduced CLEN 24 h after stroke onset to avoid early disruption of the inflammatory response, but the result was still in line with that of Kendra J’s. It seemed that pre-stroke CLEN would get a better outcome than post-stroke application. It is of interest that in the study of Zhu Y, pretreatment of CLEN at 1.0 mg/kg 3 h before transient forebrain ischemia reduced neuronal damage, while administered immediately after ischemic insult, the neuroprotective effect of CLEN was abolished.29 Disparities in the outcomes may be explained by differences in the administration time point of CLEN. Pre-stroke application may down-regulate β2-ARs in the brain,30 hence reducing pro-inflammatory gene expression and infarct size, which was demonstrated on β2-ARs knockout mice model.11 Post-stroke CLEN application exacerbates brain injury. The detailed mechanism needs further study to clarify since the inflammatory response was considered indispensable in the effective resolution of ischemic injured tissue, the exaggerated anti-inflammatory response, which unbalanced immune response in the brain, may be the culprit.31
CLEN expanded infarct volume but counteracted the spleen reduction induced by MCAO. It is well established that the SNS is activated under global or extensive brain infarction, and the NE level rises sharply to stimulate β2-ARs.27,32,33 NE had been shown to induce significant splenic atrophy, a temporary phenomenon that occurred 24 to 48 h after stroke and returned to normal levels by 96 h post-MCAO,34 via activation of α1-adrenergic receptors (α1ARs).35,36 In this study, we observed a dramatic shrink of the spleen size in the M-saline group and thought it was due to elevated emission of the NE transmitter, which resulted in blood expulsion and immune cell migration from the spleen to the cerebral injury site. The inhibited reduction of spleen size in the M-CLEN group may result from the declined level of NE. Our results showed that CLEN counteracted the spleen reduction induced by MCAO but enlarged infarct volume, in agreement with the previous report showing no correlation between spleen size reduction and infarct volume.37
It was proposed that splenic atrophy is an essential feature of stroke-induced peripheral immunodepression, and spleen size reduction is correlated to immunodepression.38,39 To testify whether CLEN influences the immune system consistent with the change of spleen size, we first analyzed the effects of CLEN on adaptive immunity, represented by the percentage of CD3+ total T cell, B cells, and CD3+CD4+, CD3+CD8+ T cells. Then, we tested the effects of CLEN on innate immunity, represented by a population of neutrophils, macrophages, NK cells, and dendritic cells. However, no immunodepression occurred in mice of the M-saline group since no statistical difference was found in the percentage of CD3+ total T cell and percentages of CD3+CD4+, CD3+CD8+ T cells when compared with the SHAM group. The absence of immune deficiency may be due to that infarct volume in the 60 minutes MCAO mice was not large enough to induce immune deficiency. It was reported that rodents with 90 minutes MCAO compared with 30 minutes MCAO, or with infarction volume 54.99 ± 10.03% of the total brain compared with 32.59 ± 6.89%, were more prone to present an immunodepression state.33,40 The mean infarct volume in the M-saline group was only 35.0 ± 7.1%. The results presented here show that percentages of CD3+ total T cells and CD3+CD4+ T cells were reduced, and the percentage of CD3+CD8+ was increased in mice subjected to CLEN treatment, suggesting that CLEN treatment, in general, resulted in an immunosuppressive effect. Our result contradicts the previous result that spleen size reduction is correlated with immunodepression. The discrepancy may result from the increased level of NE detected in the M-CLEN group since treatment with prazosin, an alpha-1 receptor blocker, can prevent the post-stroke reduction in spleen size.37 Our results further showed that splenic atrophy showed little evidence for impairment of normal immune responses, proposed by Wardrop in a clinical study.41
Similarly, the B cell population was dropped sharply in the M-CLEN group. Statistical significance was seen compared with the M-saline group. NA nerves, particularly rich in T-cell zones, are poorly innervated in follicular and nodular zones where most B lymphocytes are located.42 Unlike T lymphocytes, the post-stroke changes in B cells remain elusive due to contradictory results.29,43 The percentage of B lymphocytes in our study declined after MCAO, and CLEN application resulted in a further decrease. It was suggested that B cell was neuroprotective, and the cell count of B cells was positively correlated with infarct outcome.44,45 A more considerable drop found in the B cell counts may be associated with the worse infarct outcome in the mice of the M-CELN group. Conversely, NK cells were detected increased in the M-saline group, and CLEN application resulted in a more profound increase in the NK cell population. It has been demonstrated that cell-intrinsic β-adrenergic signaling is required for adaptive NK cell expansion and memory, and activation of β-adrenergic signaling may potentiate their cytolytic and anti-viral/tumor effector functions.46,47 NK cell numbers in the ischemic brain of mice subjected to MCAO peaked during the early phase of stroke onset, then gradually declined by 3 to 10 d later.48 In mice with large infarcts induced by MCAO, NK cells promoted local inflammation and neuronal hyperactivity, lost tolerance to neurons and exacerbated brain infarction.49,50 Hence, the vast recruitment of NK cells may account for the exacerbation of ischemic injury by CLEN.
Up to date, there is still no consensus on reasons for the high incidence of pneumonia in patients sustaining a stroke. It was suggested that aspiration was a critical factor in pneumonia and mortality in stroke patients. However, aspiration alone cannot explain the high incidence of stroke-associated pneumonia.51 Aspiration occurs in healthy adults during sleep without inducing pneumonia.52 Recently, more and more studies believed that immunodepression induced by sympathetic hyperactivity was essential for the progression of bacterial aspiration to pneumonia.1 Phan TG’s study concluded that stroke severity status, but not dysphagia or dysphagia screening, contributed to the decision tree model of post-stroke pneumonia.53 In our experiment, though there was no immunodepression presented in the M-saline group, all the mice in this group appeared pneumonia. Consistent with our results, a recent clinical study found a higher incidence of post-stroke infection was found in patients with spleen volume reduction. Nevertheless, there was no decrease in lymphocytes after application of Bonferroni correction in the group with spleen volume reduction, indicating that spleen size reduction, but not immunodepression, was highly related to the incidence of post-stroke infection.54 SNS activation (elevated NE levels), induced by brain inflammatory immune response, was regarded as associated with an impaired immune function and a higher susceptibility to post-stroke infections.55 The data above indicated that CLEN, though lowered the sympathetic tone of (decreased NE levels) stroked mice, still played a role in augmenting the immune/inflammatory suppressive response after stroke. We supposed that CLEN administration would increase the incidence of pneumonia or aggravate pneumonia. Short of our expectation, all mice in the M-saline group developed spontaneous pneumonia 3 d after ischemia, with slight or severe signs of inflammation in lung tissues, while only one mouse in five had signs of pneumonia in the M-CLEN group. We have excluded the data since it is statistically shown as an outlier. The reason may be that the mice have not received enough CLEN. It seemed that CLEN administration would reduce the vulnerability of MCAO mice to pneumonia. As an anti-inflammatory agent, demonstrated in our experiment with inhibition of pro-inflammatory cytokines TNF-α, IL-1β, and IFN-γ, and promotion of anti-inflammatory cytokines IL-10, IL-4, and TGF-β1, CLEN was proved to exert its effects by inhibiting pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, and provide protection in various organs, such as the brain, liver, and renal.56–58 Hence, another explanation may be that CLEN administration 24 h after MCAO stroke, a time point of post-stroke infection initiation, contained the initial lung infection. Though larger samples are required to determine an accurate incidence of pneumonia in MCAO mice with or without CLEN administration, it is compelling that CLEN served a beneficial role in reducing inflammation in lung tissues. Collectively, these data indicated that activation of β2-ARs may exert a protective effect in the setting of inflammation against lung injury. Further evidence provided by cytokine analysis of the lung tissues showed TNF-α, indicating as an early inflammatory cytokine, was down-regulated in the M-CLEN group compared with the M-saline group. At the same time, IL-10, which acts as a mediator for the resolution of inflammation, was elevated in the M-CLEN group compared with the M-saline group, consistent with the two groups’ pathological changes.
Limits
This study has some limitations, including that the sample size performed is too small to determine the accurate incidence of pneumonia after CLEN treatment in MCAO mice as well as no detailed mechanisms of how β2-ARs agonist CLEN exaggerated infarct volume are characterized. Thus, further studies are needed.
Conclusion
Our study is the first to evaluate both central and peripheral effects of β2-adrenergic receptor stimulation. Our work shows that increasing β2-adrenergic receptor stimulation after ischemic stroke suppresses the peripheral immune response, inhibits or limits post-stroke pneumonia but leads to larger stroke volume. Given that many stroke patients are prescribed sympathomimetic adrenergic agonists to maintain blood pressure and cerebral perfusion, this work highlights the role of the β2-adrenergic signaling pathway on post-stroke immune responses and outcomes.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Grant No. 82172143).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Prass K, Braun JS, Dirnagl U, et al. Stroke propagates bacterial aspiration to pneumonia in a model of cerebral ischemia. Stroke. 2006;37(10):2607–2612. doi:10.1161/01.STR.0000240409.68739.2b
2. Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40(5):1849–1857. doi:10.1161/STROKEAHA.108.534503
3. Martín-Cordero L, García JJ, Hinchado MD, et al. Influence of exercise on NA- and Hsp72-induced release of IFN γ by the peritoneal suspension of macrophages and lymphocytes from genetically obese Zucker rats. J Physiol Biochem. 2013;69(1):125–131. doi:10.1007/s13105-012-0196-5
4. Bosmann M, Grailer JJ, Zhu K, et al. Anti-inflammatory effects of β2 adrenergic receptor agonists in experimental acute lung injury. FASEB J. 2012;26(5):2137–2144. doi:10.1096/fj.11-201640
5. Hervé J, Haurogné K, Bacou E, et al. β2-adrenergic stimulation of dendritic cells favors IL-10 secretion by CD4+ T cells. Immunol Res. 2017;65(6):1156–1163. doi:10.1007/s12026-017-8966-3
6. Ağaç D, Estrada LD, Maples R, et al. The β2-adrenergic receptor controls inflammation by driving rapid IL-10 secretion. Brain Behav Immun. 2018;74:176–185. doi:10.1016/j.bbi.2018.09.004
7. Culmsee C, Semkova I, Krieglstein J. NGF mediates the neuroprotective effect of the beta2-adrenoceptor agonist clenbuterol in vitro and in vivo: evidence from an NGF-antisense study. Neurochem Int. 1999;35(1):47–57. doi:10.1016/S0197-0186(99)00032-7
8. Culmsee C, Stumm RK, Schäfer MK, et al. Clenbuterol induces growth factor mRNA, activates astrocytes, and protects rat brain tissue against ischemic damage. Eur J Pharmacol. 1999;379(1):33–45. doi:10.1016/S0014-2999(99)00452-5
9. Lechtenberg KJ, Meyer ST, Doyle JB, et al. Augmented β2-adrenergic signaling dampens the neuroinflammatory response following ischemic stroke and increases stroke size. J Neuroinflammation. 2019;16(1):112. doi:10.1186/s12974-019-1506-4
10. Han RQ, Ouyang YB, Xu L, et al. Postischemic brain injury is attenuated in mice lacking the beta2-adrenergic receptor. Anesth Analg. 2009;108(1):280–287. doi:10.1213/ane.0b013e318187ba6b
11. White RE, Palm C, Xu L, et al. Mice lacking the β2 adrenergic receptor have a unique genetic profile before and after focal brain ischaemia. ASN Neuro. 2012;4:343–356. doi:10.1042/AN20110020
12. Winklewski PJ, Radkowski M, Demkow U. Cross-talk between the inflammatory response, sympathetic activation and pulmonary infection in the ischemic stroke. J Neuroinflammation. 2014;11:213. doi:10.1186/s12974-014-0213-4
13. Katzan IL, Cebul RD, Husak SH, et al. The effect of pneumonia on mortality among patients hospitalized for acute stroke. Neurology. 2003;60:620–625. doi:10.1212/01.WNL.0000046586.38284.60
14. Liu T, Yang L, Han X, et al. Local sympathetic innervations modulate the lung innate immune responses. Sci Adv. 2020;6(20):eaay1497. doi:10.1126/sciadv.aay1497
15. Prass K, Meisel C, Höflich C, et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. 2003;198(5):725–736. doi:10.1084/jem.20021098
16. Wong CHY, Jenne CN, Lee WY, et al. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science. 2011;334(6052):101–105. doi:10.1126/science.1210301
17. Cardoso-Sousa L, Aguiar EMG, Caixeta DC, et al. Effects of salbutamol and phlorizin on acute pulmonary inflammation and disease severity in experimental sepsis. PLoS One. 2019;14(9):e0222575. doi:10.1371/journal.pone.0222575
18. Chiang T, Messing RO, Chou WH. Mouse model of middle cerebral artery occlusion. J Vis Exp. 2011;(48):2761. doi:10.3791/2761
19. Liang X, Hu Q, Li B, et al. Follistatin-like 1 attenuates apoptosis via disco-interacting protein 2 homolog A/Akt pathway after middle cerebral artery occlusion in rats. Stroke. 2014;45(10):3048–3054. doi:10.1161/STROKEAHA.114.006092
20. Bieber M, Werner RA, Tanai E, et al. Stroke-induced chronic systolic dysfunction driven by sympathetic overactivity. Ann Neurol. 2017;82(5):729–743. doi:10.1002/ana.25073
21. Gendron A, Teitelbaum J, Cossette C, et al. Temporal effects of left versus right middle cerebral artery occlusion on spleen lymphocyte subsets and mitogenic response in Wistar rats. Brain Res. 2002;955(1–2):85–97. doi:10.1016/S0006-8993(02)03368-1
22. Myers MG, Norris JW, Hachniski VC, et al. Plasma norepinephrine in stroke. Stroke. 1981;12(2):200–204. doi:10.1161/01.STR.12.2.200
23. D’Alessio FR, Tsushima K, Aggarwal NR, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest. 2009;119(10):2898–2913. doi:10.1172/JCI36498
24. Sander D, Winbeck K, Klingelhöfer J, et al. Prognostic relevance of pathological sympathetic activation after acute thromboembolic stroke. Neurology. 2001;57(5):833–838. doi:10.1212/WNL.57.5.833
25. Bergquist J, Tarkowski A, Ewing A, et al. Catecholaminergic suppression of immunocompetent cells. Immunol Today. 1998;19(12):562–567. doi:10.1016/S0167-5699(98)01367-X
26. Felten SY, Olschowka J. Noradrenergic sympathetic innervation of the spleen: II. Tyrosine hydroxylase (TH)-positive nerve terminals form synaptic like contacts on lymphocytes in the splenic white pulp. J Neurosci Res. 1987;18(1):37–48. doi:10.1002/jnr.490180108
27. Hug A, Dalpke A, Wieczorek N, et al. Infarct volume is a major determiner of post-stroke immune cell function and susceptibility to infection. Stroke. 2009;40(10):3226–3232. doi:10.1161/STROKEAHA.109.557967
28. Semkova I, Schilling M, Henrich-Noack P, et al. Clenbuterol protects mouse cerebral cortex and rat hippocampus from ischemic damage and attenuates glutamate neurotoxicity in cultured hippocampal neurons by induction of NGF. Brain Res. 1996;717:44–54. doi:10.1016/0006-8993(95)01567-1
29. Zhu Y, Culmsee C, Semkova I, et al. Stimulation of β 2-adrenoceptors inhibits apoptosis in rat brain after transient forebrain ischemia. J Cereb Blood Flow Metab. 1998;18(9):1032–1039. doi:10.1097/00004647-199809000-00013
30. Newman-Tancredi A, Verrièle L, Chaput C, et al. Down-regulation of rat beta-adrenoceptors by clenbuterol or desipramine does not require chronic treatment: [3H] CGP-12177 binding reveals rapid (24 hour) modulation. Brain Res Bull. 1996;41(2):93–96.
31. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi:10.1038/nm.2399
32. Liesz A, Rüger H, Purrucker J, et al. Stress mediators and immune dysfunction in patients with acute cerebrovascular diseases. PLoS One. 2013;8(9):e74839. doi:10.1371/journal.pone.0074839
33. Liesz A, Hagmann S, Zschoche C, et al. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke. 2009;40(8):2849–2858. doi:10.1161/STROKEAHA.109.549618
34. Seifert HA, Hall AA, Chapman CB, et al. A transient decrease in spleen size following stroke corresponds to splenocyte release into systemic circulation. J Neuroimmune Pharmacol. 2012;7(4):1017–1024. doi:10.1007/s11481-012-9406-8
35. Gillespie JS, Hamilton DN. Binding of noradrenaline to smooth muscle cells in the spleen. Nature. 1966;212(5061):524–525. doi:10.1038/212524a0
36. Aboud R, Shafii M, Docherty JR. Investigation of the subtypes of alpha 1-adrenoceptor mediating contractions of rat aorta, vas deferens and spleen. Br J Pharmacol. 1993;109(1):80–87. doi:10.1111/j.1476-5381.1993.tb13534.x
37. Ajmo CT, Collier LA, Leonardo CC, et al. Blockade of adrenoreceptors inhibits the splenic response to stroke. Exp Neurol. 2009;218:47–55. doi:10.1016/j.expneurol.2009.03.044
38. Felten DL, Felten SY, Carlson SL, et al. Noradrenergic and peptidergic innervation of lymphoid tissue. J Immunol. 1985;135(2 Suppl):755s–765s.
39. Yan FL, Zhang JH. Role of the sympathetic nervous system and spleen in experimental stroke-induced immunodepression. Med Sci Monit. 2014;20:2489–2496. doi:10.12659/MSM.890844
40. Wang H, Deng QW, Peng AN, et al. β-arrestin2 functions as a key regulator in the sympathetic-triggered immunodepression after stroke. J Neuroinflammation. 2018;15(1):102. doi:10.1186/s12974-018-1142-4
41. Wardrop CA, Dagg JH, Lee FD, et al. Immunological abnormalities in splenic atrophy. Lancet. 1975;2(7923):4–7. doi:10.1016/S0140-6736(75)92949-9
42. Offner H, Subramanian S, Parker SM, et al. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006;176(11):6523–6531. doi:10.4049/jimmunol.176.11.6523
43. Haeusler KG, Schmidt WUH, Föhring F, et al. Cellular immunodepression preceding infectious complications after acute ischemic stroke in humans. Cerebrovasc Dis. 2008;25(1–2):50–58. doi:10.1159/000111499
44. Chen Y, Bodhankar S, Murphy SJ, et al. Intrastriatal B-cell administration limits infarct size after stroke in B-cell deficient mice. Metab Brain Dis. 2012;27(4):487–493. doi:10.1007/s11011-012-9317-7
45. Ren X, Akiyoshi K, Dziennis S, et al. Regulatory B cells limit CNS inflammation and neurologic deficits in murine experimental stroke. J Neurosci. 2011;31(23):8556–8563. doi:10.1523/JNEUROSCI.1623-11.2011
46. Tarr AJ, Powel D, Reader BF, et al. β-adrenergic receptor mediated increases in activation and function of natural killer cells following repeated social disruption. Brain Behav Immun. 2012;26(8):1226–1238. doi:10.1016/j.bbi.2012.07.002
47. Diaz-Salazar C, Bou-Puerto R, Mujal AM, et al. Cell-intrinsic adrenergic signaling controls the adaptive NK cell response to viral infection. J Exp Med. 2020;217(4):e20190549. doi:10.1084/jem.20190549
48. Liu Q, Jin WN, Liu Y, et al. Brain ischemia suppresses immunity in the periphery and brain via different neurogenic innervations. Immunity. 2017;46(3):474–487. doi:10.1016/j.immuni.2017.02.015
49. Gan Y, Liu Q, Wu W, et al. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc Natl Acad Sci U S A. 2014;111(7):2704–2709. doi:10.1073/pnas.1315943111
50. Zhang Y, Gao Z, Wang D, et al. Accumulation of natural killer cells in ischemic brain tissues and the chemotactic effect of IP-10. J Neuroinflammation. 2014;11:79. doi:10.1186/1742-2094-11-79
51. Perry L, Love CP. Screening for dysphagia and aspiration in acute stroke: a systematic review. Dysphagia. 2001;16(1):7–18. doi:10.1007/PL00021290
52. Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med. 2001;344(9):665–671. doi:10.1056/NEJM200103013440908
53. Phan TG, Kooblal T, Matley C, et al. Stroke severity versus dysphagia screen as driver for post-stroke pneumonia. Front Neurol. 2019;10:16. doi:10.3389/fneur.2019.00016
54. Nous A, Peeters I, Nieboer K, et al. Post-stroke infections associated with spleen volume reduction: a pilot study. PLoS One. 2020;15(5):e0232497. doi:10.1371/journal.pone.0232497
55. Harms H, Reimnitz P, Bohner G, et al. Influence of stroke localization on autonomic activation, immunodepression, and post-stroke infection. Cerebrovasc Dis. 2011;32(6):552–560. doi:10.1159/000331922
56. O’Neill E, Yssel JD, McNamara C, et al. Pharmacological targeting of β 2-adrenoceptors is neuroprotective in the LPS inflammatory rat model of Parkinson’s disease. Br J Pharmacol. 2020;177(2):282–297. doi:10.1111/bph.14862
57. Izeboud CA, Hoebe KHN, Grootendorst AF, et al. Endotoxin-induced liver damage in rats is minimized by beta 2-adrenoceptor stimulation. Inflamm Res. 2004;53(3):93–99. doi:10.1007/s00011-003-1228-y
58. Nakamura A, Johns EJ, Imaizumi A, et al. Beta (2)-adrenoceptor agonist suppresses renal tumour necrosis factor and enhances interleukin-6 gene expression induced by endotoxin. Nephrol Dial Transplant. 2000;15(12):1928–1934. doi:10.1093/ndt/15.12.1928
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.