Back to Journals » Drug Design, Development and Therapy » Volume 11
Cisplatin induces protective autophagy through activation of BECN1 in human bladder cancer cells
Authors Lin JF ![]() , Lin YC, Tsai TF
, Lin YC, Tsai TF ![]() , Chen HE, Chou KY, Hwang TIS
, Chen HE, Chou KY, Hwang TIS
Received 3 November 2016
Accepted for publication 14 February 2017
Published 16 May 2017 Volume 2017:11 Pages 1517—1533
DOI https://doi.org/10.2147/DDDT.S126464
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Manfred Ogris
Ji-Fan Lin,1 Yi-Chia Lin,2 Te-Fu Tsai,2,3 Hung-En Chen,2 Kuang-Yu Chou,2,3 Thomas I-Sheng Hwang2–4
1Central Laboratory, Shin Kong Wu Ho-Su Memorial Hospital, Taipei, 2Division of Urology, School of Medicine, Fu-Jen Catholic University, New Taipei, 3Division of Urology, Department of Surgery, Shin Kong Wu Ho-Su Memorial Hospital, 4Department of Urology, Taipei Medical University, Taipei, Taiwan
Purpose: Cisplatin-based chemotherapy is the first line treatment for several cancers including bladder cancer (BC). Autophagy induction has been implied to contribute to cisplatin resistance in ovarian cancer; and a high basal level of autophagy has been demonstrated in human bladder tumors. Therefore, it is reasonable to speculate that autophagy may account for the failure of cisplatin single treatment in BC. This study investigated whether cisplatin induces autophagy and the mechanism involved using human BC cell lines.
Materials and methods: Human BC cells (5637 and T24) were used in this study. Cell viability was detected using water soluble tetrazolium-8 reagents. Autophagy induction was detected by monitoring the levels of light chain 3 (LC3)-II and p62 by Western blot, LC3-positive puncta formation by immunofluorescence, and direct observation of the autophagolysosome (AL) formation by transmission electron microscopy. Inhibitors including bafilomycin A1 (Baf A1), chloroquine (CQ), and shRNA-based lentivirus against autophagy-related genes (ATG7 and ATG12) were utilized. Apoptosis level was detected by caspase 3/7 activity and DNA fragmentation.
Results: Cisplatin decreased cell viability and induced apoptosis of 5637 and T24 cells in a dose- and time-dependent manner. The increased LC3-II accumulation, p62 clearance, the number of LC3-positive puncta, and ALs in cisplatin-treated cells suggested that cisplatin indeed induces autophagy. Inhibition of cisplatin-induced autophagy using Baf A1, CQ, or ATG7/ATG12 shRNAs significantly enhanced cytotoxicity of cisplatin toward BC cells. These results indicated that cisplatin induced protective autophagy which may contribute to the development of cisplatin resistance and resulted in treatment failure. Mechanistically, upregulation of beclin-1 (BECN1) was detected in cisplatin-treated cells, and knockdown of BECN1 using shRNA attenuated cisplatin-induced autophagy and subsequently enhanced cisplatin-induced apoptosis.
Conclusion: Collectively, the study results indicated that cisplatin-induced autophagy is mediated by BECN1 in BC cells. Therefore, combinative treatment using cisplatin and autophagy inhibitors could potentially overcome cisplatin resistance related to autophagy induction.
Keywords: autophagy inhibition, autophagy related genes, apoptosis, cisplatin resistance, human urinary bladder urothelial carcinoma, lentiviral-based shRNA
Introduction
Bladder cancer (BC) is the seventh most common neoplasia in males worldwide.1 In Taiwan, BC was the ninth leading cause of cancer-related death in men in 2011.2 BC remains one of the most expensive cancers with regard to treatment and the monitoring of cytological changes, such as surveillance cystoscopy and periodic imaging. However, despite the existence of appropriate therapies, patients are continually under the threat of ongoing recurrence and muscle invasion. Therefore, the development of new treatment strategies to reduce the risk of recurrence and progression is strongly desired.
Cisplatin, a coordination complex of platinum which was discovered in early 1960s, has been developed as a single agent or in combination with other anticancer drugs to be an important chemotherapeutic drug for the treatment of many cancers, including BC.3,4 However, the efficacy of cisplatin-based combination chemotherapy is limited due to drug resistance or the development of cellular resistance and severe side effects during treatment. Hence, an increased survival rate cannot always be expected.
Autophagy is known to protect cells from metabolic stress-induced cell death, such as starvation, hypoxia, and endoplasmic reticulum stress.5 The process involves the formation of a double-membraned vesicle that encapsulates cytoplasm and organelles, fusing with lysosome to degrade the contents of the vesicle.6 Autophagy has been demonstrated to play important roles in the development of numerous diseases, including infections and neurodegenerative and cardiovascular diseases.7 Cancer cells have been reported to induce autophagy against anticancer treatments by helping cells to evade apoptotic pathway.8 Several signaling molecules have been involved in the regulation of autophagy, including mammalian target of rapamycin (mTOR), 5′-AMP-activated protein kinase (AMPK), and extracellular signal regulated kinase.9 mTOR kinase activation in the presence of growth factors or nutrition-rich conditions results in inhibition of autophagy, while AMPK activation in response to low energy or nutrient deprivation induces autophagy.10 Autophagic process involves subcellular rearrangements that include de novo membrane formation, autophagosomes formation, and fusion of lysosomes to autophagosomes for degradation or reuse of engulfed macromolecules.11 Recent studies have indicated that cisplatin treatment induces protective autophagy in many types of cancer cells, leading to cisplatin resistance.12–14 However, the role of autophagy in cisplatin resistance in BC cells is still not clear.
This study hypothesized that cisplatin induces autophagy as a resistant mechanism for cancer cell survival in human BC cells. The cooperative inhibition of autophagy that is a vital strategy against cisplatin resistance was investigated in human BC cells.
Materials and methods
Chemicals
Cisplatin was purchased from Sigma (St Louis, MO, USA), and the stock solution was prepared at a concentration of 2 mM in 0.95% NaCl solution, pH 7.4; aliquots were stored at −20°C. All other chemicals, unless otherwise stated, were from Sigma. Antibodies against light chain 3 (LC3), p62, cleaved caspase 3 (c-Casp3), poly(adenosine diphosphate ribose) polymerase (PARP), caspase 9 (Casp9), cleaved caspase 8 (c-Casp8), ATG7, ATG12, and beclin-1 (BECN1) were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against B-cell lymphoma 2 (Bcl-2) and β-actin were from Sigma. All primary antibodies were diluted (1:10,000 dilution for β-actin and 1:1,000 for others) in ECL Advance Blocking Solution (Sigma), according to the manufacturer’s instructions.
Cell culture
The human cancer cell lines 5637 (ATCC#HTB-9) and T24 (ATCC#HTB-4) were purchased from Bioresource Collection and Research Center (Hsinchu, Taiwan) and cultured in RPMI-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA). Media were supplemented with 10% fetal bovine serum, 2 mM GlutaMAX-1, 100 units/mL penicillin, and 100 μg/mL streptomycin. Cells were treated with the indicated concentrations of cisplatin. In some experiments, autophagy inhibitors, bafilomycin A1 (Baf A1) or chloroquine (CQ), were added 1 hour prior to the administration of cisplatin. Control cells received an equal volume of dimethyl sulfoxide (DMSO) when treating with Baf A1 that dissolved in DMSO. The final concentration of DMSO was <0.1%.
Cell viability assays
Cell viability in cells treated with the indicated concentration of cisplatin with or without the 2-hour pretreatment of autophagy inhibitors (Baf A1 or CQ) was detected by using water soluble tetrazolium (WST)-8 reagents (Sigma-Aldrich) as described.15
DNA fragmentation assays
The level of DNA fragmentation in cisplatin-treated cells was determined by using APO-DIRECT TUNEL assay kit from BD Biosciences (San Jose, CA, USA) as described.15
Detection of autophagy
To detect autophagy in cisplatin-treated human BC cells, (A) LC3-II accumulation and p62 degradation were detected by Western blot, (B) LC3-positive puncta were detected by using immunofluorescent detection, and (C) the formation of autophagolysosomes (ALs) was directly monitored in control and cisplatin-treated cells by using transmission electron microscopy (TEM).
Detection of LC3-II accumulation and p62 degradation
Cells were treated with different concentration of cisplatin for 24 hours and subjected to protein extraction. Western blot detection of LC3-II and p62 was performed as described.16,17
Immunofluorescent detection of LC3-positive puncta
To determine the LC3-positive puncta in cisplatin-treated cells, cells were seeded in chamber slides and treated with indicated concentrations of cisplatin for 24 hours. Immunofluorescent detection was then performed as previously described.16
TEM
The T24 cells were seeded in 10 cm dishes overnight prior to the cisplatin treatment with or without the 1 hour pretreatment of 25 μM CQ. Subsequently, cells were collected with trpysin treatment and washed three times with cold phosphate-buffered saline. They were then fixed in fixative buffer (2.5% glutaraldehyde and 4% formaldehyde) and postfixed in 1% osmium tetroxide. The fixed cells were then dehydrated by gradually replacing water with an increasing concentration of ethanol. After the cells reached 100% ethanol, they were infiltrated with a plastic monomer, Spurr resin kit (Electron Microscopy Sciences [EMS], Hatfield, PA, USA; EMS #14300) and cut at 75 nm by using an ultramicrotome (EM-UC7; Leica Microsystems, Wetzlar, Germany). Photos were taken by using TEM (JEOL JEM-1400), 100 kVA.
Detection of apoptosis
Apoptosis induction in cisplatin-treated cells with or without the inhibition of autophagy (by Baf A1 treatment or ATG7/ATG12 knockdown by lentiviral shRNAs) was accessed by activation of Casp3/7 activity; detection of PARP, Casp3, Casp8, and Casp9 cleavage by Western blot; and DNA fragmentation assay by using flow cytometry as previously described.15
Generation of shRNA lentivirus against ATG7 and ATG12
To examine the cisplatin-induced autophagy in human BC cells, lentiviral vectors expressing shRNAs targeting ATG7 and ATG12 were employed. The four pLKO.1-shRNA vectors (obtained from the RNAi Core Facility, Academia Sinica, Taipei, Taiwan) used for ATG7 and ATG12 knockdown are the following: TRCN0000007584 (psh7-1), TRCN0000007587 (psh7-2), TRCN0000007391 (psh12-1), and TRCN0000007392 (psh12-2). pLKO_TRC001 (ASN0000000002) containing scramble shRNA sequences was used as control plasmid (pS). The generation of lentiviral particles was conducted as previously described.15 The expression level of ATG7 and ATG12 in lentivirus-transduced cells was detected to confirm successful knockdown prior to the detection of autophagy or apoptosis level (data not shown).
Statistical analysis
All experiments were performed at least three times for each condition. Data were expressed as the mean ± standard deviation. Student’s t-test was performed and P-values <0.05 were considered to indicate statistical significance.
Results
Cisplatin induces autophagy in BC cells
Cisplatin has been demonstrated to induce apoptosis in T24 BC cells.18 To determine the cytotoxicity of cisplatin on 5637 and T24 cells, cell viability was first detected by using a WST-8 assay. The results showed that cisplatin decreased cell viability in 5637 and T24 cells in a dose- and time-dependent manner (Figure 1A and B), while T24 cells were more resistant to cisplatin than 5637 cells. Next, apoptosis induction in cisplatin-treated cells was determined by the detection of DNA fragmentation. As shown in Figure 1C, 25 μM cisplatin induces significant amount of apoptosis in BC cells. Again, the induced apoptosis is more significant in 5637 compared to T24 cells. In addition to apoptosis, cisplatin has been reported to induce autophagy in lung, esophageal, and ovarian cancer cells.19–21 It has been previously shown that RAD001, an mTOR inhibitor, induces protective autophagy in human BC cell lines.22 Therefore, this analysis focused on the detection of autophagy induction in cisplatin-treated BC cells to investigate whether autophagy contributes to cisplatin-induced cytotoxicity in human BC cells. As shown in Figure 2A, cisplatin induced autophagy in BC cells as a result of a dose-dependent increase in LC3-II level. In addition, the expression level of p62 protein which is known as sequestosome 1, SQSTM1, binding directly to LC3 was significantly reduced (Figure 2A). Furthermore, increased number of LC3 puncta was observed in cisplatin-treated 5637 and T24 cells, showing LC3 aggregation during the formation of autophagosomes (Figure 2B). Cisplatin-induced autophagy in BC cells was examined by detecting formation of ALs in T24 cells treated with or without 25 μM cisplatin for 24 hours by using TEM. As illustrated in Figure 2C, cisplatin treatment resulted in an increased number and area of double-membraned ALs in cisplatin-treated T24 cells.
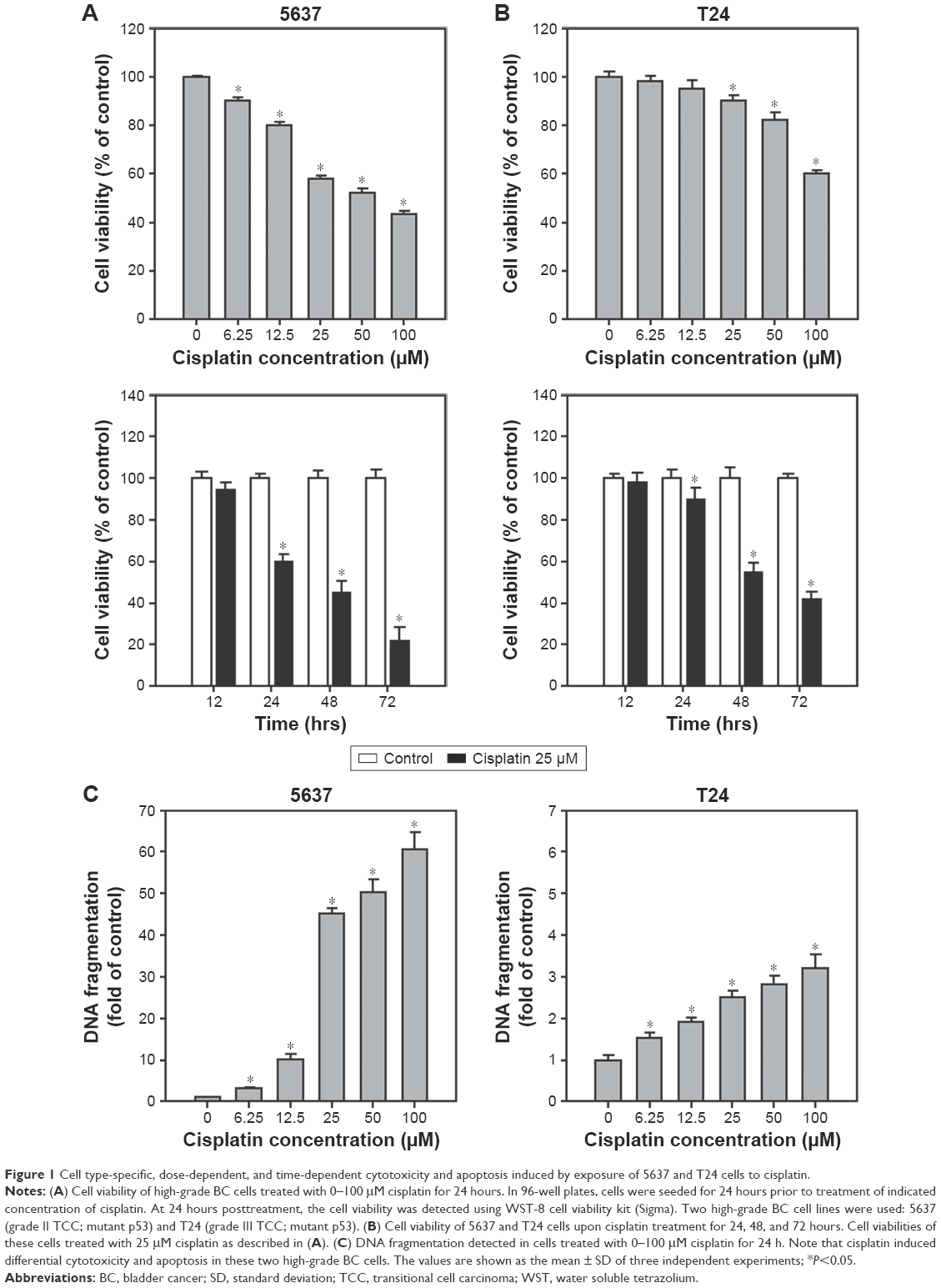 | Figure 1 Cell type-specific, dose-dependent, and time-dependent cytotoxicity and apoptosis induced by exposure of 5637 and T24 cells to cisplatin. |
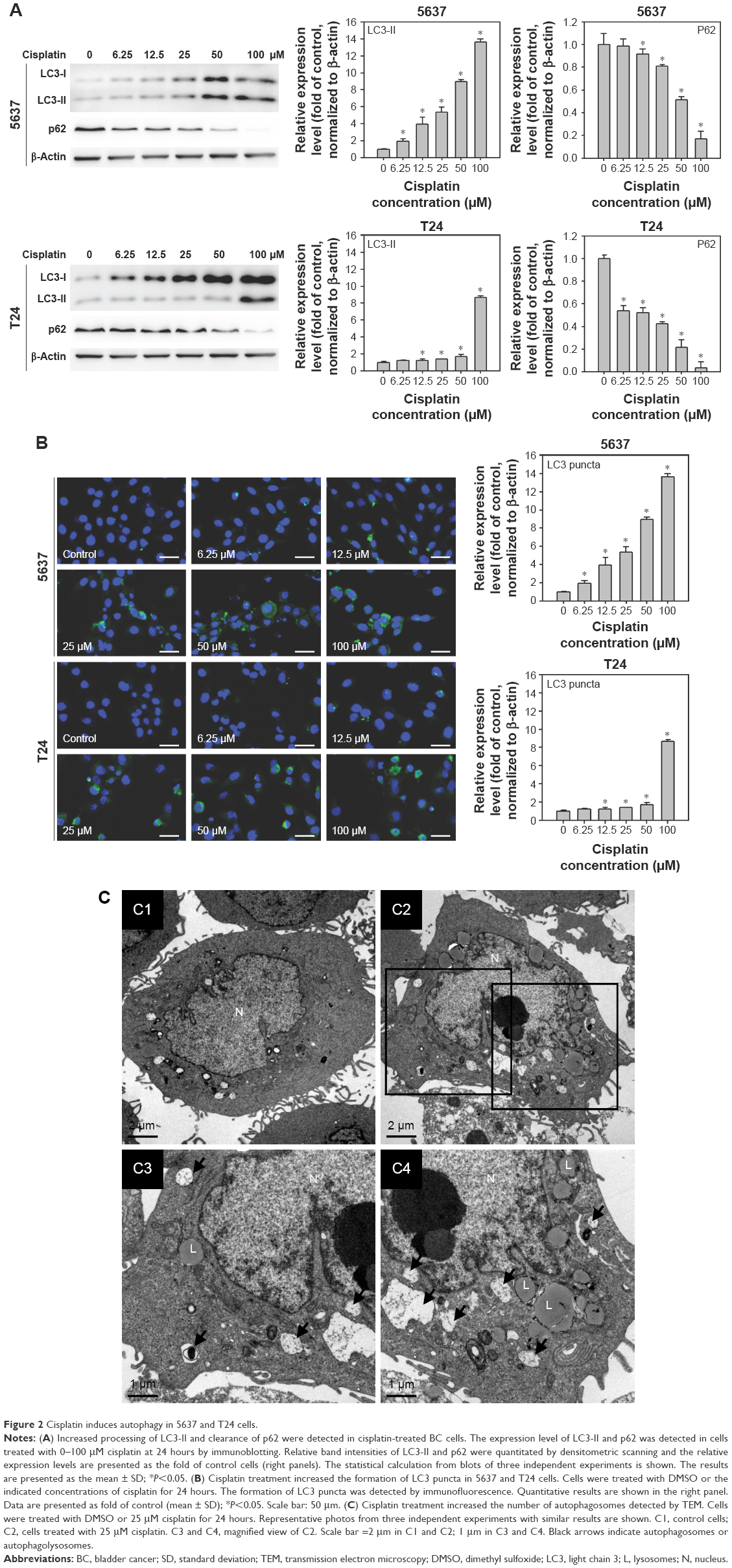 | Figure 2 Cisplatin induces autophagy in 5637 and T24 cells. |
Detection of cisplatin-induced autophagic flux by autophagy inhibitors
Autophagy is a highly dynamic and complex process regulated by multiple steps. Autophagic flux was investigated in cisplatin-treated BC cells by introducing autophagy inhibitors: Baf A1, which is an inhibitor of vacuolar H+ ATPase, or CQ, which is a lysosomotropic agent that blocks the late phase of autophagy by inhibiting fusion between autophagosomes and lysosomes.23 As shown in Figure 3A, a significant amount of LC3-II accumulation was detected in cisplatin-treated BC cells with 200 nM Baf A1 or 25 μM CQ pretreatment. Furthermore, accumulated LC3 puncta in cisplatin-treated cells with the autophagy inhibitors were detected by immunofluorescent (Figure 3B). In addition, increased number of autophagosomes and lysosomes was detected directly in cisplatin-treated T24 cells with the pretreatment of CQ by TEM (Figure 3C). The results showed an increased number of autophagosomes in cisplatin-treated T24 cells (Figure 3C2) compared to control cells (Figure 3C1). CQ, a well-known lysosomotropic agent, inhibits autophagy in its late phase by disrupting the fusion of autophagosomes with lysosomes, resulting in the accumulation of acidic vesicular organelles in the cytoplasm.24 As shown in Figure 3C3, CQ alone increased the number of autophagosomes while the number was further increased in cisplatin-treated T24 cells with the pretreatment of CQ (Figure 3C4). It was also observed that several autophagosomes were closely beneath the lysosomes in cisplatin-treated cells with CQ pretreatment (asterisk mark in Figure 3C4-1 and C4-2). These results demonstrated that Baf A1 and CQ were able to disrupt cisplatin-induced autophagy by blocking the fusion of autophagosomes with lysosomes, and thus compromised the recycling phases of autophagy progress.
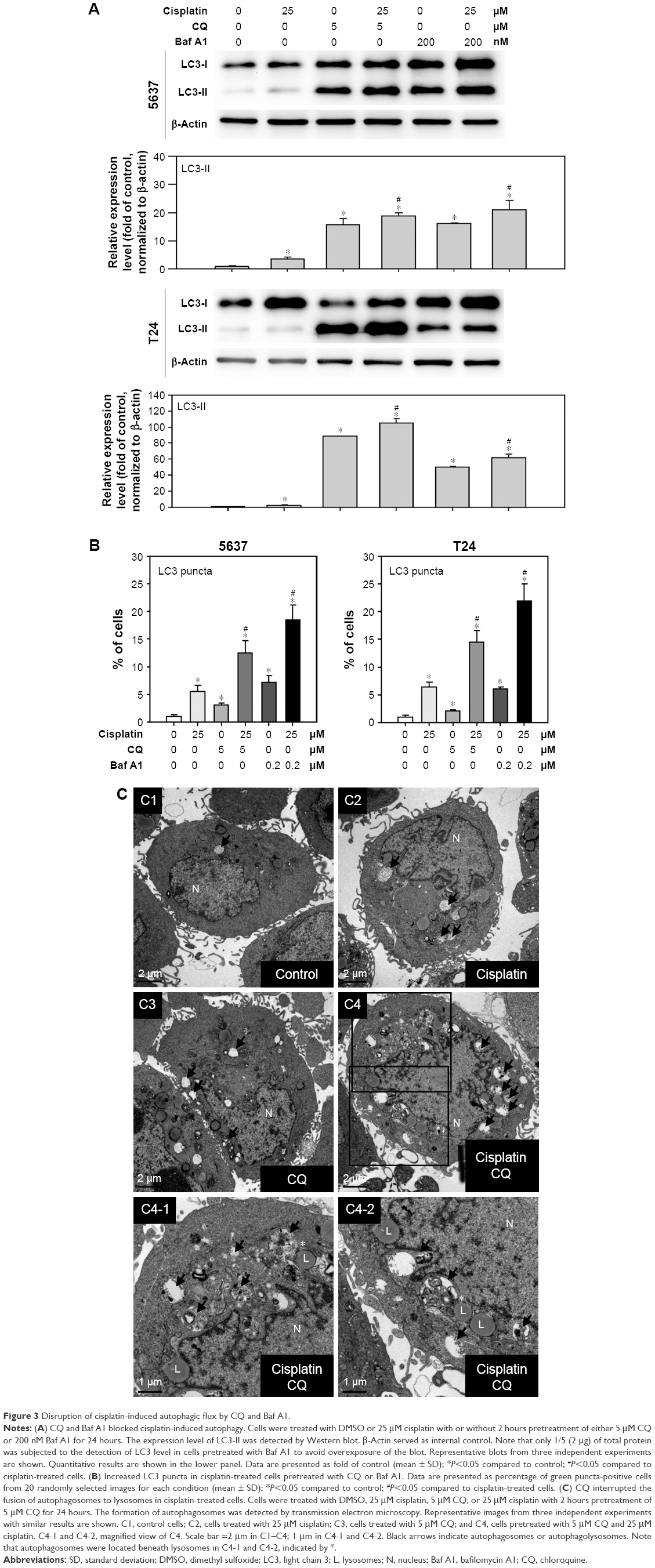 | Figure 3 Disruption of cisplatin-induced autophagic flux by CQ and Baf A1. |
Inhibition of cisplatin-induced autophagy-enhanced apoptosis and reduced cell viability
To further understand the role of cisplatin-induced autophagy in BC cells, the apoptotic markers were first detected in cisplatin-treated BC cells with or without the inhibition of autophagy using Baf A1 or CQ. As shown in Figure 4A, apoptotic proteins including c-Casp3, c-PARP, c-Casp9, and c-Casp8 were increased in 25 μM cisplatin-treated BC cells. Pretreatment of 5 μM CQ or 200 nM Baf A1 further enhanced the activation of these proapoptotic proteins. Consistent with the observed proapoptotic protein profile, DNA fragmentation (Figure 4B) and Casp3/7 activities (Figure 4C) were increased in cisplatin-treated cells pretreated with autophagy inhibitors. In addition, pretreatment of Baf A1 or CQ further reduced cell viability in cisplatin-treated cells, suggesting that cisplatin induces protective autophagy (Figure 4D).
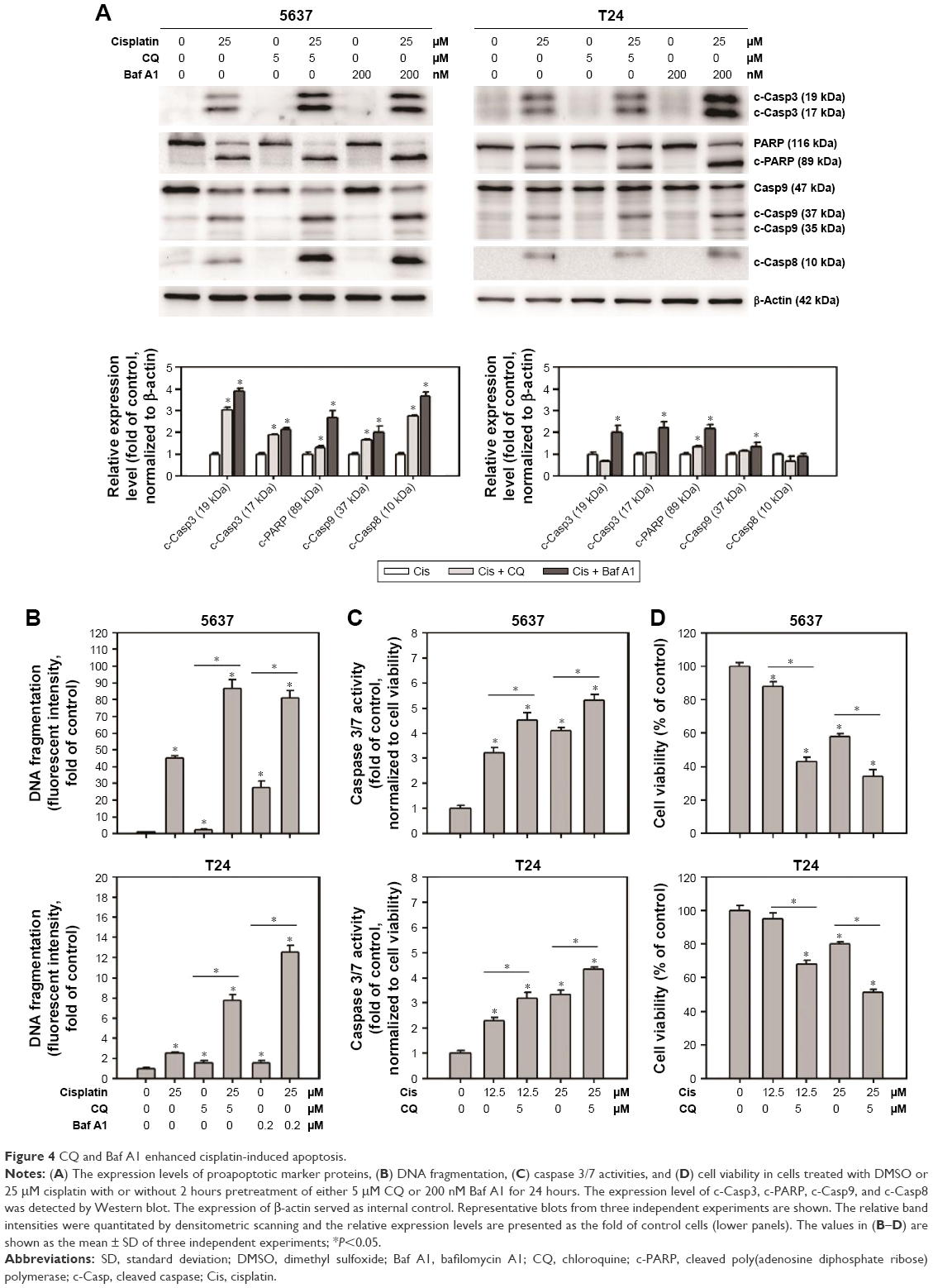 | Figure 4 CQ and Baf A1 enhanced cisplatin-induced apoptosis. |
To further elucidate the inhibition of autophagy-enhanced apoptosis, BC cells were treated with cisplatin followed by transducing lentivirus expressing shRNAs against ATG7 or ATG12, which are involved in the elongation stage of autophagy process.25 As shown in Figure 5, knocking down the expression levels of ATG7 or ATG12 decreased the formation of LC3-II in cisplatin-treated cells and enhanced the processing of c-Casp3. Taken together, these results demonstrated that inhibition of cisplatin-induced autophagy-enhanced apoptotic induction and reduced cell viability in human BC cells.
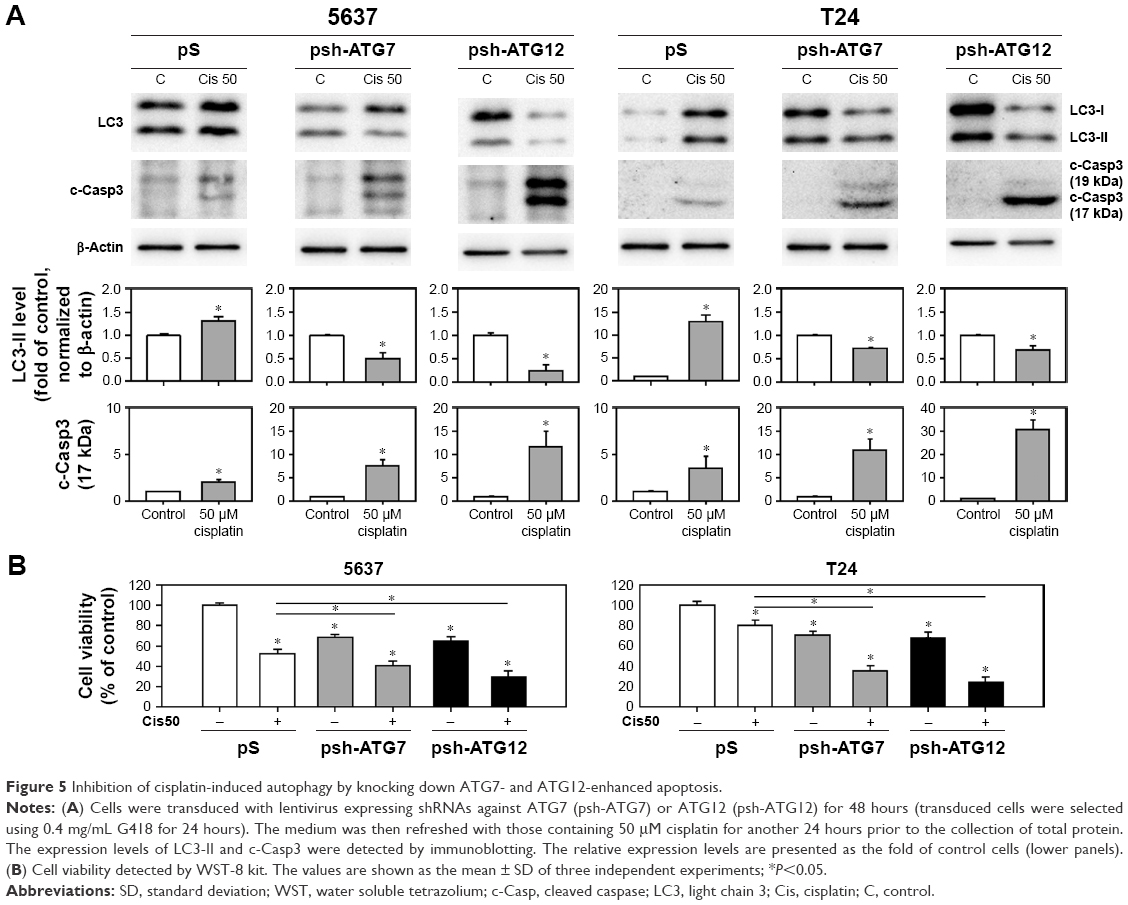 | Figure 5 Inhibition of cisplatin-induced autophagy by knocking down ATG7- and ATG12-enhanced apoptosis. |
BECN1 was upregulated in cisplatin-treated cells and responsible for cisplatin-induced autophagy
BECN1 is suggested to be involved in the sensitivity of chemotherapeutic agents on cisplatin-resistant ovarian cancer cells.26,27 Therefore, this study focused on the expression level of BECN1 in cisplatin-treated human BC cells. As shown in Figure 6A, the expression of BECN1 increased in a dose-dependent manner in 5637 and T24 cells upon cisplatin treatment. Since the interaction of BECN1 and antiapoptotic Bcl-2 is considered to be an important regulatory mechanism of autophagy induction, the expression of Bcl-2 in cisplatin-treated cells was examined. The results showed that Bcl-2 increased in cisplatin-treated cells, but still resulted in an increased level of BECN1/Bcl-2, suggesting a tendency of autophagy induction (Figure 6A). To investigate the role of BECN1 in cisplatin-induced autophagy, lentiviral vector expressing shRNA against BECN1 was employed. In Figure 6B, the expression of BECN1 and LC3-II was increased in scramble shRNA (pS)-transduced cells. In cells transduced with BECN1 shRNA (psh-BECN1), the expression of BECN1 was almost abolished and the processing of LC3-II was inhibited upon cisplatin treatment.
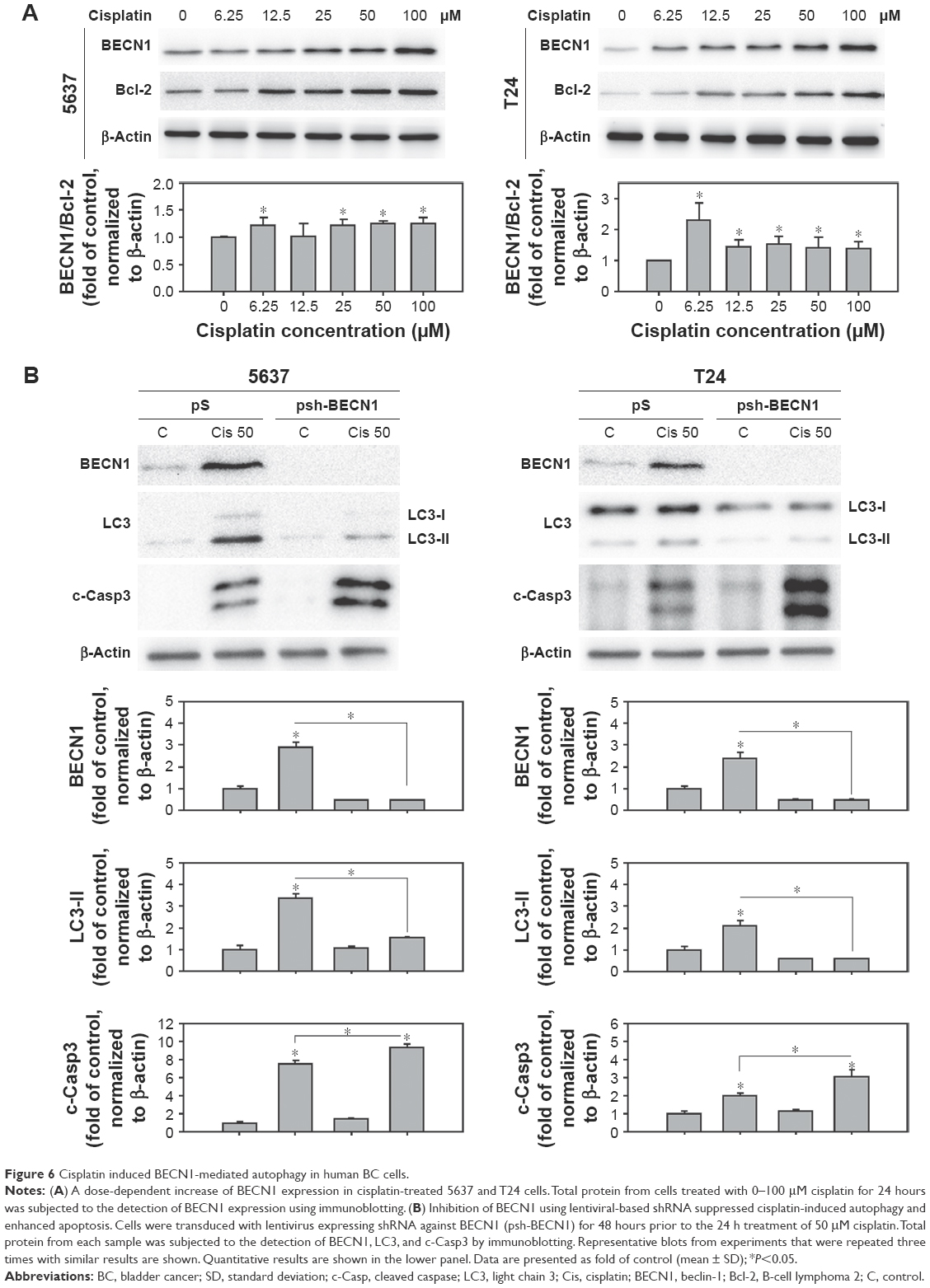 | Figure 6 Cisplatin induced BECN1-mediated autophagy in human BC cells. |
Discussion
Platinum-based (eg, cisplatin-based) chemotherapy remains the first-line treatment for muscle-invasive BC despite much effort to study the genetic alteration in the past decade. However, cisplatin resistance is a major problem in the successful treatment of cancer. Therefore, a better understanding of the mechanism of cisplatin resistance is of clinical significance. This study showed that cisplatin treatment induces a protective autophagic response that may counteract cisplatin-induced cytotoxicity toward human BC cells. Pharmacological inhibition of autophagic activity using Baf A1/CQ or molecular knockdown of ATG7/12 using shRNA-enhanced cisplatin-induced apoptosis. Moreover, this study showed that cisplatin-induced autophagy was mediated by the increased level of BECN1. Knocking down cisplatin-induced BECN1 decreased autophagy induction and enhanced apoptosis.
Over the past decade, mechanisms involving intrinsic or acquired resistance have been intensively studied with the hope to overcome cisplatin resistance.3 To date, known mechanisms determining cisplatin resistance include the regulation of pro and antiapoptotic proteins, drug transport, detoxification, and DNA repair.28 Investigating resistant mechanisms in BC is essential for developing therapeutic strategies for improving cancer therapy.
In 2009, Konstantakou et al had demonstrated that cisplatin induces apoptosis in human BC cells and is associated with p53-dependent and independent responses.18 They showed that cisplatin induces apoptosis in RT4 (low grade, with wild-type p53) and T24 (high grade, with mutant p53) cells, albeit in a dose-specific and grade-dependent manner. In the present study, 5637 (high grade, with mutant p53) and T24 cells were used, because cisplatin-based chemotherapy is usually administrated in muscle-invasive (high grade) BC. As shown in Figure 1, however, differential response to cisplatin was still observed between 5637 and T24 cells (both with mutant p53; the T24 cells carried an in-frame deletion of tyrosine 126, while 5637 cells carried the p53R280T mutant).29,30 One possibility is the p53-independent responses that remained to be investigated further, and the other may due to the differential basal autophagic activity in these two cell types reported previously.15 It was demonstrated here that cisplatin induces autophagy in BC cells (Figures 2 and 3) and may represent a cisplatin-resistant mechanism as seen in other types of cancer.
A recent study showed that primary cultured BC cells derived from cisplatin-resistant patient and T24 cells have higher basal autophagic flux.31 A previous study also demonstrated a grade-dependent increase of basal autophagy among human BC cells.15 It is reasonable to conclude that cisplatin induces protective autophagy in human BC cells, and the induced autophagy may represent a cisplatin-resistant mechanism. To disrupt the autophagic flux, autophagy inhibitors were utilized, and it was demonstrated that inhibition of cisplatin-induced autophagy-enhanced apoptosis (Figure 4). Autophagy inhibitors can be divided into two categories based on their inhibitory action to disrupt the multistep autophagy process: early stage inhibitors and late-stage inhibitors.32 CQ, Baf A1, and lysosomal protease inhibitors are late-stage inhibitors that exhibit suppressive effect downstream of autophagosome formation. These inhibitors inhibit the fusion of autophagosomes and lysosomes and/or block the degradation of autophagic cargo.33 The present study results demonstrated that Baf A1 and CQ block the degradation of LC3-II, subsequently leading to enhanced apoptosis. In addition, TEM images clearly showed that the fusion of autophagosomes and lysosomes is disrupted by CQ treatment in cisplatin-treated T24 cells (Figure 3C). On the other hand, wortmannin, LY294002, and 3-methyladenine (3-MA) that inhibit class III PI3-kinase and block the formation of autophagosomes are early stage autophagy inhibitors. However, these inhibitors were not specific to autophagy inhibition. For example, 3-MA was shown to have a dual role in the modulation of autophagy.34 To avoid misinterpretation of results using these early-stage inhibitors, a molecular approach of using shRNAs against ATG7 and ATG12 that are involved in the formation of autophagosomes to block cisplatin-induced autophagy was chosen. Consistent with the results obtained by using Baf A1 and CQ, knocking down the expression level of ATG7/12 significantly attenuated cisplatin-induced autophagy in human BC cells (Figure 5A). In Figure 5A, the increased LC3 levels in psh-ATG7/12 control (CM) may have resulted from the G418 selection which poses metabolic stress to the transduced cells.
BECN1, a component of the phosphatidylinositol-3-kinase complex, is the mammalian orthologue of yeast ATG6 that plays a central role in autophagy. It has been demonstrated that BECN1 induces autophagy by promoting the formation of core complexes (BECN1-Vps34-Vps15). In contrast, the BH3 domain of BECN1 was shown to bind and inhibit by Bcl-2 or Bcl-xL.35 The interaction of BECN1 and Bcl-2 was considered as an important switch between autophagy and apoptosis.36 Recently, BECN1 was considered as a target to overcome chemotherapy resistance (including cisplatin) in many cancers. For example, inhibition of BECN1 in human ovarian, cervical, and melanoma cancers enhanced cisplatin-induced apoptosis.37–39 The expression level of BECN1 in cisplatin-treated 5637 and T24 cells was dose dependent (Figure 6). In addition, cisplatin-induced BECN1 expression in BC cells was restored in response to shRNA treatment, resulting in enhanced apoptosis. It is suggested that regulation of BECN1 expression in autophagy is associated with nuclear factor (NF) kappa B (NFκB), which is responsible for cisplatin resistance in many cancer types, including BC.40 NFκB has been involved in the regulation of BECN1 transcription and autophagy,41 suggesting that cisplatin activates NFκB incorporating with BECN1 expression in BC cells. However, the relationship between cisplatin treatment and BECN1 upregulation requires further investigation. In addition, this study did not detect the expression of cleaved BECN1. According to a report by Zhu et al, BECN1 is a substrate of Casp3.42 It is possible that activated Casp3 may inactivate autophagy by degrading BECN1 and promote apoptosis in cells upon cisplatin treatment. The results suggest that BECN1 contributes to cisplatin-induced autophagy in human BC cells.
Conclusion
This study demonstrated that cisplatin induces BECN1-mediated autophagy through ATG7/12 pathway. The results suggest that utilizing a lower dose of cisplatin is feasible when combined with autophagy inhibitors as an adjuvant treatment, thereby improving both the dose-dependent toxicity, as well as cisplatin resistance in high-grade BC cells.
Acknowledgments
This work was supported by Shin Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan (SKH-8302-103-0201 to YCL, SKH-8302-103-0201 to TISH, and SKH-8302-102-0202 to JFL) and by the Ministry of Science and Technology, Taiwan (NSC102-2314-B-341-003-MY3 to JFL).
Disclosure
The authors report no conflicts of interest in this work.
References
Dobruch J, Daneshmand S, Fisch M, et al. Gender and bladder cancer: a collaborative review of etiology, biology, and outcomes. Eur Urol. 2015;69(2):300–310. | ||
Lisiak N, Toton E, Rybczynska M. [Autophagy, new perspectives in anticancer therapy]. Postepy Hig Med Dosw (Online). 2014;68:925–935. Polish. | ||
Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573–584. | ||
Cerbone L, Sternberg CN, Sengelov L, et al. Results from a phase I study of lapatinib with gemcitabine and cisplatin in advanced or metastatic bladder cancer: EORTC trial 30061. Oncology. 2015;90(1):21–28. | ||
Lum JJ, Bauer DE, Kong M, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120(2):237–248. | ||
Rosenfeldt MT, Ryan KM. The role of autophagy in tumour development and cancer therapy. Expert Rev Mol Med. 2009;11:e36. | ||
Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(19):1845–1846. | ||
Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(9):726–734. | ||
Klionsky DJ. Coming soon to a journal near you – the updated guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2014;10(10):1691. | ||
Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–937. | ||
Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873. | ||
O’Donovan TR, O’Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7(5):509–524. | ||
Sirichanchuen B, Pengsuparp T, Chanvorachote P. Long-term cisplatin exposure impairs autophagy and causes cisplatin resistance in human lung cancer cells. Mol Cell Biochem. 2012;364(1–2):11–18. | ||
Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem. 2014;289(24):17163–17173. | ||
Lin YC, Lin JF, Wen SI, et al. Inhibition of high basal level of autophagy induces apoptosis in human bladder cancer cells. J Urol. 2016;195(4P1):1126–1135. | ||
Lin JF, Lin YC, Lin YH, et al. Zoledronic acid induces autophagic cell death in human prostate cancer cells. J Urol. 2011;185(4):1490–1496. | ||
Lin JF, Tsai TF, Liao PC, et al. Benzyl isothiocyanate induces protective autophagy in human prostate cancer cells via inhibition of mTOR signaling. Carcinogenesis. 2013;34(2):406–414. | ||
Konstantakou EG, Voutsinas GE, Karkoulis PK, Aravantinos G, Margaritis LH, Stravopodis DJ. Human bladder cancer cells undergo cisplatin-induced apoptosis that is associated with p53-dependent and p53-independent responses. Int J Oncol. 2009;35(2):401–416. | ||
Wu HM, Jiang ZF, Ding PS, Shao LJ, Liu RY. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci Rep. 2015;5:12291. | ||
Xu Y, Yu H, Qin H, et al. Inhibition of autophagy enhances cisplatin cytotoxicity through endoplasmic reticulum stress in human cervical cancer cells. Cancer Lett. 2012;314(2):232–243. | ||
Yu L, Gu C, Zhong D, et al. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett. 2014;355(1):34–45. | ||
Lin JF, Lin YC, Yang SC, et al. Autophagy inhibition enhances RAD001-induced cytotoxicity in human bladder cancer cells. Drug Des Devel Ther. 2016;10:1501–1513. | ||
Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23(1):33–42. | ||
Tasdemir E, Galluzzi L, Maiuri MC, et al. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol. 2008;445:29–76. | ||
Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12. | ||
Sun Y, Liu JH, Jin L, Sui YX, Han LL, Huang Y. Effect of autophagy-related beclin 1 on sensitivity of cisplatin-resistant ovarian cancer cells to chemotherapeutic agents. Asian Pac J Cancer Prev. 2015;16(7):2785–2791. | ||
Bao L, Jaramillo MC, Zhang Z, et al. Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol Med Rep. 2015;11(1):91–98. | ||
Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265–7279. | ||
Cooper MJ, Haluschak JJ, Johnson D, et al. p53 mutations in bladder carcinoma cell lines. Oncol Res. 1994;6(12):569–579. | ||
Rieger KM, Little AF, Swart JM, et al. Human bladder carcinoma cell lines as indicators of oncogenic change relevant to urothelial neoplastic progression. Br J Cancer. 1995;72(3):683–690. | ||
Ojha R, Singh SK, Bhattacharyya S. JAK-mediated autophagy regulates stemness and cell survival in cisplatin resistant bladder cancer cells. Biochim Biophys Acta. 2016;1860(11):2484–2497. | ||
Yang YP, Hu LF, Zheng HF, et al. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin. 2013;34(5):625–635. | ||
Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6(4):304–312. | ||
Wu YT, Tan HL, Shui G, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285(14):10850–10861. | ||
Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. | ||
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752. | ||
Sun Y, Liu JH, Jin L, Sui YX, Lai L, Yang Y. Inhibition of Beclin 1 expression enhances cisplatin-induced apoptosis through a mitochondrial-dependent pathway in human ovarian cancer SKOV3/DDP cells. Oncol Res. 2014;21(5):261–269. | ||
Sun Y, Liu JH, Jin L, et al. Beclin 1 influences cisplatin-induced apoptosis in cervical cancer CaSki cells by mitochondrial dependent pathway. Int J Gynecol Cancer. 2012;22(7):1118–1124. | ||
Del Bello B, Toscano M, Moretti D, Maellaro E. Cisplatin-induced apoptosis inhibits autophagy, which acts as a pro-survival mechanism in human melanoma cells. PLoS One. 2013;8(2):e57236. | ||
Ito Y, Kikuchi E, Tanaka N, et al. Down-regulation of NF kappa B activation is an effective therapeutic modality in acquired platinum-resistant bladder cancer. BMC Cancer. 2015;15:324. | ||
Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol. 2009;29(10):2594–2608. | ||
Zhu Y, Zhao L, Liu L, et al. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell. 2010;1(5):468–477. |
Supplementary materials
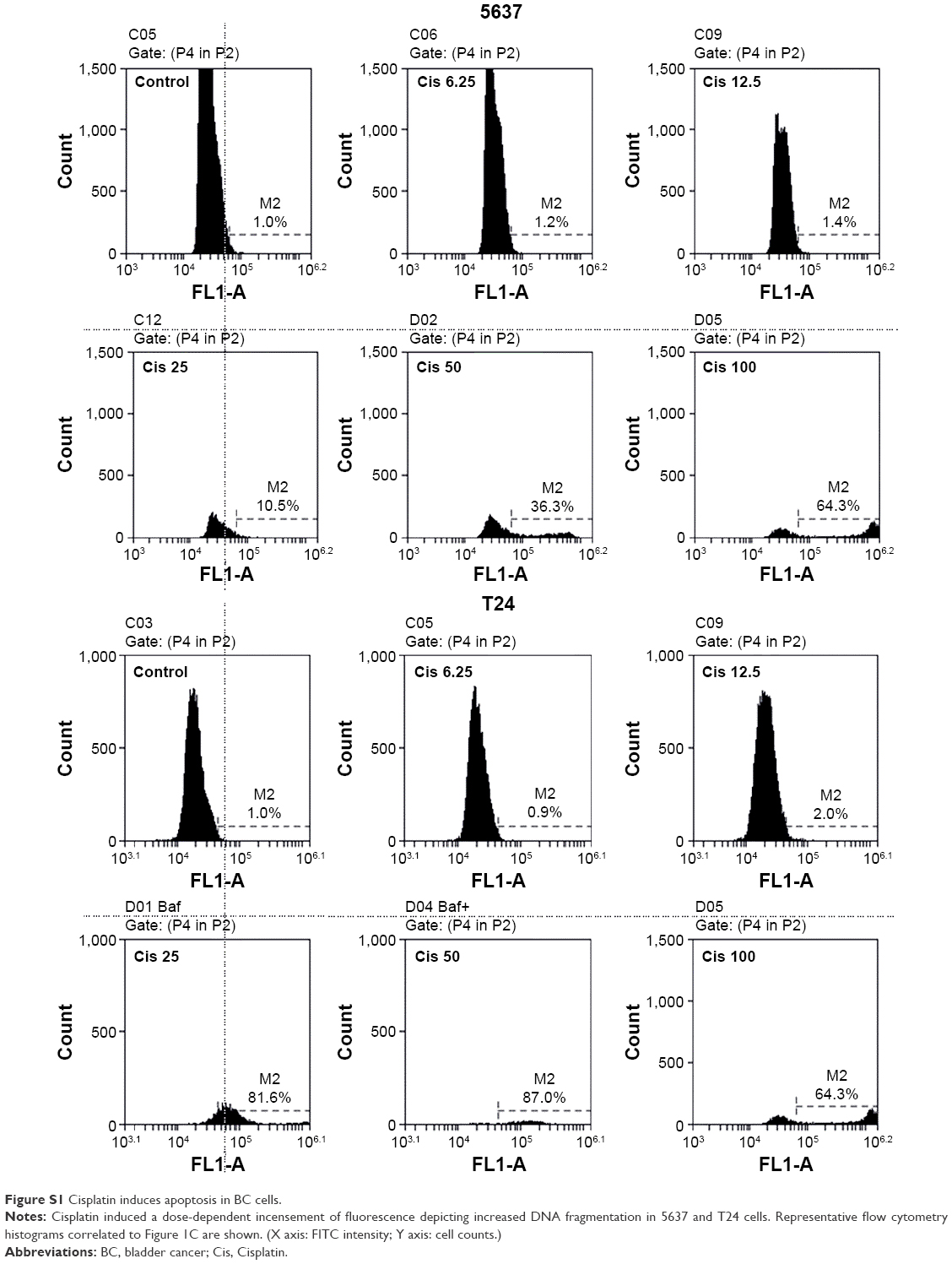 | Figure S1 Cisplatin induces apoptosis in BC cells. |
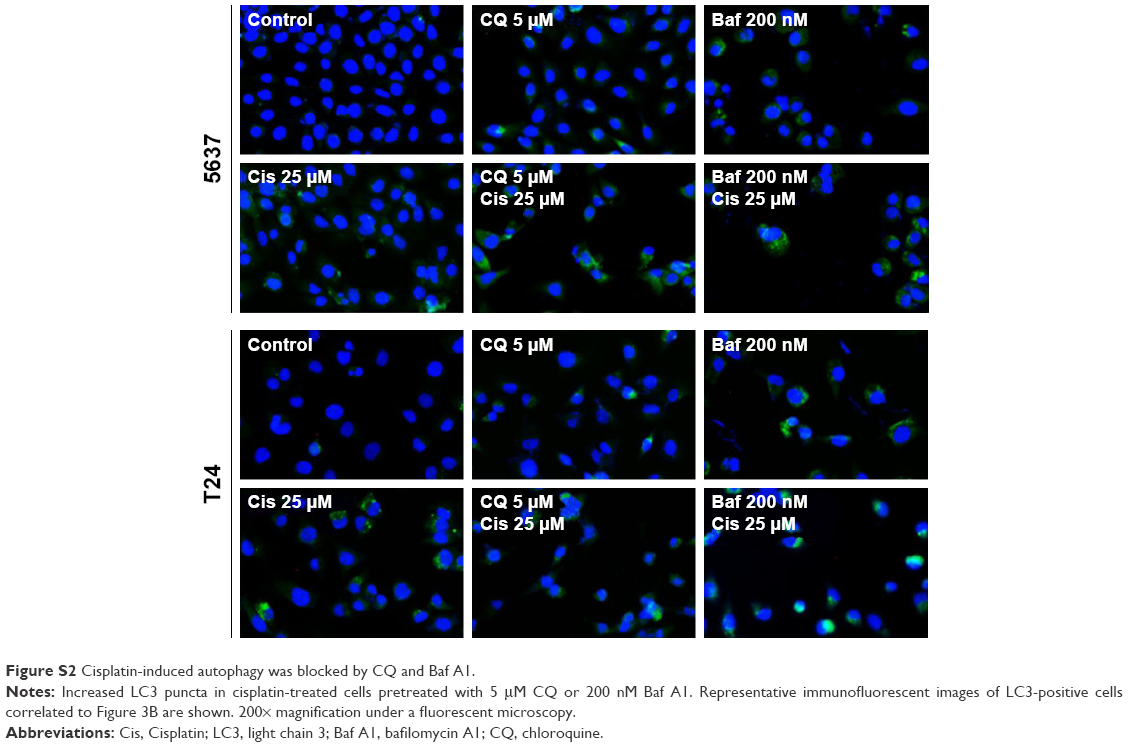 | Figure S2 Cisplatin-induced autophagy was blocked by CQ and Baf A1. |
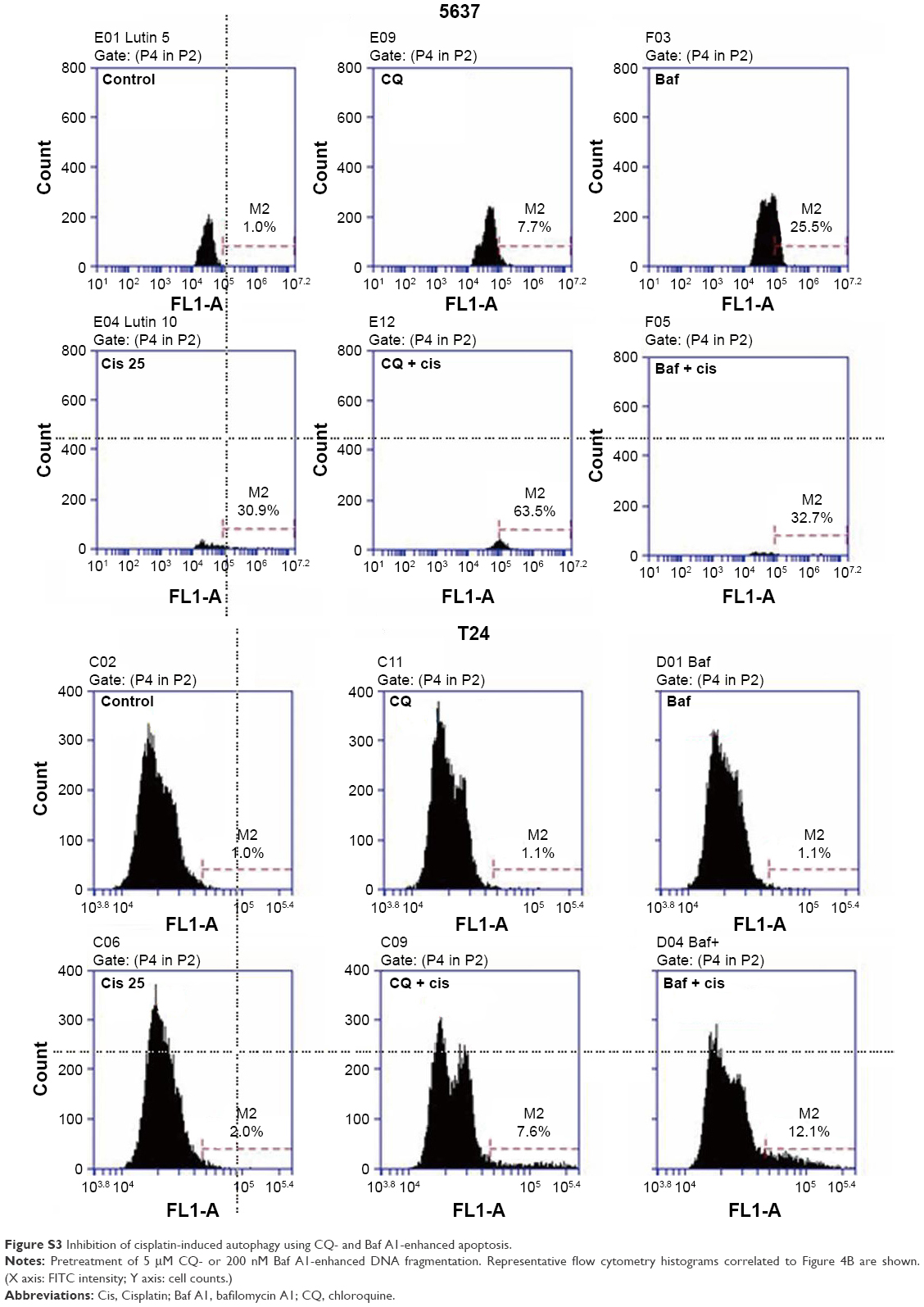 | Figure S3 Inhibition of cisplatin-induced autophagy using CQ- and Baf A1-enhanced apoptosis. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.