Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 20
Cigarette Smoking-Induced Glucose Metabolic Reprogramming in Chronic Obstructive Pulmonary Disease: Mechanisms and Therapeutic Implications
Authors Ji T ![]() , Gu YY, Gao YM, Xie Y, Liu Z, Cheng A, Zhou X
, Gu YY, Gao YM, Xie Y, Liu Z, Cheng A, Zhou X ![]() , Ailifeire A, Wang M, Song Q, Shi Y, Shi S, He J, Zhao L, Xiao D, Wang C
, Ailifeire A, Wang M, Song Q, Shi Y, Shi S, He J, Zhao L, Xiao D, Wang C
Received 28 July 2025
Accepted for publication 24 November 2025
Published 2 December 2025 Volume 2025:20 Pages 3855—3866
DOI https://doi.org/10.2147/COPD.S556362
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Vanesa Bellou
Tingfen Ji,1– 6 Yang-Yang Gu,7 Ying-Man Gao,2– 6,8 Ying Xie,2– 6,8 Zhao Liu,2– 6 Anqi Cheng,2– 6 Xinmei Zhou,2– 6,9 Aihemaiti Ailifeire,2– 6,8 Min Wang,2– 6,8 Qingqing Song,1– 6 Yuxin Shi,2– 6,8 Shunyi Shi,2– 6,9 Jiahui He,1– 6 Liang Zhao,2– 6 Dan Xiao,2– 6,8 Chen Wang2– 6,8
1China-Japan Friendship School of Clinical Medicine, Capital Medical University, Beijing, People’s Republic of China; 2Department of Tobacco Control and Prevention of Respiratory Diseases, China-Japan Friendship Hospital, Beijing, People’s Republic of China; 3WHO Collaborating Center for Tobacco Cessation and Respiratory Diseases Prevention, Beijing, People’s Republic of China; 4National Clinical Research Center for Respiratory Diseases, Beijing, People’s Republic of China; 5Institute of Respiratory Medicine, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 6National Center for Respiratory Medicine, Beijing, People’s Republic of China; 7Department of Respiratory & Critical Care Medicine, Jiaxing Second Hospital, The Second Affiliated Hospital of Jiaxing University, Jiaxing, People’s Republic of China; 8Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 9School of Health Policy and Management, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, People’s Republic of China
Correspondence: Dan Xiao, Department of Tobacco Control and Prevention of Respiratory Diseases, China-Japan Friendship Hospital, No. 2 Yinghua East Street, Chaoyang District, Beijing, 100029, People’s Republic of China, Email [email protected]
Abstract: Chronic obstructive pulmonary disease (COPD) is a complex and heterogeneous respiratory disorder that arises from interactions between genetic susceptibility and environmental exposures, with cigarette smoking being the primary modifiable risk factor. Cigarette smoke reprograms pulmonary glucose metabolism, a process recognized as an early molecular event driving disease progression. Prolonged exposure is associated with enhanced glycolysis, suppression of the tricarboxylic acid cycle and oxidative phosphorylation, mitochondrial dysfunction, and excessive production of mitochondrial reactive oxygen species. These metabolic disturbances converge to form a pathological axis linking metabolism, inflammation, and immunity, leading to immune dysregulation, chronic airway inflammation, and tissue remodeling. This review summarizes the characteristics and molecular mechanisms of cigarette smoke–induced glucose metabolic reprogramming in COPD while highlighting the therapeutic potential of targeting glucose metabolism. Particular emphasis is placed on comprehensive strategies aimed at restoring metabolic homeostasis. A deeper understanding of glucose metabolic reprogramming in COPD associated with smoking may provide novel insights into disease pathogenesis and contribute to the development of individualized therapies. Nevertheless, clinical evidence remains limited, underscoring the need for translational studies targeting glucose metabolism in COPD.
Keywords: chronic obstructive pulmonary disease, glucose metabolism reprogramming, cigarette smoke, mitochondrial dysfunction, targeted metabolic intervention
Introduction
Chronic obstructive pulmonary disease (COPD) is a prevalent and heterogeneous respiratory disorder characterized by irreversible airflow limitation and chronic respiratory symptoms, with primary clinical phenotypes including chronic bronchitis and emphysema.1,2 In 2022, the Lancet COPD Commission categorized COPD into five types based on the major risk factors and emphasized that distinct pathophysiological mechanisms driven by these risk factors may translate into differentiated approaches to diagnosis, prognosis, and treatment.3
Cigarette smoking is the primary risk factor for COPD. Combustible cigarettes release substantial amounts of oxidants, tar, nicotine, and fine particulate matter during combustion, forming a complex mixture of harmful substances.4 Because combustible cigarettes contain thousands of toxic chemicals with well-established health hazards, so-called “novel tobacco products” have been marketed as alternatives, but accumulating evidence indicates that they are far from harmless.4 Among these, e-cigarettes produce inhalable aerosols by heating liquids containing propylene glycol, glycerin, flavorings, and nicotine.5 Heat-not-burn products(HTPs), conversely, heat tobacco material itself at lower temperatures to release nicotine-containing aerosols.4 Currently, there is insufficient epidemiological and mechanistic evidence to establish a link between e-cigarettes or HTPs and COPD, despite their capacity to induce oxidative stress, inflammatory responses, and lung function impairment.6
Compared with non-smoking-related COPD, the smoking-related subtype exhibits more pronounced differences in clinical phenotype, underlying mechanisms, and disease progression. Studies have demonstrated that individuals who smoke exhibit a higher prevalence of respiratory symptoms and pulmonary function abnormalities, a more rapid annual decline in forced expiratory volume in one second (FEV1), increased frequency of acute COPD exacerbations, and elevated mortality rates.7,8 Furthermore, genetic susceptibility plays a significant role in the onset and progression of COPD.9 Exposure to cigarette smoke (CS) promotes the development of COPD by altering gene expression and signaling pathways, while emerging evidence suggests that dysregulated glucose metabolism may represent another key mechanism in this process10,11(Figure 1). This review aims to outline a perspective that considers the metabolism–inflammation–immunity interplay under CS exposure. We systematically summarizes how CS alters pulmonary glucose metabolism and clarify its contribution to COPD pathogenesis. In addition, the potential of targeting glucose metabolic remodeling as a therapeutic strategy will also be discussed.
 |
Figure 1 Metabolic reprogramming links genetic susceptibility and environmental exposures to COPD development. |
Glucose Metabolism in Lung
The concept of glucose metabolism reprogramming was first recognized in cancer biology, where it represents a hallmark metabolic alteration that distinguishes tumor cells from their normal counterparts.12 This phenomenon, commonly referred to as the “Warburg effect”, describes the propensity of cancer cells to favor glycolysis for lactate production even under normoxic conditions.12
Interestingly, despite sufficient oxygen availability, lung tissue demonstrates aerobic glycolysis, whereby substantial amounts of glucose are converted to lactate through the glycolytic pathway even under normoxic conditions.13 On the one hand, this glycolysis-favored pattern may help reduce local oxygen consumption, thereby allowing greater oxygen distribution to peripheral tissues; on the other hand, lactate may act as a metabolic coupling factor, being taken up by neighboring lung cells via monocarboxylate transporters and subsequently converted to pyruvate for mitochondrial oxidation, thus facilitating intercellular metabolic cooperation within the lung microenvironment.14,15 In addition to glycolysis, glucose can be metabolized via the pentose phosphate pathway (PPP) to generate nicotinamide adenine dinucleotide phosphate (NADPH, reduced form) and ribose-5-phosphate, which are essential for maintaining redox homeostasis and synthesizing nucleic acids, respectively, thereby supporting antioxidant defense and cell proliferation.16 Furthermore, pyruvate can be converted into acetyl-CoA by pyruvate dehydrogenase complex, entering the mitochondria to supply the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS), providing an efficient energy source for alveolar epithelial cells, alveolar macrophages, and other immune cells.17
Importantly, aerobic glycolysis in the healthy lung represents a physiological adaptation that coexists with intact mitochondrial respiration and preserved redox homeostasis. This should be distinguished from a pathological Warburg-like program that emerges under injurious conditions and is characterized by reduced OXPHOS and disrupted cellular redox balance.
Clinical Impact of Glucose Dysregulation in COPD
Glucose dysregulation represents a clinically relevant but underrecognized feature of COPD. Clinical observational evidence has shown that patients with severe COPD had increased endogenous glucose production and clearance, along with elevated pyruvate flux and oxidation, suggesting dysregulated glucose metabolism contributes to pulmonary metabolic stress.10 The triglyceride–glucose index has been strongly associated with all-cause mortality in patients with severe COPD.18 In acute exacerbations complicated by ventilatory insufficiency, hyperglycemia correlates with worse clinical outcomes.19 Subsequent studies have demonstrated that aberrant glucose metabolism may not only reflect comorbid conditions but also actively participate in COPD progression through metabolic and inflammatory crosstalk.11,18 Importantly, such glucose-related disturbances are not limited to moderate or severe COPD. Large-scale population-based studies have shown that a higher dietary inflammatory index and the presence of diabetes are both significantly associated with reduced lung function.20,21 A cross-sectional study conducted in a Japanese population undergoing medical examinations revealed that individuals with elevated glycated hemoglobin (HbA1c ≥ 5.6%) exhibited significantly lower FEV1/FVC ratios, further underscoring the link between impaired glucose homeostasis and pulmonary dysfunction.22
Diabetes itself may contribute to pulmonary impairment through several interrelated mechanisms. Chronic hyperglycemia promotes the accumulation of advanced glycation end-products, which trigger systemic and airway inflammation and enhance oxidative stress, thereby damaging lung tissue structure and elasticity.23,24 Persistent inflammation exacerbates insulin resistance and metabolic imbalance, further weakening pulmonary metabolic homeostasis and repair capacity.25 In addition, diabetes-related microvascular complications can involve the pulmonary circulation, leading to thickening of the alveolar–capillary basement membrane and impairment of gas diffusion, contributing to reduced lung function.26
Beyond disease-specific mechanisms, systemic factors also contribute to altered glucose metabolism in COPD. Insulin resistance and metabolic syndrome, which are common comorbidities in COPD patients, can exacerbate dysregulated glucose homeostasis and thereby confound the interpretation of metabolic findings.27 Moreover, widely used pharmacological treatments such as systemic corticosteroids may induce hyperglycemia, while β-agonists and anticholinergics can influence glucose handling through effects on glycogenolysis, insulin secretion, and cellular uptake.27,28
Cigarette Smoke, Glucose Metabolism Reprogramming and COPD
CS exerts complex, multilayered effects on pulmonary metabolism and immune function in both active smokers and those with passive exposure, with such effects potentially varying by age, sex, and mode of exposure, thereby complicating the understanding of smoking-related COPD pathogenesis. Numerous observational clinical studies have indicated that patients with smoking-related COPD exhibit glucose metabolic dysregulation.21,29 However, given the substantial heterogeneity across clinical populations and external factors, many studies have used cellular or animal models to minimize variability and elucidate the mechanistic role of smoking-induced alterations in glucose metabolism during COPD progression. These studies collectively suggest that CS induced glucose metabolic reprogramming serves as a critical node for amplifying inflammation and shaping immune adaptation, thereby contributing to the pathological progression of COPD through the axis of metabolism, inflammation and immunity.
Shift Toward Glycolysis: Impaired TCA Cycle and OXPHOS
CS exposure can induce alterations in glucose metabolism within pulmonary cells. In smoking-related COPD, a prominent metabolic feature is increased glucose uptake accompanied by enhanced glycolytic activity, whereas mitochondrial oxidative metabolism, including the TCA cycle and OXPHOS, is impaired.30,31 It is noteworthy that this metabolic reprogramming occurs not only in airway epithelial cells but also in pulmonary immune cells, and is closely associated with persistent inflammation.
Clinical metabolomic studies have shown that lung tissues from COPD patients exhibit significantly elevated levels of glycolytic intermediates such as lactate and pyruvate, while key TCA cycle metabolites such as citrate and α-ketoglutarate are markedly reduced. These alterations suggest that glycolytic flux is enhanced whereas mitochondrial oxidative metabolism is impaired.32,33 Such metabolic changes are closely linked to lung function decline and chronic inflammation in COPD.
Animal and cellular studies further corroborate these clinical observations. In a murine COPD model, prolonged CS exposure (12 weeks) markedly increased the expression of glucose transporter 3 (GLUT-3) in lung tissue, whereas GLUT-3 knockout significantly alleviated airway remodeling and lung function impairment, and inhibited epithelial–mesenchymal transition (EMT) in bronchial epithelial cells.34,35 Consistently, mice subjected to long-term CS exposure exhibited substantial lactate accumulation in lung tissue accompanied by reduced activity of key TCA cycle enzymes.36 In vitro, BEAS-2B bronchial epithelial cells exposed to cigarette smoke extract(CSE)(CSE: 1%-12.5%; 6-months) similarly showed GLUT-3 upregulation, along with enhanced glycolytic flux, decreased oxygen consumption rate (OCR), reduced ATP synthesis, and excessive lactate release.34,37 To compensate for this energy deficit, cells further increased glucose uptake and reinforced glycolysis, accompanied by upregulation of glucose metabolism–related genes such as RPL13A and ATP5B.32 In in vitro bronchial epithelial cell models, pyruvate kinase M2 (PKM2) upregulation is closely associated with enhanced inflammatory responses and EMT, whereas PKM2 knockdown significantly reduces the release of pro-inflammatory cytokines, lowers reactive oxygen species(ROS) levels, and suppresses EMT.38
Of note, the regulatory patterns of glucose metabolism differ depending on the duration of CS exposure. In a murine model subjected to whole-body CS exposure for 4 weeks, lung glycolytic flux exhibited a decreasing trend, accompanied by enhanced activity of the PPP and downregulation of key glycolytic enzymes such as glyceraldehyde-3-phosphate dehydrogenase. Concurrently, the expression and activity of electron transport chain(ETC) complexes were significantly upregulated, suggesting that acute CS exposure may induce a metabolic adaptation primarily driven by oxidative stress rather than by inflammation.39 Short-term CS exposure may primarily trigger oxidative stress adaptation, whereas chronic exposure leads to inflammation-driven metabolic imbalance.
Mitochondrial Dysfunction and mtROS Accumulation
Mitochondria serve as the central site for cellular energy metabolism, hosting the primary locations for the TCA cycle and OXPHOS reactions. Exposure to CS significantly damages the structure and function of pulmonary cell mitochondria, leading to metabolic homeostasis disruption and excessive production of ROS. In particular, mitochondria-derived ROS (mtROS) play a crucial role in the development and progression of COPD.
Morphological analysis of bronchial epithelial cells in patients with COPD revealed typical mitochondrial morphological abnormalities, including loss of cristae, swelling, and fragmentation.40 Animal studies and cell models have also yielded similar findings. In a CS-exposed mouse model (5 days/week for 4–6 months), genes associated with the TCA cycle, fatty acid oxidation, and redox regulation were compensatorily upregulated in pulmonary epithelial cells, accompanied by mitochondrial morphological and functional abnormalities including reduced cristae number, enlarged volume, decreased membrane potential, and reduced OCR.39,41 In the BEAS-2B bronchial epithelial cell model, CSE stimulation (CSE: 1%-12.5%; 6-months or 24-hours) revealed characteristic ultrastructural alterations such as mitochondrial debranching, swelling, and cristae depletion.32,42 Furthermore, CSE inhibits the activity of key complexes in the ETC, such as complexes I and II, leading to diminished NADH reoxidation capacity, disruption of the NAD+/NADH ratio, and ultimately suppression of dehydrogenase reactions in the TCA cycle.43
These structural and functional abnormalities converge on impaired ETC efficiency and electron leakage.44 When electrons combine with oxygen, they generate excessive mitochondrial mtROS, including superoxide anion (O2−·), hydrogen peroxide (H2O2), and hydroxyl radical (·OH).44,45 These mtROS not only directly cause oxidative damage to DNA, lipids, and proteins but also act as signaling molecules to activate inflammation-related pathways such as the NLRP3 inflammasome and NF-κB.44 Accumulating evidence further suggests that mtROS act upstream of signaling pathways such as STAT3 and PINK1-Parkin, thereby promoting EMT and small airway remodeling.46,47 Together, these effects amplify chronic inflammatory responses in lung tissue and accelerate disease progression.44–47
Therefore, CS-induced mitochondrial structural disruption and dysfunction not only impair cellular energy metabolism but also drive chronic inflammation and lung tissue injury through mtROS-mediated signaling pathways, making it one of the key molecular events in the development of COPD (Figure 2).
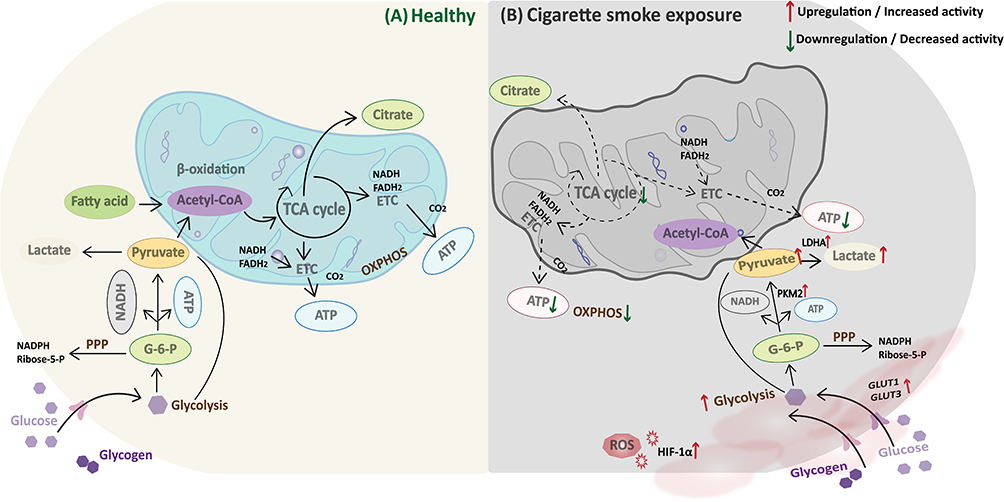 |
Figure 2 Altered glucose metabolism in lungs exposed to tobacco smoke. Note: (A) In the healthy state, lung cells efficiently generate ATP via the glycolysis-TCA cycle-OXPHOS pathway, supplemented by the PPP to maintain metabolic homeostasis. (B) Under cigarette smoke exposure, GLUT1 and GLUT3 expression is upregulated and glucose uptake is increased. Activation of HIF-1α and PKM2 promotes a rise in glycolytic flux and an increase in lactic acid production, whereas TCA cycle flux and OXPHOS activity decrease. Mitochondrial dysfunction, decreased ATP production, and ROS accumulation promote a chronic inflammatory process. |
Crosstalk Between Metabolism and Immunity in COPD
Disrupted glucose metabolism represents not only changes in cellular energetics but also a key driver of immune dysregulation. Metabolic intermediates and signaling molecules serve as “bridges” for immune regulation, establishing intricate metabolic-immune interaction networks between immune cells and structural cells (Figure 3). This interaction is particularly pronounced in the context of CS exposure, driving chronic inflammatory responses and airway remodeling.
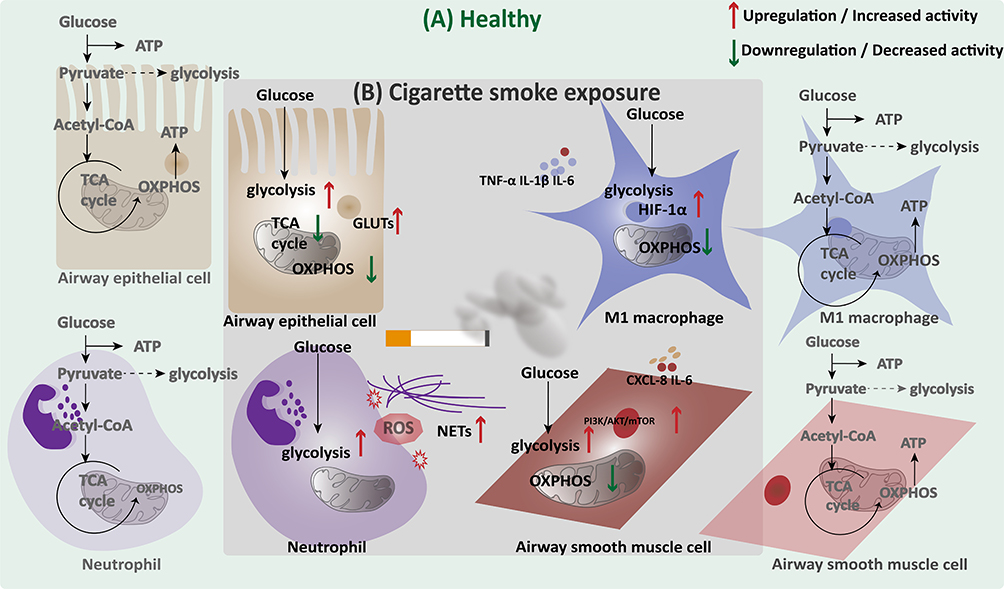 |
Figure 3 Cell-specific glucose metabolic reprogramming in the lung under cigarette smoke exposure. Note: (A) Under normal conditions, airway epithelial cells, macrophages, neutrophils, and airway smooth muscle cells mainly utilize OXPHOS through the TCA cycle for efficient ATP generation. (B) Cigarette smoke exposure disrupts this balance, promoting glycolysis while suppressing mitochondrial OXPHOS in multiple lung cell types. These metabolic shifts are accompanied by increased glucose uptake and inflammatory activation in epithelial and immune cells, as well as altered energy metabolism in airway smooth muscle cells. |
Under chronic CS exposure, pulmonary macrophages exhibit a phenotypic shift predominantly toward classical activation (M1) polarization, regulated by an imbalance between glycolysis and mitochondrial metabolism.48,49 M1 macrophages primarily rely on glycolysis for rapid energy production, promoting the release of proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and IL-6. In contrast, alternatively activated macrophages (M2) predominantly utilize OXPHOS and fatty acid oxidation to drive anti-inflammatory responses and tissue repair.49 In a mouse COPD model, continuous exposure to CS for three months led to enhanced macrophage glycolysis, impaired mitochondrial function, and suppressed TCA cycle activity. This disrupts the metabolic equilibrium between M1 and M2 macrophages, promoting a shift toward the M1 phenotype.50 Secondary metabolites like succinate stabilize HIF-1α expression, further enhancing glycolytic activity and pro-inflammatory factor production, thereby perpetuating chronic pulmonary inflammation and airway remodeling.51,52
Under physiological conditions, neutrophils are highly dependent on glycolysis to sustain effector functions such as chemotaxis, phagocytosis, and oxygen bursts.53 Persistent CS exposure further enhances their glycolytic activity, leading to increased release of inflammatory mediators, elevated reactive ROS production, and excessive release of extracellular trap networks. This process amplifies airway inflammation while directly exacerbating epithelial injury and mucus secretion, thereby driving disease progression.54 Furthermore, disrupted glycogen cycling mechanisms, including gluconeogenesis, glycogen synthesis and glycogen breakdown, in COPD neutrophils may contribute to unstable energy metabolism and imbalanced inflammatory responses.55
Beyond immune cells, airway smooth muscle cells (ASMCs) also participate in COPD airway remodeling through glycometabolic reprogramming.40 In COPD animal models, CS disrupts mitochondrial structure in ASMCs and activates the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mechanistic target of rapamycin (mTOR) signaling pathway, and leading to oxidative stress and metabolic dysfunction. The PI3K/AKT/mTOR pathway serves as a central hub regulating cellular growth, survival, and metabolism by promoting glucose uptake, stimulating glycolysis, and inhibiting autophagy.56 This ultimately leads to elevated glycolytic activity and mitochondrial dysfunction.57,58 Hollins et al observed upregulation of NADPH oxidase 4 (NOX4) in bronchial biopsy specimens from COPD patients. In vitro stimulation models revealed that CS exposure to COPD-derived ASMCs induces excessive ROS production, partially reversible by NOX4 inhibitors.59 These metabolic abnormalities enhance ASMCs contractility and further drive airway hyperresponsiveness and structural remodeling by promoting the release of inflammatory mediators such as IL-6 and CXCL-8.59 Studies also indicate that metabolic reprogramming in ASMCs is not entirely dependent on exogenous CS exposure but may reflect intrinsic metabolic abnormalities inherent to COPD.60,61
Overall, immune cells and structural cells form a self-amplifying inflammatory loop in COPD through metabolic-immune interactions. This loop not only sustains chronic airway inflammation but also drives tissue injury and structural remodeling. This evidence currently derived primarily from animal and in vitro studies.
Intervention Strategies for COPD Targeting Glucose Metabolism Regulation
Smoking Cessation and Glucose Reduction
Smoking cessation is the most fundamental and well-established intervention for slowing lung function decline in individuals with COPD.62 As early as the 20th century, the Lung Health Study showed in a 5-year follow-up of 5,887 smokers with early COPD that the annual rate of decline in FEV1 was −0.33% per year in complete quitters, significantly lower than that of intermittent smokers and continuous smokers.63 Similar results have been obtained in subsequent studies.64,65 In addition to reducing chronic inflammation and oxidative stress, smoking cessation may partially reverse smoking-induced disturbances in glucose metabolism. A meta-analysis involving 98,978 individuals with diabetes showed that HbA1c levels were 0.61% lower in non-smokers compared to smokers, and that HbA1c levels were similarly low among those who had quit smoking within the previous 10 years. Smoking cessation does not lead to an increase in HbA1c in the long term and may reduce the risk of vascular complications in diabetes by improving the lipid profile.66 An observational study of 12 smokers also found a significant increase in insulin sensitivity and a decrease in fasting insulin levels immediately after 1–2 weeks of smoking cessation.67 Together, these findings suggest that smoking cessation improves both pulmonary and metabolic health, thereby reducing COPD progression and enhancing quality of life.
Potential Therapeutic Agents Targeting Glucose Metabolism
In COPD patients with concomitant diabetes mellitus, rational glucose-lowering therapy is associated with improved prognosis. Classical hypoglycemic agents primarily consist of biguanides (eg, metformin), sulfonylureas (eg, glimepiride), glinides (eg, repaglinide), and α-glucosidase inhibitors (eg, acarbose), which exert glucose-lowering effects by inhibiting hepatic glucose output, facilitating insulin secretion, or delaying glucose absorption.68 Novel hypoglycemic agents, on the other hand, include dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists, sodium-glucose cotransporter-2 (SGLT-2) inhibitors, and modified insulin analogs, which not only provide effective glycemic control but also confer additional benefits such as weight reduction and cardiorenal protection.69 Specifically, DPP-4 inhibitors and GLP-1 receptor agonists enhance insulin secretion and suppress glucagon by amplifying the incretin effect. SGLT-2 inhibitors increase urinary glucose excretion by inhibiting renal glucose reabsorption.70
Multiple real-world studies have evaluated the role of novel antidiabetic drugs in COPD patients with concomitant diabetes. SGLT-2 inhibitors consistently demonstrate a reduction in the risk of acute exacerbations, hospitalizations, and mortality in COPD patients with diabetes, with their efficacy validated across large population studies in multiple countries.71–73 Furthermore, retrospective cohort studies have further confirmed that SGLT-2 inhibitor use significantly reduces these risks of emergency department visits, hospitalizations, and mortality.74,75 In contrast, GLP-1 receptor agonists offer limited benefits, while DPP-4 inhibitors show no significant improvement in most studies and even demonstrate increased acute exacerbation risk in some investigations.71–73 A meta-analysis including nine observational studies demonstrated that, compared with sulfonylureas, SGLT-2 inhibitors were associated with a significantly reduced risk of COPD exacerbations (OR = 0.64, 95% CI: 0.52–0.79). Similarly, GLP-1 receptor agonists showed benefit (OR = 0.66, 95% CI: 0.49–0.89), whereas DPP-4 inhibitors did not provide consistent improvement.76 However, meta-analyses of randomized controlled trials (RCTs) are still lacking for metabolic-targeted therapies in COPD and further studies are needed.77–79
However, it is worth noting whether glucose-lowering agents then benefit in COPD patients without diabetes? Animal models have shown that metformin inhibits epithelial sodium channel activity and significantly improves COPD-associated emphysema and lung dysfunction without adverse effects on non-pulmonary parameters.80 However, metformin does not currently show clinical benefit in COPD patients without diabetes, and the results of a multicenter RCT that included 52 nondiabetic patients with acute exacerbations of COPD showed that metformin (up to 2 g/day for one month) failed to improve glycemia in hospitalized nondiabetic patients, and did not have a significant impact on validation metrics or clinical symptoms.81 The primary endpoint was mean in-hospital blood glucose concentration, with secondary endpoints including fructosamine, C-reactive protein, and COPD Assessment Test scores. These negative results may reflect the short exposure period, the acute disease setting, and potential confounding from comorbidities and concomitant medications such as systemic corticosteroids and inhaled bronchodilators, all of which could obscure metabolic or clinical signals. Novel hypoglycemic agents, SGLT2i and GLP1RA have also shown anti-inflammatory, antioxidant, mucus secretion reduction and airway remodeling effects in animal models, but there are no clinical studies to demonstrate their role in COPD patients without diabetes.82,83
In COPD management, combining smoking cessation, dietary intervention and metabolic homeostasis regulation to construct an integrated intervention strategy is expected to delay disease progression more effectively. A summary of the main glucose metabolism–targeting agents and their population-specific effects in COPD is provided in Table 1.
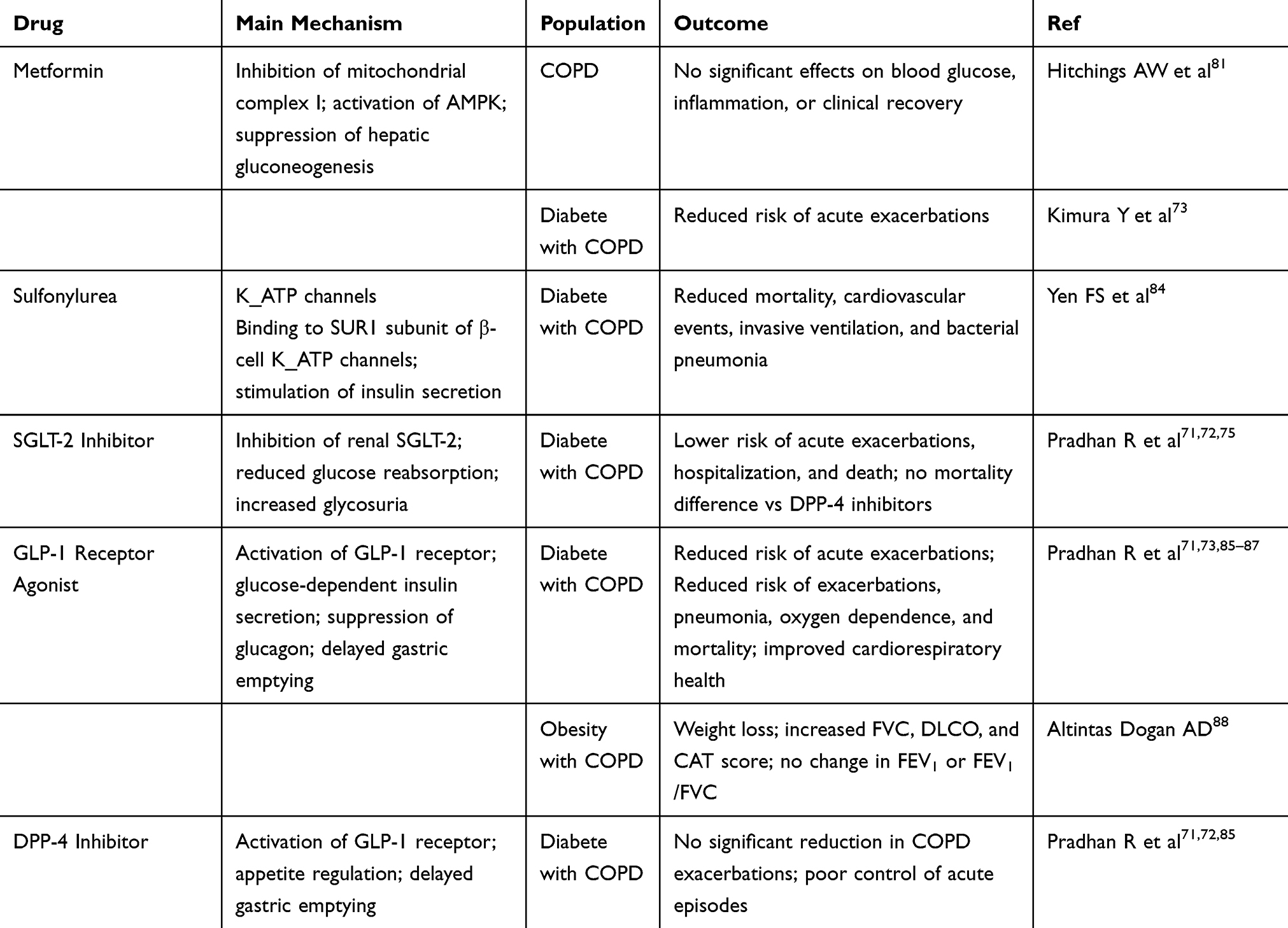 |
Table 1 Potential Effects and Population-Specific Outcomes of Agents Targeting Glucose Metabolism in COPD Treatment |
Conclusions and Challenges
In recent years, much attention has been paid to the role of metabolic reprogramming in the pathogenesis of COPD, and in particular, abnormalities in glucose metabolism, characterized by enhanced glycolysis and impaired OXPHOS, have been recognized as an important metabolic remodeling phenomenon driven by CS exposure. In this review, we summarize the specific alterations of glucose metabolism in different cells of the lung and how they are involved in airway remodeling, mucus secretion disturbances, immune imbalance, and structural damage in COPD by affecting cellular energy homeostasis, autophagy regulation, immune responses, and inflammatory signaling. Our review highlights that smoking-induced glucose metabolic reprogramming should not be regarded as isolated abnormalities in glycolysis, the TCA cycle, or OXPHOS. Instead, these changes converge into a pathological metabolism–inflammation–immunity axis, which serves as a central mechanism linking energy imbalance with chronic inflammation, immune dysfunction, and structural damage in COPD. Moreover, when interpreting associations between glycemic markers or dietary indices and lung outcomes, potential confounding factors should be considered. Comorbid metabolic diseases, systemic corticosteroid use, bronchodilator therapies, as well as BMI and physical activity may all influence glucose metabolism and lung function, and could partly account for the heterogeneity across studies.
However, current research still faces several challenges. First, metabolic alterations across different COPD subtypes remain poorly characterized, highlighting the need for subtype-specific investigations. Second, although experimental studies provide strong evidence linking altered glucose metabolism to COPD pathogenesis, the causal relationship with clinical phenotypes has not been fully established, and clinical validation remains limited. Third, while some glucose-lowering agents have shown protective effects in COPD patients with comorbid diabetes, their potential application in non-diabetic COPD populations requires well-designed, large-scale randomized controlled trials. Finally, most available metabolomic data are derived from tissue samples, lacking single-cell resolution and spatial mapping. Future studies integrating multi-omics approaches, including metabolomics, transcriptomics, epigenomics, proteomics, and single-cell technologies, will be essential to build a more precise framework of “metabolic typing,” capture cellular heterogeneity and individual variability, and ultimately guide personalized interventions.
Abbreviations
COPD, chronic obstructive pulmonary disease; HTP, Heat-not-burn product; FEV1, forced expiratory volume in one second; CS, cigarette smoke; PPP, pentose phosphate pathway; NADPH, nicotinamide adenine dinucleotide phosphate; TCA, tricarboxylic acid; OXPHOS, oxidative phosphorylation; GLUT-3, glucose transporter-3; EMT, epithelial–mesenchymal transition; CSE, cigarette smoke extract; OCR, oxygen consumption rate; ETC, electron transport chain; ROS, oxygen species; PKM2, pyruvate kinase M2; TNF-α, tumor necrosis factor-alpha; IL-1β, interleukin-1β; ASMC, airway smooth muscle cell; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; mTOR, mechanistic target of rapamycin; NOX4, NADPH oxidase 4; T2DM, type-2 diabetes mellitus; GLP-1, Glucagon-like peptide 1; DPP-4, Dipeptidyl peptidase-4; SGLT-2, sodium-glucose cotransporter-2; RCT, randomized controlled trial.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, design, literature search, analysis, or interpretation; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the following funding sources: Medical and Health Science and Technology Innovation Project of Chinese Academy of Medical Sciences (2021-I2M-1-010); Noncommunicable Chronic Diseases-National Science and Technology Major Project (2023ZD0506400).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lareau SC, Fahy B, Meek P, Wang A. Chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med. 2019;199(1):P1–P2. doi:10.1164/rccm.1991P1
2. Ferrera MC, Labaki WW, Han MK. Advances in chronic obstructive pulmonary disease. Annu Rev Med. 2021;72:119–134. doi:10.1146/annurev-med-080919-112707
3. Stolz D, Mkorombindo T, Schumann DM, et al. Towards the elimination of chronic obstructive pulmonary disease: a Lancet Commission. Lancet. 2022;400(10356):921–972. doi:10.1016/S0140-6736(22)01273-9
4. Allbright K, Villandre J, Crotty Alexander LE, et al. The paradox of the safer cigarette: understanding the pulmonary effects of electronic cigarettes. Eur Respir J. 2024;63(6):2301494. doi:10.1183/13993003.01494-2023
5. Xue Z, Orr-Souza E, Nargis N, Patel M, Nighbor T. Nicotine and toxicant exposure among individuals using both combustible cigarettes and E-cigarettes based on level of product use. Nicotine Tob Res. 2025;27(9):1591–1599. doi:10.1093/ntr/ntaf053
6. Błach J, Siedliński M, Sydor W. Immunology in COPD and the use of combustible cigarettes and heated tobacco products. Eur J Med Res. 2023;28(1):397.
7. Park H, Jo SM, Jin KN, et al. Distinct risks of exacerbation and lung function decline between never-smokers and ever-smokers with COPD. BMC Pulm Med. 2025;25(1):138. doi:10.1186/s12890-025-03604-1
8. Kohansal R, Martinez-Camblor P, Agustí A, Buist AS, Mannino DM, Soriano JB. The natural history of chronic airflow obstruction revisited: an analysis of the Framingham offspring cohort. Am J Respir Crit Care Med. 2009;180(1):3–10. doi:10.1164/rccm.200901-0047OC
9. Werder RB, Zhou X, Cho MH, Wilson AA. Breathing new life into the study of COPD with genes identified from genome-wide association studies. Eur Respir Rev. 2024;33(172):240019. doi:10.1183/16000617.0019-2024
10. Kao CC, Hsu JW, Bandi V, Hanania NA, Kheradmand F, Jahoor F. Glucose and pyruvate metabolism in severe chronic obstructive pulmonary disease. J Appl Physiol. 2012;112(1):42–47. doi:10.1152/japplphysiol.00599.2011
11. Wu TD, Fawzy A, Brigham E, et al. Association of triglyceride-glucose index and lung health: a population-based study. Chest. 2021;160(3):1026–1034. doi:10.1016/j.chest.2021.03.056
12. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–337. doi:10.1038/nrc3038
13. Liu G, Summer R. Cellular Metabolism in Lung Health and Disease. Annu Rev Physiol. 2019;81:403–428. doi:10.1146/annurev-physiol-020518-114640
14. O’Neil JJ, Tierney DF. Rat lung metabolism: glucose utilization by isolated perfused lungs and tissue slices. Am J Physiol. 1974;226(4):867–873. doi:10.1152/ajplegacy.1974.226.4.867
15. Faubert B, Li KY, Cai L, et al. Lactate metabolism in human lung tumors. Cell. 2017;171(2):358–371.e9. doi:10.1016/j.cell.2017.09.019
16. Chettimada S, Gupte R, Rawat D, Gebb SA, McMurtry IF, Gupte SA. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2015;308(3):L287–300. doi:10.1152/ajplung.00229.2014
17. Cai L, Hammond NG, Tasdogan A, et al. High glucose contribution to the TCA cycle is a feature of aggressive non-small cell lung cancer in patients. Cancer Discov. 2025;15(4):702–716. doi:10.1158/2159-8290.CD-23-1319
18. Zhou WQ, Song X, Dong WH, Chen Z. Independent effect of the triglyceride-glucose index on all-cause mortality in critically ill patients with chronic obstructive pulmonary disease and asthma: a retrospective cohort study. Chron Respir Dis. 2024;21:14799731241245424. doi:10.1177/14799731241245424
19. Chakrabarti B, Angus RM, Agarwal S, Lane S, Calverley PM. Hyperglycaemia as a predictor of outcome during non-invasive ventilation in decompensated COPD. Thorax. 2009;64(10):857–862. doi:10.1136/thx.2008.106989
20. Zheng Y, Liu W, Zhu X, Xu M, Lin B, Bai Y. Associations of dietary inflammation index and composite dietary antioxidant index with preserved ratio impaired spirometry in US adults and the mediating roles of triglyceride-glucose index: NHANES 2007–2012. Redox Biol. 2024;76:103334. doi:10.1016/j.redox.2024.103334
21. Chen C, Huang Z, Liu L, Su B, Feng Y, Huang Y. Lung function impairment and risks of incident cardiovascular diseases and mortality among people with type 2 diabetes: a prospective cohort study. Diabetes Care. 2025;48(5):728–736. doi:10.2337/dc24-2188
22. Baba S, Takashima T, Hirota M, Kawashima M, Horikawa E. Relationship between pulmonary function and elevated glycated hemoglobin levels in health checkups: a cross-sectional observational study in Japanese participants. J Epidemiol. 2017;27(11):511–515. doi:10.1016/j.je.2016.10.008
23. Haider SH, Oskuei A, Crowley G, et al. Receptor for advanced glycation end-products and environmental exposure related obstructive airways disease: a systematic review. Eur Respir Rev. 2019;28(151):180096. doi:10.1183/16000617.0096-2018
24. Mulrennan S, Baltic S, Aggarwal S, et al. The role of receptor for advanced glycation end products in airway inflammation in CF and CF related diabetes. Sci Rep. 2015;5:8931. doi:10.1038/srep08931
25. Fuso L, Pitocco D, Antonelli-Incalzi R. Diabetic lung, an underrated complication from restrictive functional pattern to pulmonary hypertension. Diabetes Metab Res Rev. 2019;35(6):e3159. doi:10.1002/dmrr.3159
26. Mauricio D, Gratacòs M, Franch-Nadal J. Diabetic microvascular disease in non-classical beds: the hidden impact beyond the retina, the kidney, and the peripheral nerves. Cardiovasc Diabetol. 2023;22(1):314. doi:10.1186/s12933-023-02056-3
27. Cho JH, Suh S. Glucocorticoid-induced hyperglycemia: a neglected problem. Endocrinol Metab. 2024;39(2):222–238. doi:10.3803/EnM.2024.1951
28. Alrabbaie H, Al-Wardat M, Etoom M, Beauchamp M, Goldstein R, Brooks D. The prevalence of metabolic syndrome in chronic obstructive pulmonary disease: a systematic review and meta-analysis. Chron Respir Dis. 2025;22:14799731251346194. doi:10.1177/14799731251346194
29. Zaigham S, Tanash H, Nilsson PM, Muhammad IF. Triglyceride-glucose index is a risk marker of incident COPD events in women. Int J Chron Obstruct Pulmon Dis. 2022;17:1393–1401. doi:10.2147/COPD.S360793
30. Gan PXL, Zhang S, Fred Wong WS. Targeting reprogrammed metabolism as a therapeutic approach for respiratory diseases. Biochem Pharmacol. 2024;228:116187. doi:10.1016/j.bcp.2024.116187
31. Wu W, Li Z, Wang Y, Huang C, Zhang T, Zhao H. Advances in metabolomics of chronic obstructive pulmonary disease. Chin Med J Pulm Crit Care Med. 2023;1(4):223–230. doi:10.1016/j.pccm.2023.10.001
32. Wang Y, Ninaber DK, Faiz A, et al. Acute cigarette smoke exposure leads to higher viral infection in human bronchial epithelial cultures by altering interferon, glycolysis and GDF15-related pathways. Respir Res. 2023;24(1):207. doi:10.1186/s12931-023-02511-5
33. Xue M, Zeng Y, Lin R, et al. Metabolomic profiling of anaerobic and aerobic energy metabolic pathways in chronic obstructive pulmonary disease. Exp Biol Med. 2021;246(14):1586–1596. doi:10.1177/15353702211008808
34. Ding Y, Wang Z, Zhang Z, You R, Wu Y, Bian T. GLUT3-mediated cigarette smoke-induced epithelial-mesenchymal transition in chronic obstructive pulmonary disease through the NF-kB/ZEB1 pathway. Respir Res. 2024;25(1):158. doi:10.1186/s12931-024-02785-3
35. Li J, Li Y, Chen G, et al. GLUT1 promotes NLRP3 inflammasome activation of airway epithelium in lipopolysaccharide-induced acute lung injury. Am J Pathol. 2024;194(7):1185–1196. doi:10.1016/j.ajpath.2024.03.003
36. Feng Y, Xie M, Liu Q, et al. Changes in targeted metabolomics in lung tissue of chronic obstructive pulmonary disease. J Thorac Dis. 2023;15(5):2544–2558. doi:10.21037/jtd-22-1731
37. Hoffmann RF, Zarrintan S, Brandenburg SM, et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res. 2013;14(1):97. doi:10.1186/1465-9921-14-97
38. Li D, Shen C, Liu L, et al. PKM2 regulates cigarette smoke-induced airway inflammation and epithelial-to-mesenchymal transition via modulating PINK1/Parkin-mediated mitophagy. Toxicology. 2022;477:153251. doi:10.1016/j.tox.2022.153251
39. Agarwal AR, Zhao L, Sancheti H, Sundar IK, Rahman I, Cadenas E. Short-term cigarette smoke exposure induces reversible changes in energy metabolism and cellular redox status independent of inflammatory responses in mouse lungs. Am J Physiol Lung Cell Mol Physiol. 2012;303(10):L889–98. doi:10.1152/ajplung.00219.2012
40. Agarwal AR, Kadam S, Brahme A, et al. Systemic Immuno-metabolic alterations in chronic obstructive pulmonary disease (COPD). Respir Res. 2019;20(1):171. doi:10.1186/s12931-019-1139-2
41. Cloonan SM, Glass K, Laucho-Contreras ME, et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. 2016;22(2):163–174. doi:10.1038/nm.4021
42. Aghapour M, Tulen CBM, Abdi Sarabi M, et al. Cigarette smoke extract disturbs mitochondria-regulated airway epithelial cell responses to pneumococci. Cells. 2022;11(11):1771. doi:10.3390/cells11111771
43. Ryter SW, Rosas IO, Owen CA, et al. Mitochondrial dysfunction as a pathogenic mediator of chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 2018;15(Suppl 4):S266–S272. doi:10.1513/AnnalsATS.201808-585MG
44. Li F, Xu M, Wang M, et al. Roles of mitochondrial ROS and NLRP3 inflammasome in multiple ozone-induced lung inflammation and emphysema. Respir Res. 2018;19(1):230. doi:10.1186/s12931-018-0931-8
45. Rosa CP, Belo TCA, Santos NCM, et al. Reactive oxygen species trigger inflammasome activation after intracellular microbial interaction. Life Sci. 2023;331:122076. doi:10.1016/j.lfs.2023.122076
46. Tsubouchi K, Araya J, Kuwano K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm Regen. 2018;38:18. doi:10.1186/s41232-018-0077-6
47. Wei Y, Li Q, He K, et al. Mechanism of cigarette smoke in promoting small airway remodeling in mice via STAT 3 / PINK 1-Parkin / EMT. Free Radic Biol Med. 2024;224:447–456. doi:10.1016/j.freeradbiomed.2024.08.036
48. Pervizaj-Oruqaj L, Ferrero MR, Matt U, Herold S. The guardians of pulmonary harmony: alveolar macrophages orchestrating the symphony of lung inflammation and tissue homeostasis. Eur Respir Rev. 2024;33(172):230263. doi:10.1183/16000617.0263-2023
49. Liu Y, Xu R, Gu H, et al. Metabolic reprogramming in macrophage responses. Biomark Res. 2021;9(1):1. doi:10.1186/s40364-020-00251-y
50. Zhao H, Dennery PA, Yao H. Metabolic reprogramming in the pathogenesis of chronic lung diseases, including BPD, COPD, and pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2018;314(4):L544–L554. doi:10.1152/ajplung.00521.2017
51. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi:10.1038/nature11986
52. Ogger PP, Byrne AJ. Macrophage metabolic reprogramming during chronic lung disease. Mucosal Immunol. 2021;14(2):282–295. doi:10.1038/s41385-020-00356-5
53. Jeon JH, Hong CW, Kim EY, Lee JM. Current understanding on the metabolism of neutrophils. Immune Netw. 2020;20(6):e46. doi:10.4110/in.2020.20.e46
54. Ye Y, Wang Y, Xu Q, et al. In vitro study: HIF-1α-dependent glycolysis enhances NETosis in hypoxic conditions. Front Immunol. 2025;16:1583587. doi:10.3389/fimmu.2025.1583587
55. Sadiku P, Willson JA, Ryan EM, et al. Neutrophils fuel effective immune responses through gluconeogenesis and glycogenesis. Cell Metab. 2021;33(2):411–423.e4. doi:10.1016/j.cmet.2020.11.016
56. Ding T, Zhao S, Gu Y, et al. IL-17A regulates airway remodelling in COPD through the PI3K/AKT/mTOR pathway. Sci Rep. 2025;15(1):16546. doi:10.1038/s41598-025-00458-9
57. Aravamudan B, Kiel A, Freeman M, et al. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2014;306(9):L840–54. doi:10.1152/ajplung.00155.2013
58. Athari SS. Targeting cell signaling in allergic asthma. Signal Transduct Target Ther. 2019;4:45. doi:10.1038/s41392-019-0079-0
59. Hollins F, Sutcliffe A, Gomez E, et al. Airway smooth muscle NOX4 is upregulated and modulates ROS generation in COPD. Respir Res. 2016;17(1):84. doi:10.1186/s12931-016-0403-y
60. Michaeloudes C, Kuo CH, Haji G, et al. Metabolic re-patterning in COPD airway smooth muscle cells. Eur Respir J. 2017;50(5):1700202. doi:10.1183/13993003.00202-2017
61. Li Y, Zhang L, Polverino F, et al. Hedgehog interacting protein (HHIP) represses airway remodeling and metabolic reprogramming in COPD-derived airway smooth muscle cells. Sci Rep. 2021;11(1):9074.
62. Pezzuto A, Ricci A, D’Ascanio M, et al. Short-term benefits of smoking cessation improve respiratory function and metabolism in smokers. Int J Chron Obstruct Pulmon Dis. 2023;18:2861–2865. doi:10.2147/COPD.S423148
63. Murray RP, Anthonisen NR, Connett JE, et al. Effects of multiple attempts to quit smoking and relapses to smoking on pulmonary function. Lung Health Study Res Group J Clin Epidemiol. 1998;51(12):1317–1326.
64. Chaudhuri R, Livingston E, McMahon AD, et al. Effects of smoking cessation on lung function and airway inflammation in smokers with asthma. Am J Respir Crit Care Med. 2006;174(2):127–133. doi:10.1164/rccm.200510-1589OC
65. Dhariwal J, Tennant RC, Hansell DM, et al. Smoking cessation in COPD causes a transient improvement in spirometry and decreases micronodules on high-resolution CT imaging. Chest. 2014;145(5):1006–1015. doi:10.1378/chest.13-2220
66. Kar D, Gillies C, Zaccardi F, et al. Relationship of cardiometabolic parameters in non-smokers, current smokers, and quitters in diabetes: a systematic review and meta-analysis. Cardiovasc Diabetol. 2016;15(1):158.
67. Bergman BC, Perreault L, Hunerdosse D, et al. Novel and reversible mechanisms of smoking-induced insulin resistance in humans. Diabetes. 2012;61(12):3156–3166. doi:10.2337/db12-0418
68. Chan JCN, Yang A, Chu N, Chow E. Current type 2 diabetes guidelines: individualized treatment and how to make the most of metformin. Diabetes Obes Metab. 2024;26(Suppl 3):55–74. doi:10.1111/dom.15700
69. Konings LAM, Miguelañez-Matute L, Boeren AMP, et al. Pharmacological treatment options for metabolic dysfunction-associated steatotic liver disease in patients with type 2 diabetes mellitus: a systematic review. Eur J Clin Invest. 2025;55(4):e70003.
70. Giugliano D, Longo M, Signoriello S, et al. The effect of DPP-4 inhibitors, GLP-1 receptor agonists and SGLT-2 inhibitors on cardiorenal outcomes: a network meta-analysis of 23 CVOTs. Cardiovasc Diabetol. 2022;21(1):42. doi:10.1186/s12933-022-01474-z
71. Pradhan R, Lu S, Yin H, et al. Novel antihyperglycaemic drugs and prevention of chronic obstructive pulmonary disease exacerbations among patients with type 2 diabetes: population based cohort study. BMJ. 2022;
72. Chang TC, Liang YC, Lai CC, et al. Comparison of SGLT2 and DPP4 inhibitors on clinical outcomes in COPD patients with diabetes: a nationwide cohort study. Diabet Res Clin Pract. 2025;223:112122. doi:10.1016/j.diabres.2025.112122
73. Kimura Y, Jo T, Inoue N, et al. Association of novel antihyperglycaemic drugs versus metformin with COPD exacerbations. ERJ Open Res. 2025;11(3):00757–2024. doi:10.1183/23120541.00757-2024
74. Shanmugavel Geetha H, Teo YX, Ravichandran S, et al. Use of Sodium-glucose cotransporter 2 (SGLT 2) inhibitor is associated with reduced emergency room visits and hospitalizations in patients with chronic obstructive pulmonary disease (COPD) and type 2 diabetes mellitus. Respir Med. 2024;234:107819.
75. Yen FS, Wei JC, Huang YH, et al. SGLT-2 inhibitors and the risk of chronic obstructive pulmonary disease exacerbations and mortality in chronic obstructive pulmonary disease patients. Ann Am Thorac Soc. 2025;22(6):846–854.
76. Pirera E, Di Raimondo D, D’Anna L, Tuttolomondo A. Efficacy of SGLT2 inhibitors, GLP-1 receptor agonists, DPP-4 inhibitors, and sulfonylureas on moderate-to-severe COPD exacerbations among patients with type 2 diabetes: a systematic review and network meta-analysis. Pharmaceuticals. 2025;18(9):1337. doi:10.3390/ph18091337
77. Liu P, Meng J, Xiong Y, Wu Y, Xiao Y, Gao S. Contraception with levonorgestrel-releasing intrauterine system versus copper intrauterine device: a meta-analysis of randomized controlled trials. EClinicalMedicine. 2024;78:102926. doi:10.1016/j.eclinm.2024.102926
78. Meng J, Li X, Xiong Y, Wu Y, Liu P, Gao S. The role of vitamin D in the prevention and treatment of tuberculosis: a meta-analysis of randomized controlled trials. Infection. 2025;53(3):1129–1140. doi:10.1007/s15010-024-02446-z
79. Liu P, Meng J, Tang H, et al. Association between bariatric surgery and outcomes of total joint arthroplasty: a meta-analysis. Int J Surg. 2025;111(1):1541–1546. doi:10.1097/JS9.0000000000002002
80. Nakashima R, Nohara H, Takahashi N, et al. Metformin suppresses epithelial sodium channel hyperactivation and its associated phenotypes in a mouse model of obstructive lung diseases. J Pharmacol Sci. 2022;149(2):37–45. doi:10.1016/j.jphs.2022.03.002
81. Hitchings AW, Lai D, Jones PW, Baker EH, Metformin in COPD Trial Team. Metformin in severe exacerbations of chronic obstructive pulmonary disease: a randomised controlled trial. Thorax. 2016;71(7):587–593. doi:10.1136/thoraxjnl-2015-208035
82. Rykova EY, Klimontov VV, Shmakova E, Korbut AI, Merkulova TI, Kzhyshkowska J. Anti-inflammatory effects of SGLT2 inhibitors: focus on macrophages. Int J Mol Sci. 2025;26(4):1670. doi:10.3390/ijms26041670
83. Wang W, Mei A, Qian H, et al. The role of glucagon-like peptide-1 receptor agonists in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2023;18:129–137. doi:10.2147/COPD.S393323
84. Yen FS, Wei JC, Yu TS, Hsu CY, Hsu CC, Hwu CM. Sulfonylurea use in patients with type 2 diabetes and COPD: a Nationwide Population-Based Cohort Study. Int J Environ Res Public Health. 2022;19(22):15013. doi:10.3390/ijerph192215013
85. Foer D, Strasser ZH, Cui J, et al. Association of GLP-1 receptor agonists with chronic obstructive pulmonary disease exacerbations among patients with type 2 diabetes. Am J Respir Crit Care Med. 2023;208(10):1088–1100. doi:10.1164/rccm.202303-0491OC
86. See XY, Xanthavanij N, Lee YC, et al. Pulmonary outcomes of incretin-based therapies in COPD patients receiving single-inhaler triple therapy. ERJ Open Res. 2025;11(2):00803–2024. doi:10.1183/23120541.00803-2024
87. Yen FS, Hsu CC, Wei JC, et al. Glucagon-like peptide-1 receptor agonists may benefit cardiopulmonary outcomes in patients with COPD. Thorax. 2024;79(11):1017–1023. doi:10.1136/thorax-2023-221040
88. Altintas Dogan AD, Hilberg O, Hess S, Jensen TT, Bladbjerg EM, Juhl CB. Respiratory effects of treatment with a glucagon-like peptide-1 receptor agonist in patients suffering from obesity and chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2022;17:405–414. doi:10.2147/COPD.S350133
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Integrating Multi-Omics Data to Uncover Causal Links Between Mitochondria-Related Genes and Chronic Obstructive Pulmonary Disease: A Mendelian Randomization Study
Hong E, Mao J, Ke Z, Wu Y
International Journal of Chronic Obstructive Pulmonary Disease 2026, 21:553092
Published Date: 17 January 2026