Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
Cigarette Smoking Contributes to Th1/Th2 Cell Dysfunction via the Cytokine Milieu in Chronic Obstructive Pulmonary Disease
Received 8 July 2023
Accepted for publication 6 September 2023
Published 12 September 2023 Volume 2023:18 Pages 2027—2038
DOI https://doi.org/10.2147/COPD.S426215
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Gang Chen,1 Qing Mu,1 Zhao-Ji Meng2
1Department of Respiratory and Critical Care Medicine, Henan Provincial People’s Hospital, People’s Hospital of Zhengzhou University, Zhengzhou, Henan, People’s Republic of China; 2Department of Immune Allergy, Henan Provincial People’s Hospital, People’s Hospital of Zhengzhou University, Zhengzhou, Henan, People’s Republic of China
Correspondence: Gang Chen, Department of Respiratory and Critical Care Medicine, Henan Provincial People’s Hospital, People’s Hospital of Zhengzhou University, 7 Weiwu Road, Zhengzhou, Henan, 450003, People’s Republic of China, Email [email protected]
Background: Dysregulation and pyroptosis of T-helper (Th) cells and inflammatory cytokines have been implicated in the pathogenesis of chronic obstructive pulmonary disease (COPD). However, the immune response mechanisms as a consequence of tobacco smoke exposure are not fully understood. We hypothesized that cigarette smoke-induced inflammation could be modulated through the cytokine milieu and T-cell nicotinic acetylcholine receptors (nAChRs).
Methods: The proportions of peripheral blood Th1 and Th2 cells from patients with COPD, smokers without airway obstruction and healthy nonsmokers were analyzed using flow cytometry. The levels of plasma proinflammatory cytokines and their potential association with pulmonary function were also measured. The influence of cigarette smoke extract (CSE) on the conditioned differentiation of T helper cell subsets was further examined in vitro.
Results: Significantly higher Th1 cell and plasma IFN-γ and IL-18 levels but lower levels of Th2 cells were found in the peripheral blood from patients with COPD. The increased plasma levels of IFN-γ and IL-18 were negatively correlated with pulmonary function (FEV1% predicted value). Pyroptosis participates in COPD development probably through the activation of the NLRP3 inflammasome upon exposure to CSE. CSE does not directly induce the differentiation of T helper cells; however, under conditioned medium, CSE promotes Th1 development through α 7 nAChR modification, while it does not substantially interfere with Th2 differentiation.
Conclusion: The differences in the cytokine milieu play a key role in the effects of CSE on the immune response in patients with COPD.
Keywords: chronic obstructive pulmonary disease, COPD, cigarette smoke, T helper 1, Th1 cells, Th2, nicotinic acetylcholine receptors, nAChRs
Introduction
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death worldwide and is characterized by local and systemic inflammation.1 In addition to innate immunity, an increasing number of studies suggest that that adaptive immune responses play key roles in the pathogenesis of COPD, as COPD is typically characterized by the propagation of autoreactive T cells and B cells.1,2 These pathogenic lymphocyte cells exert a variety of functions by releasing various mediators and recruiting and activating other immune and parenchymal effector cells. Tobacco Smoke skews immune responses to promote the development of COPD, and smoking cessation can change the natural history of COPD.3–5
T-helper (Th) cells are a subtype of T cells that participates in the immune response; IFN-γ-producing Th1 cells are responsible for cellular immunity and tissue injury in autoimmune and infectious diseases, while IL-4-producing Th2 cells are mainly involved in the humoral response through enhancing antibody secretion by B cells.6 Previous studies have attempted to characterize the lymphocyte and cytokine production profile in COPD patients, but the results are conflicting. The majority of studies indicated that patients with COPD showed a predominant Th1 pattern,7–9 whereas others reported a predominant Th2 phenotype.10,11 Moreover, the characteristics of intrapulmonary and circulating lymphocytes in COPD patients are notable for often discrepant results.8 Tobacco smoke might contain antigens that induce Th1 responses in susceptible individuals, but not all smokers develop COPD and, unlike the findings in humans, inflammatory and disease parameters of lung emphysema attenuate after smoke cessation in mice.12 Pyroptosis is commonly induced by the NLRP3 inflammasome and is accompanied by the release of inflammatory cytokines such as IL-1β and IL-18.13 However, the role of pyroptosis in the pathogenesis of COPD remains unknown.
Recent studies have shown that nicotinic acetylcholine receptors (nAChRs) play a pivotal part in development and differentiation of T cells.14,15 Inflammatory cytokines can change α7 nAChRs expressed by immune cells, and these receptors display immunosuppressive function by inhibiting the release of inflammatory lymphocytes/cytokines.16,17 Moreover, we have previously reported that the expression of anti-inflammatory α7 nAChR in CD4+ T cells was reduced in patients with COPD.18 We therefore hypothesized that the different effects of cigarette exposure on Th1/Th2 dysregulation might suggest differences in the inflammatory microenvironment, which may further change α7 nAChR expression in CD4+ T cells.
In this study, we measured the proportion of Th1 and Th2 cells in the peripheral blood from patients with COPD. Moreover, we characterized the plasma levels of IFN-γ, IL-4, IL-1β and IL-18 and analyzed the potential association of cytokines with pulmonary function in those patients. Notably, we investigated the interaction of cigarette smoke extract (CSE) and the cytokine milieu on the specific differentiation of T helper cell subsets in vitro, especially on the expression of immunosuppressive α7 nAChR in CD4+ T cells. We present the following article in accordance with the MDAR reporting checklist.
Materials and Methods
Subjects
The study protocol was approved by the Ethics Committee of People’s Hospital of Zhengzhou University (#2021-52), and written informed consent was obtained from each subject. Based upon the Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria,19 22 patients with stable COPD who were free of exacerbation for more than 4 weeks were recruited (Table 1). At the same time, a cohort of healthy nonsmoking controls with normal lung function (HC; n = 20) and healthy asymptomatic smokers with normal lung function (HS; n = 18) were matched on age and sex to patients. COPD patients and HS individuals had a matched smoking history of more than 10 packs/year. Individuals with asthma, malignant tumors, inflammatory bowel disease, or other immune-related diseases were excluded.
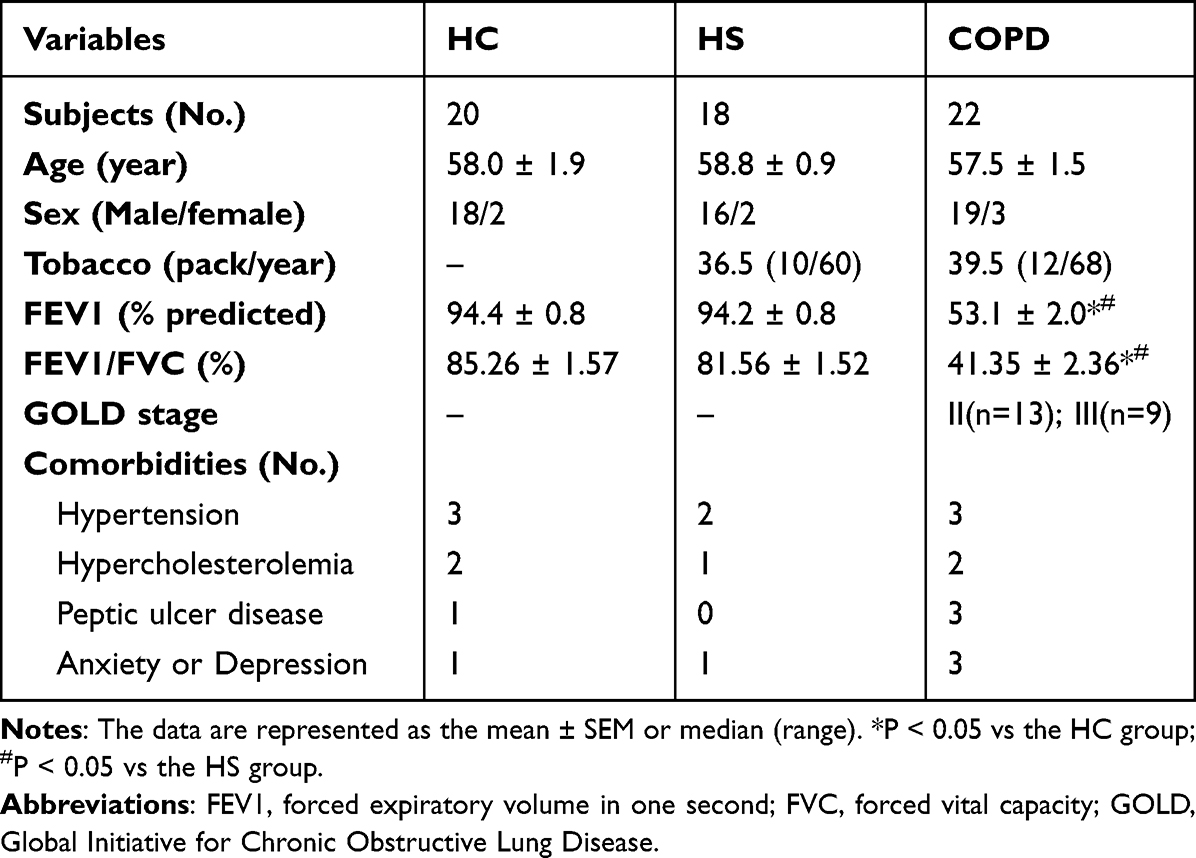 |
Table 1 Demographics and Clinical Characteristics of All Participants |
Sample Collection and Processing
Peripheral blood samples from each subject were collected in heparin-treated tubes (BD Biosciences, San Diego, USA). The blood samples were immersed in ice immediately and were then centrifuged at 2000 rpm for 10 min. The plasma aliquots were immediately frozen at −80°C for later determination of cytokine levels. Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Hypaque density-gradient centrifugation (#50494, MP Biomedicals, Illkirch, France).
Flow Cytometry
The expression of markers on T cells from peripheral blood was analyzed using flow cytometry as previously described18 after surface or intracellular staining with anti-human-specific antibodies (Abs) conjugated with FITC, PerCP-cy5.5, PE, or PE-CY7. These human Abs included anti-CD3 (#45-0037-41), anti-CD8 (#555366), anti-IFN-γ (#12-7319-42), anti-IL-4 (#25-7049-82) and isotype mAbs, which were purchased from BD Biosciences or eBioscience (San Diego, USA). To explore the expression of intracellular cytokines, T cells were stimulated with PMA (#P8139, 50 ng/mL; Sigma‒Aldrich, St. Louis, USA) and ionomycin (#I3909, 1 μg/mL; Sigma‒Aldrich) in the presence of GolgiStop (#554724, BD Biosciences) for 5 h and were then stained with the corresponding mAbs conjugated to fluoresceins after fixation and permeabilization (#88-8824, permeabilization kit, eBioscience) according to the manufacturer’s instructions. Flow cytometry was performed on a FACS Canto II (BD Biosciences) and analyzed with BD FACSDiva Software and FlowJo_V10 software (TreeStar, San Carlos, USA).
Cell Isolation
CD4+ T cells were isolated from PBMCs by magnetic-activated cell sorting (MACS) based on negative selection using the CD4+ T-cell isolation kit (#130-096-533, Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. The purity of CD4+ T cells was > 97%, as measured by flow cytometry.
Quantitative Real-Time PCR
Quantitative gene expression was determined by quantitative real-time PCR (qRT‒PCR) as previously described.18 Briefly, total RNA from CD4+ T cells was extracted using RNAiso plus (#9108, TaKaRa, Dalian, China) and reverse transcribed into cDNA using the PrimeScriptTM RT Reagent Kit (#RR036A, TaKaRa). Subsequently, qRT‒PCR was performed in a StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, USA) using gene-specific primers and SYBR® Premix Ex TaqTM (#DRR420A, TaKaRa). The qRT‒PCR data were determined by StepOne software v2.3 (Applied Biosystems). The relative expression values of each gene were normalized to GAPDH expression by the 2-∆Ct method (∆Ct = Ct target gene - Ct GADPH).20 The sequences of the primers were forward 5’-GCACCGTCAAGGCTGAGAAC-3’ and reverse 5’-TGGTGAAGACGCCAGTGGA-3’ for GAPDH; forward 5’-TGGCCAGATTTGGAAACCAGA-3’ and reverse 5’-AGTGTGGAATGTGGCGTCAAAG-3’ for α7 nAChR; and forward 5’- CCGCCATGTACTACCTGCT-3’ and reverse 5’- ACCCCTTTTCGAATTTGCCAT −3’ for NLRP3.
Measurement of Cytokine Levels
The plasma levels of IFN-γ (#EK0373), IL-4 (#EK0404), IL-1β (#EK0392) and IL-18 (#EK0864) were measured using sandwich ELISA kits according to the manufacturer’s protocols (all kits were purchased from Boster, Wuhan China). All samples were assayed in duplicate. The limits of detection for IFN-γ, IL-4, IL-1β and IL-18 were 1 pg/mL, 1.5 pg/mL, 3.9 pg/mL and 31.2 pg/mL, respectively.
CSE Preparation
CSE was prepared as previously described18 by infusing smoke from 1 cigarette (Huanghelou, Wuhan, China) into 2.5 mL of serum-free RPMI-1640. This solution was considered to be 100% CSE and was further filtered with a 0.22-μm filter (Millipore, Pittsburgh, USA) to remove bacteria and large particles. The 100% CSE was freshly prepared and diluted to the final working concentration 30 min prior to each experiment.
Differentiation of Th1/Th2 Cells in CSE-Conditioned Medium
Purified CD4+ T cells (1 × 106) were cultured in 200 μL of complete medium containing human IL-2 (10 ng/mL) in 96-well plates and stimulated with soluble anti-CD3 (#16-0037-38, 3 μg/mL; eBioscience) and anti-CD28 mAbs (#16-0289-85 μg/mL; eBioscience) for 5 d. To investigate the contribution of cigarette smoke and the cytokine milieu to the differentiation of CD4+ T cells, different concentrations of CSE with or without 20 ng/mL IL-12 (#200-12) or IL-4 (#200-04) (both from PeproTech, Rocky Hill, USA) were simultaneously added at the initiation of culture. Cells were harvested, and intracellular staining for IFN-γ and IL-4 was performed using flow cytometry as described above. The expression of the NLRP3 gene was quantified at the mRNA level using qRT‒PCR as described above.
In vitro Cytotoxicity Assay
Purified CD4+ T cells were seeded in 96-well plates as described above, and cells were stimulated with serial dilutions of CSE from 0.025% to 0.4%. Cell viability was assessed 5 d later using a Cell-Counting Kit-8 (#CK04, CCK-8; Dojindo Laboratories, Kumamoto, Japan) assay according to the manufacturer’s protocol.
Cytokine Stimulation Experiments
CD4+ T cells were incubated in the presence of anti-CD3/CD28 alone or with CSE, IL-12 (20 ng/mL), IL-4 (20 ng/mL) or GTS-21 (#ab120560, an agonist for the α7 nAChR; Abcam; Cambridge, USA, 10 μmol/l), and then α7 nAChR expression and Th1/Th2 differentiation were evaluated.
Statistics
Data are expressed as the mean ± SEM unless otherwise indicated. Comparisons of the data between different groups were performed using the Kruskal‒Wallis test followed by Dunn’s post hoc test. The correlations between variables were calculated by Spearman’s rank correlation coefficients. Analysis was completed with GraphPad Prism 6 software (GraphPad Software, La Jolla, USA), and p values of less than 0.05 were considered statistically significant.
Results
Patients with COPD Exhibit Imbalance in Circulating Th1/Th2 Cells
We first performed flow cytometry on PBMCs with gating on CD3+ and CD8− T cells to identify Th1 and Th2 cells (Figure 1A and B). We noted that the PBMCs from COPD patients contained an increased proportion of IFN-γ+ CD4+ T cells and a decreased proportion of IL-4+ CD4+ T cells compared with those from HC and HS subjects (Figure 1C and D). In addition, no significant differences in Th1 and Th2 levels were observed between HC and HS subjects. Thus, the accumulation of Th1 cells may play an important role in COPD immune dysregulation.
 |
Figure 1 Imbalance of circulating Th1 and Th2 cells in patients with chronic obstructive pulmonary disease (COPD). (A) Lymphocytes were gated on forward scatter area (FSC-A) versus side scatter area (SSC-A) plots, and CD4+ T cells were identified based on their expression of CD3, not CD8. (B) Representative flow cytometric dot plots of Th1 and Th2 cells in peripheral blood. Comparisons of Th1 (C) and Th2 (D) cell percentages in the peripheral blood from healthy controls (HC, n = 20), healthy smokers (HS, n = 18) and COPD patients (n = 22). The data are represented as the mean ± SEM; a value of P < 0.05 (2-tailed) was considered statistically significant. |
Increased Production of a Proinflammatory Th1-Associated Cytokine in the Plasma of COPD Patients
The presence of a Th1/Th2 imbalance prompted us to examine the cytokine milieu in patients with COPD. Indeed, we found significantly increased plasma levels of the proinflammatory Th1-associated cytokine IFN-γ in patients with COPD (Figure 2A). Although IL-4 levels did not significantly differ among the tested groups, we found a declining trend in IL-4 plasma levels in the COPD group (Figure 2B). Therefore, our results demonstrated that the plasma cytokine milieu in patients with COPD was skewed toward a Th1-associated proinflammatory phenotype. However, no significant differences in the levels of Th1/Th2 cells and related cytokines were observed between HC and HS subjects (Figures 1 and 2), suggesting that tobacco smoke exposure alone is not sufficient for the immune disorder. Moreover, as key cytokines for pyroptosis, the levels of IL-1β and IL-18 in patients with COPD were also evaluated. Although no significant difference in IL-1β levels was observed among the different groups, we found significantly increased IL-18 levels in the plasma of patients with COPD (Figure 2C and D).
 |
Figure 2 Disorder of pro-inflammatory cytokines in the plasma from patients with chronic obstructive pulmonary disease (COPD). Standardized sandwich ELISAs of plasma from healthy controls (HC, n = 20), healthy smokers (HS, n = 18) and COPD patients (n = 22) were performed to assess the levels of IFN-γ (A), IL-4 (B), IL-1β (C) and IL-18 (D). The data are represented as the mean ± SEM; a value of P < 0.05 (2-tailed) was considered statistically significant. |
Correlation of Plasma Cytokines with Pulmonary Function in COPD Patients
To examine the potential association of plasma cytokines with pulmonary function in COPD patients, single regression analysis was performed. Notably, unlike IL-4, increased levels of the proinflammatory cytokine IFN-γ exhibited a negative correlation with FEV1 (% predicted value, r = - 0.457, p < 0.05), suggesting that the predominant Th1 cytokine pattern worsens lung function in patients with COPD (Figure 3A and B). Notably, we observed that the IL-18 level displayed a negative correlation with FEV1 (% predicted) (r = - 0.464, p < 0.05) (Figure 3C), supporting an essential role for pyroptosis in airway inflammation. However, the level of IL-1β did not correlate with pulmonary function (Figure 3D).
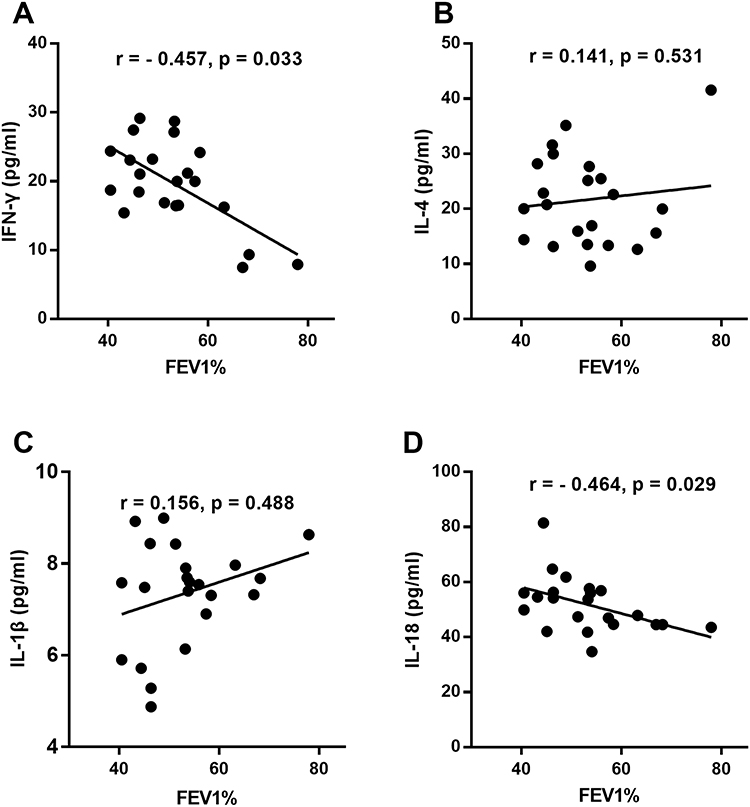 |
Figure 3 Correlation between plasma cytokine levels and lung function in patients with chronic obstructive pulmonary disease (COPD). Correlation of plasma IFN-γ (A), IL-4 (B), IL-1β (C) and IL-18 (D) levels with FEV1% predicted value in COPD patients (n = 22). Each symbol represents one individual COPD patient (black dots); a value of P < 0.05 (2-tailed) was considered statistically significant. |
Cytotoxicity of CSE on CD4+ T Cells
Since high concentrations of CSE are known to be cytotoxic, we performed a CCK-8 assay based on dehydrogenase activity evaluation in viable cells to ensure that the concentrations of CSE we used had no toxic effects on human CD4+ T cells. As shown in Figure 4, CSE at a concentration of ≥ 0.2% substantially reduced viability; therefore, we used 0.025% to 0.1% CSE as stimulation in subsequent experiments.
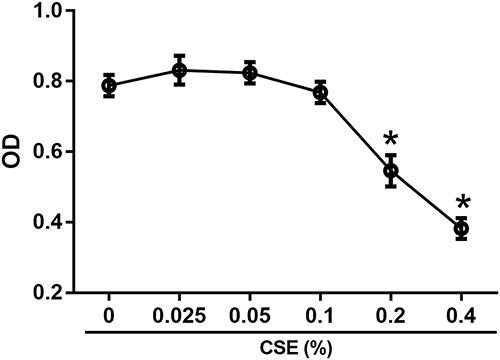 |
Figure 4 The effect of cigarette smoke extract (CSE) on cell viability. The cytotoxicity of CSE based on the dehydrogenase activity of viable cells was measured by CCK-8 assay at an absorbance of 450 nm. The CD4+ T cells were stimulated with serial dilutions of CSE for 5 d (n = 4). The data are represented as the mean ± SEM; *P < 0.05 vs 0% CSE. Abbreviation: OD, optical density. |
Contribution of the Cytokine Milieu and CSE-Conditioned Medium to the T Helper Cell Differentiation in vitro
Since the immune response mechanisms stimulated by tobacco smoke exposure are involved in COPD,21 we analyzed the immunomodulatory effects of CSE on alterations in T helper cell differentiation. We exposed purified CD4+ T cells from healthy nonsmokers to different concentrations of CSE for 5 d and then evaluated changes in the levels of Th1/Th2 cells by flow cytometry. However, the results showed that CSE did not directly induce the differentiation of either Th1 or Th2 cells (Figure 5A and B).
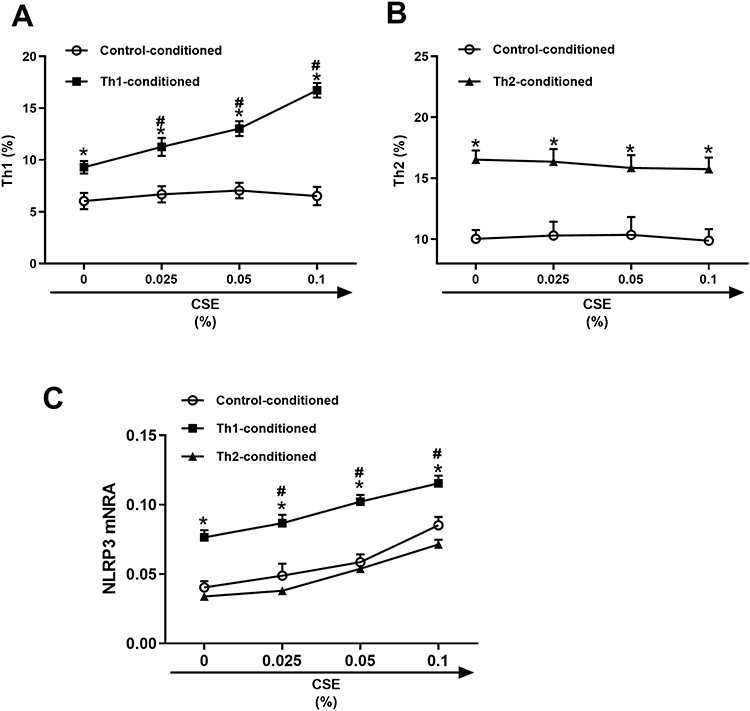 |
Figure 5 The contribution of IL-12 and IL-4 on cigarette smoke extract (CSE)-regulated Th1 and Th2 differentiation. The CD4+ T cells from healthy nonsmokers were stimulated with IL-2/anti-CD3/CD28 (control -conditioned) in the presence of serial concentrations of cigarette smoke extract (CSE) with and without 20 ng/mL IL-12 (Th1-conditioned) or IL-4(Th2-conditioned) for 5 d. The expression of Th1/Th2 was analyzed using flow cytometry (A and B), and NLRP3 mRNA levels were analyzed using qRT‒PCR (C) (n = 4). The data are represented as the mean ± SEM; *P<0.05 vs control-conditioned; #P<0.05 vs 0% CSE. |
It has been ascertained that the cytokine milieu of COPD patients differs from that of healthy individuals, with abnormally increased IL-12 levels and abnormally decreased IL-4 levels. It is also known that the development and differentiation of Th1/Th2 cells can both affect and be affected by cytokine milieu.6 Therefore, we hypothesized that differences in the inflammatory cytokines could contribute to the various effects of CSE treatment on T helper cell development. To verify this hypothesis, we further established the unique features of COPD using IL-12 stimulation (model) or IL-4 stimulation (control) in vitro. Notably, unlike IL-4, the presence of IL-12 renewed the ability of various concentrations of CSE (0.025% to 0.1%) to significantly increase the number of IFN-γ-producing CD4+ T cells in a concentration-dependent manner (Figure 5A and B). However, CSE did not substantially interfere with the number of Th2 cells in the cytokine milieu (Figure 5B).
Pyroptosis and inflammation are involved in the development of COPD.13 Increased mRNA levels of NLRP3 was observed after exposure to CSE (Figure 5C). Unlike the Th2-conditioned milieu, the Th1-conditioned milieu further promoted NLRP3 inflammasome activation induced by CSE (Figure 5C).
Cytokines Modulate α7 nAChR Expression in CD4+ T Cells
It has been reported that cytokine milieu, but not nicotinic stimulation, can modify α7 nAChR expression by immune cells, and α7 nAChRs exhibit anti-inflammatory functions.16,17 Therefore, the diverse effects of CSE on Th1/Th2 differentiation might reflect α7 nAChR expression differences in CD4+ T cells modulated by the local cytokine milieu.
To test this hypothesis, we determined the changes in α7 nAChR expression under the action of IL-12 and IL-4 using qRT‒PCR. Figure 6A shows that CSE did not directly induce α7 nAChR expression. In the presence of anti-CD3/CD28 stimulation, IL-12 significantly downregulated α7 nAChR mRNA expression, whereas IL-4 markedly upregulated it (Figure 6B). In addition, our study demonstrated that GTS-21, a selective α7 nAChR agonist, had the potential to reduce the levels of Th1 cells by directly acting on T cells and inhibiting their production (Figure 6C). The presence of GTS-21 did not affect Th2 cell differentiation (Figure 6D). Hence, IL-12 might reinforce the proinflammatory effects of CSE by specifically reducing expression of the immunosuppressive α7 nAChR in CD4+ T cells.
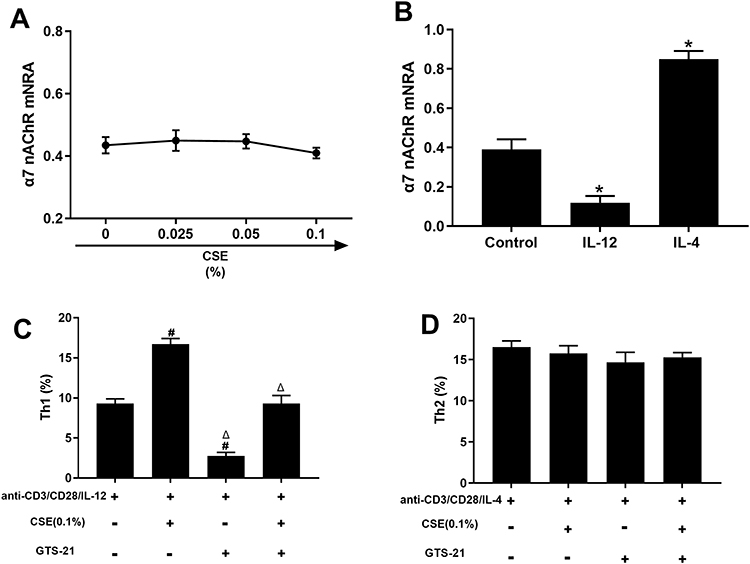 |
Figure 6 Alterations of α7 nAChR expression in CD4+ T cells by IL-12 and IL-4. The CD4+ T cells from healthy nonsmokers were incubated in the presence of IL-2/anti-CD3/CD28 alone or with cigarette smoke extract (CSE), IL-12 (20 ng/mL), IL-4 (20 ng/mL) or GTS-21 (an agonist for the α7 nAChR; 10 μmol/l) for 5 d, after which the levels of α7 nAChR mRNA (A and B) and Th1/Th2 differentiation (C and D) were analyzed (n = 4). The data are represented as the mean ± SEM; *P<0.05 vs Control; #P<0.05 vs anti-CD3/CD28/IL-12. ΔP<0.05 vs anti-CD3/CD28/IL-12 + CSE. |
Discussion
In this study, we found an imbalance in proinflammatory Th1 and Th2 responses in patients with COPD. Importantly, we report for the first time that differences in the cytokine milieu play a critical part in altering the outcome of cigarette smoke exposure on the immune response (Figure 7). The Th1-associated inflammatory milieu found in COPD patients inhibited expression of the immunosuppressive α7 nAChR in CD4+ T cells, which may partially account for the development of COPD in some smokers.
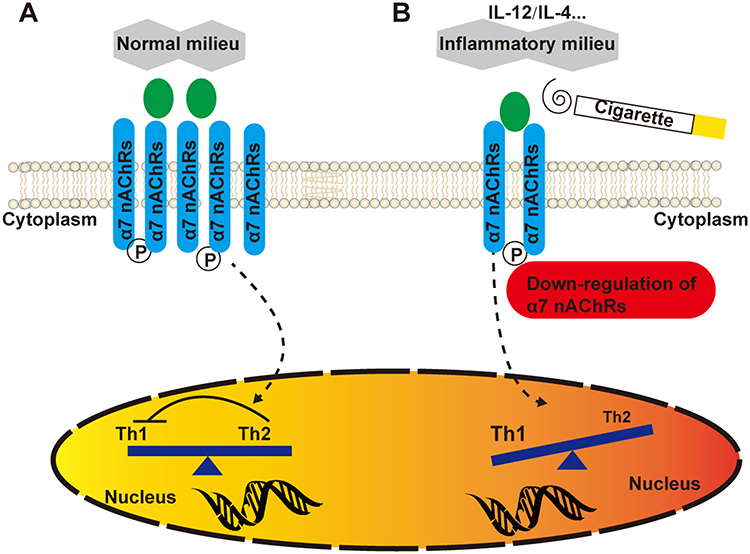 |
Figure 7 Schematic representation of Th1/Th2 cell dysfunction via the cytokine milieu. (A) In the presence of a normal milieu, appropriate α7 nAChR expression in CD4+ T cells keeps effector T cells in balance. (B) However, upon inflammatory insult or cigarette smoke extract (CSE) exposure, downregulation of α7 nAChRs by the activated immune system in susceptible individuals evokes Th1 cell-mediated proinflammatory responses. |
It is now well established that T lymphocytes actively participate in the smoke-induced inflammatory response in COPD.1 In contrast to patients with asthma, COPD patients exhibit Th1 infiltration and IFN-γ production in the airways and lung parenchyma.22 Consistent with these findings, the the expression of the Th1-associated chemokine receptor CXCR3 and its ligand IFN-γ inducible protein10 (IP-10, CXCL10) is upregulated and there is a polarized localization of IFN-γ inducible T-cell α chemoattractant (CXCL11) in the airways of patients with COPD.6,9 However, other studies have shown predominantly Th2 intrapulmonary lymphocytes in situ.10,11
It is difficult to acquire a suitable intrapulmonary specimen, and identifying reliable biomarkers that can be easily determined in peripheral blood is necessary.23 However, only a limited number of studies have investigated the presence of Th1/Th2 cells in the peripheral blood of COPD patients, and the findings of these studies are considerably heterogeneous.8,11 Majori et al found that circulating CD4+ T lymphocytes produce increased IFN-γ and decreased IL-4 levels in patients with COPD compared to smokers with normal lung function and healthy nonsmokers,24 which was consistent with our findings. The discrepancy seen between our results in peripheral blood and those reported in local intrapulmonary lymphocytes may be related to considerable differences in tissue samples, different stimuli, disease severity and other experimental methodologies.
Concordant with an earlier report,25 we detected significantly higher levels of plasma IFN-γ in COPD patients, and high IFN-γ levels were negatively correlated with lung function, which suggests that a proinflammatory immune response characterized by Th1 cytokines worsens lung function. IFN-γ or any type I cytokine can cause COPD-relevant pulmonary tissue inflammation and structural remodeling.6 The decreasing trend of plasma IL-4 levels in the patients with COPD could be considered consistent with a relative predominance of IFN-γ since IFN-γ inhibits the activity and function of Th2 cells.26 When viewed in combination, these studies suggest that the Th1-associated pro-inflammatory milieu, and/or the pathways it activates, may contribute to the development of abnormal expiratory airflow obstruction.
Although long-term exposure to tobacco smoke is regarded as the primary risk factor for the pathogenesis of COPD, only a small number of smokers develop COPD, and inflammation persists even after smoking cessation.21 Importantly, we have demonstrated that peripheral levels of Th1/Th2 cells and related cytokines from non-COPD smokers were comparable with those from healthy nonsmokers, suggesting that the development of this disorder cannot be fully explained by cigarette smoking. To reveal the mechanisms in charge of the adaptive immune response following tobacco smoke exposure in COPD, we determined the immunologic outcome of CSE on freshly isolated CD4+ T cells from healthy nonsmokers. Consistent with an earlier report,27 in vitro CSE stimulation did not induce the differentiation of CD4+ T cells, indicating that the development of Th1 and Th2 cells could not be facilitated simply through cigarette smoke.
It is also known that IL-12 and IL-4 are potent inducers of Th1 and Th2 cell synthesis, respectively, and suppress the differentiation of Th2 and Th1, respectively.26 Therefore, differences between tobacco smoke effects on smokers with and without COPD might reflect different inflammatory microenvironments (Figure 7). Since COPD patients exhibited increased IL-12 and decreased IL-4 plasma levels when compared with those in healthy controls, we wondered if these cytokines could modulate the CSE effects on T cells mediating immune inflammation in vivo. Thus, we investigated whether IL-12 (model) or IL-4 (control) modified the capacity of CSE to promote Th1/Th2 differentiation in CD4+ T cells. Although CSE did not substantially interfere with Th2 development in the cytokine milieu, the presence of IL-12 renewed the ability of CSE to significantly increase the number of IFN-γ-producing CD4+ T cells in a concentration-dependent manner (0.025% to 0.1%). In contrast, IL-4 did not modify the stimulatory effect of CSE. Hence, the impact of CSE on T-cell development depends not only on the CSE concentration but also on the cytokine milieu.
Pyroptosis is an inflammatory form of cell death that is accompanied by the release of inflammatory cytokines such as IL-1β and IL-18.13 Currently, it has been shown that CSE activates the NLRP3/caspase-1 signaling pathway and sequentially stimulates pyroptosis in bronchial epithelial cells, which causes severe lung injury.28 In this study, we found negative correlations between FEV1 predicted values and plasma IL-18 levels, supporting the hypothesis that systemic inflammation induces airflow obstruction in the pathogenesis of COPD. Notably, the Th1-conditioned milieu might further promote NLRP3 inflammasome activation and pyroptosis after exposure to CSE. Thus, the NLRP3 inflammasome is vital for the development of COPD, and blockade of NLRP3 might be a therapeutic strategy for COPD treatment.
To reveal the possible mechanisms by which cytokines affect the immunological processes in response to CSE exposure, we determined the expression of α7 nAChRs, which exhibit immunosuppressive functions by regulating inflammatory cytokines. Inflammatory cytokines themselves could regulate the function of α7 nAChR,16,17 which suggested a positive feedback loop. Additionally, there is an association between the α7 nAChR pathway and T-bet (Th1 transcription factor) expression in the T cells of colon cancer patients.29 Our current study elucidated the action of IL-12 and IL-4 to exhibit dichotomous effect on α7 nAChRs expression at the mRNA level, which is consistent with a previous study in mice.14 Consistent with the downregulation of α7 nAChR expression by IL-12, we have previously demonstrated the lack of α7 nAChRs in patients with COPD.18 Thus, the distinct outcome of cigarette exposure on Th1/Th2 generation might reflect differences in α7 nAChR expression in CD4+ T cells modulated by the local cytokine milieu. We also demonstrated the therapeutic potential of GTS-21 in COPD, which exerts anti-inflammatory actions on T cells by inhibiting the differentiation of Th1 via mechanisms that remain to be determined. However, the present study has several limitations that warrant mention. It is worth to notice IL-1β and IL-18 are not linked to pyroptosis exclusively but are significant mediators in other biological processes. Meanwhile, it would be interesting to determine cells/cytokines in the bronchoalveolar lavage fluid (BALF), which directly reflect lung condition per se. Further work is needed to demonstrate the potential roles of downstream signals. In addition, functional studies such as animal experiments and knockdown/overexpression in human T cells in vitro are warranted to clarify the underlying mechanism.
Conclusions
In conclusion, the present study indicated an imbalance between Th1 and Th2 responses in COPD patients. The results presented in our study highlight the importance of the combined action of both the internal environment (cytokine milieu) and external environment (cigarette smoke exposure) in immune responses. Moreover, our data enrich the concept of the cholinergic (nicotinic) anti-inflammatory pathway and provide a novel pathway for future therapeutic intervention in COPD.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article.
Ethics Approval and Informed Consent
The study was approved by the Ethics Committee of People’s Hospital of Zhengzhou University (#2021-52), and informed written consent was obtained from all individual participants. This study was conducted in accordance with the tenets of the Declaration of Helsinki.
Funding
This work was supported by a grant from the Key Scientific and Technological Project of Henan Province [grant number: 222102310704].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Brightling C, Greening N. Airway inflammation in COPD: progress to precision medicine. Eur Respir J. 2019;54(2):1900651. doi:10.1183/13993003.00651-2019
2. Rinaldi M, Lehouck A, Heulens N, et al. Antielastin B-cell and T-cell immunity in patients with chronic obstructive pulmonary disease. Thorax. 2012;67(8):694–700. doi:10.1136/thoraxjnl-2011-200690
3. Pezzuto A, Tonini G, Ciccozzi M, et al. Functional benefit of smoking cessation and triple inhaler in combustible cigarette smokers with severe COPD: a retrospective study. J Clin Med. 2022;12(1):234. doi:10.3390/jcm12010234
4. Pezzuto A, Stellato M, Catania G, et al. Short-term benefit of smoking cessation along with glycopirronium on lung function and respiratory symptoms in mild COPD patients: a retrospective study. J Breath Res. 2018;12(4):046007. doi:10.1088/1752-7163/aad0a8
5. Strzelak A, Ratajczak A, Adamiec A, Feleszko W. Tobacco smoke induces and alters immune responses in the lung triggering inflammation, allergy, asthma and other lung diseases: a mechanistic review. Int J Env Res Pub He. 2018;15(5):1033. doi:10.3390/ijerph15051033
6. Butcher MJ, Zhu J. Recent advances in understanding the Th1/Th2 effector choice. Fac Rev. 2021;10:30. doi:10.12703/r/10-30
7. Grumelli S, Corry DB, Song LZ, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004;1(1):e8. doi:10.1371/journal.pmed.0010008
8. Hodge G, Nairn J, Holmes M, et al. Increased intracellular T helper 1 proinflammatory cytokine production in peripheral blood, bronchoalveolar lavage and intraepithelial T cells of COPD subjects. Clin Exp Immunol. 2007;150(1):22–29. doi:10.1111/j.1365-2249.2007.03451.x
9. Porter JC, Falzon M, Hall A. Polarized localization of epithelial CXCL11 in chronic obstructive pulmonary disease and mechanisms of T cell egression. J Immunol. 2008;180(3):1866–1877. doi:10.4049/jimmunol.180.3.1866
10. Miotto D, Ruggieri MP, Boschetto P, et al. Interleukin-13 and −4 expression in the central airways of smokers with chronic bronchitis. Eur Respir J. 2003;22(4):602–608. doi:10.1183/09031936.03.00046402
11. Barcelo B, Pons J, Fuster A, et al. Intracellular cytokine profile of T lymphocytes in patients with chronic obstructive pulmonary disease. Clin Exp Immunol. 2006;145(3):474–479. doi:10.1111/j.1365-2249.2006.03167.x
12. Guerassimov A, Hoshino Y, Takubo Y, et al. The development of emphysema in cigarette smoke-exposed mice is strain dependent. Am J Resp Crit Care. 2004;170(9):974–980. doi:10.1164/rccm.200309-1270OC
13. Feng Y, Li M, Yangzhong X, et al. Pyroptosis in inflammation-related respiratory disease. J Physiol Biochem. 2022;78(4):721–737. doi:10.1007/s13105-022-00909-1
14. Galitovskiy V, Qian J, Chernyavsky AI, et al. Cytokine-induced alterations of alpha7 nicotinic receptor in colonic CD4 T cells mediate dichotomous response to nicotine in murine models of Th1/Th17- versus Th2-mediated colitis. J Immunol. 2011;187(5):2677–2687. doi:10.4049/jimmunol.1002711
15. Nakata Y, Miura K, Yamasaki N, et al. Expression and function of nicotinic acetylcholine receptors in induced regulatory T cells. Int J Mol Sci. 2022;23(3):1779. doi:10.3390/ijms23031779
16. Kondo Y, Tachikawa E, Ohtake S, et al. Inflammatory cytokines decrease the expression of nicotinic acetylcholine receptor during the cell maturation. Mol Cell Biochem. 2010;333(1–2):57–64. doi:10.1007/s11010-009-0204-4
17. Qian J, Galitovskiy V, Chernyavsky AI, Marchenko S, Grando SA. Plasticity of the murine spleen T-cell cholinergic receptors and their role in in vitro differentiation of naive CD4 T cells toward the Th1, Th2 and Th17 lineages. Genes Immun. 2011;12(3):222–230. doi:10.1038/gene.2010.72
18. Zhang MQ, Wan Y, Jin Y, et al. Cigarette smoking promotes inflammation in patients with COPD by affecting the polarization and survival of Th/Tregs through up-regulation of muscarinic receptor 3 and 5 expression. PLoS One. 2014;9(11):e112350. doi:10.1371/journal.pone.0112350
19. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Resp Crit Care. 2017;195(5):557–582. doi:10.1164/rccm.201701-0218PP
20. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C-T method. Nat Protoc. 2008;3(6):1101–1108. doi:10.1038/nprot.2008.73
21. Taylor JD. COPD and the response of the lung to tobacco smoke exposure. Pulm Pharmacol Ther. 2010;23(5):376–383. doi:10.1016/j.pupt.2010.04.003
22. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immun. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
23. Barnes PJ, Chowdhury B, Kharitonov SA, et al. Pulmonary biomarkers in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;174(1):6–14. doi:10.1164/rccm.200510-1659PP
24. Majori M, Corradi M, Caminati A, et al. Predominant TH1 cytokine pattern in peripheral blood from subjects with chronic obstructive pulmonary disease. J Allergy Clin Immun. 1999;103(3 Pt 1):458–462. doi:10.1016/S0091-6749(99)70471-9
25. Kalathil SG, Lugade AA, Pradhan V, et al. T-regulatory cells and programmed death 1+ T cells contribute to effector T-cell dysfunction in patients with chronic obstructive pulmonary disease. Am J Resp Crit Care. 2014;190(1):40–50. doi:10.1164/rccm.201312-2293OC
26. O’Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. 1998;8(3):275–283. doi:10.1016/S1074-7613(00)80533-6
27. Turowska A, Weissmann N, Baumgartl N, Garn H. Effects of cigarette smoke extract-conditioned medium on T helper cell development and differentiation in vitro. Am J Resp Crit Care. 2015;191:A2733.
28. Wang L, Meng J, Wang C, et al. Hydrogen sulfide attenuates cigarette smoke‑induced pyroptosis through the TLR4/NF-kappaB signaling pathway. Int J Mol Med. 2022;49(5). doi:10.3892/ijmm.2022.5112
29. Kikuchi H, Itoh J, Fukuda S. Chronic nicotine stimulation modulates the immune response of mucosal T cells to Th1-dominant pattern via nAChR by upregulation of Th1-specific transcriptional factor. Neurosci Lett. 2008;432(3):217–221. doi:10.1016/j.neulet.2007.12.027
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.