Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Cigarette smoke extract induces placental growth factor release from human bronchial epithelial cells via ROS/MAPK(ERK-1/2)/Egr-1 axis
Authors Wu D, Yuan Y, Lin Z, Lai T ![]() , Chen M, Li W, Lv Q, Yuan B, Li D, Wu B
, Chen M, Li W, Lv Q, Yuan B, Li D, Wu B
Received 27 August 2016
Accepted for publication 9 November 2016
Published 2 December 2016 Volume 2016:11(1) Pages 3031—3042
DOI https://doi.org/10.2147/COPD.S120849
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Dong Wu,1,* Yalian Yuan,1,* Zhixiu Lin,2,* Tianwen Lai,1 Min Chen,1 Wen Li,1 Quanchao Lv,1 Binfan Yuan,1 Dongmin Li,1 Bin Wu1
1Department of Respiratory, Institute of Respiratory Diseases, 2Department of Pharmacy, The Affiliated Hospital of Guangdong Medical University, Zhanjiang, People’s Republic of China
*These authors contributed equally to this work
Abstract: Etiological evidence demonstrates that there is a significant association between cigarette smoking and chronic airway inflammatory disease. Abnormal expression of placental growth factor (PlGF) has been reported in COPD, and its downstream signaling molecules have been reported to contribute to the pathogenesis of airway epithelial cell apoptosis and emphysema. However, the signaling mechanisms underlying cigarette smoke extract (CSE)-induced PlGF expression in airway microenvironment remain unclear. Herein, we investigated the effects of reactive oxygen species (ROS)-dependent activation of the mitogen-activated protein kinase (MAPK) (extracellular signal-regulated kinase1/2 [ERK-1/2])/early growth response-1 (Egr-1) pathway on CSE-induced PlGF upregulation in human bronchial epithelium (HBE). The data obtained with quantitative reverse transcription polymerase chain reaction, Western blot, enzyme-linked immunosorbent assay (ELISA) and immunofluorescence staining analyses showed that CSE-induced Egr-1 activation was mainly mediated through production of ROS and activation of the MAPK (ERK-1/2) cascade. The binding of Egr-1 to the PlGF promoter was corroborated by an ELISA-based DNA binding activity assay. These results demonstrate that ROS activation of the MAPK (ERK-1/2)/Egr-1 pathway is a main player in the regulatory mechanism for CSE-induced PlGF production and that the use of an antioxidant could partly abolish these effects. Understanding the mechanisms of PlGF upregulation by CSE in the airway microenvironment may provide rational therapeutic interventions for cigarette smoking-related airway inflammatory diseases.
Keywords: cigarette smoke, reactive oxygen species, airway epithelium, placental growth factor, extracellular signal-regulated kinase1/2, early growth response-1
Introduction
Epidemiologic data link cigarette smoking to many pulmonary disorders such as COPD and chronic bronchitis.1,2 Cigarette smoke (CS) contains >4,000 different kinds of constituents, including a mass of toxic oxidants.3,4 Airway epithelial cells are directly exposed to CS and serve as a major target of oxidative stress.5 Activation of the airway epithelium by oxidative stress is identified as an important pathologic process linking to chronic airway inflammatory disease.6,7
Placental growth factor (PlGF) is a member of the vascular endothelial growth factor (VEGF) family, which is composed of VEGF-A, VEGF-B, VEGF-C, VEGF-D and VEGF-E.8 PlGF-deficient mice develop normally, suggesting that PlGF is dispensable for development and health;9 however, it has been associated with tissue ischemia, malignancy, inflammation and multiple other diseases.10–12 In fact, increased expression of PlGF has been observed in the serum and bronchoalveolar lavage fluid (BALF) of patients with COPD.13 Moreover, the overexpression of PlGF in the lungs of mice has been shown to cause emphysema due to type II pneumocyte autophagy and apoptosis.14 Conversely, knocking-out PlGF protects mice against elastase-induced pulmonary emphysema.15 These phenotypes suggest a potential role for PlGF in the development of COPD and emphysema. However, the mechanism responsible for the CS-induced production and secretion of PlGF in pulmonary microenvironment remains to be determined.
In this study, we investigated the molecular mechanisms by which cigarette smoke extract (CSE) induced PlGF expression in human bronchial epithelium (HBE). We used HBE cells as a model and prepared CSE to imitate the situation of human smoking in the airway. Our data suggested that CSE-induced Egr-1 activation was mainly mediated through reactive oxygen species (ROS)-dependent activation of the mitogen-activated protein kinase (MAPK) (ERK-1/2) cascade and binding of Egr-1 to PlGF promoter. These results demonstrate that activation of an ROS, MAPK (ERK-1/2), Egr-1 cascade is the main player in the regulatory mechanism for CS-induced PlGF production and that pretreating cells with N-acetyl-L-cysteine (NAC) could partly abolish this effect. Our research advances our knowledge about the role of PlGF, which may be recognized as a therapeutic target in smoking-related airway inflammatory diseases.
Materials and methods
Cell lines and drug treatment
The human bronchial epithelial cell line (16-HBE) was obtained from a cell bank (American Type Culture Collection [ATCC], Manassas, VA, USA) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS) (Thermo Fisher Scientific), 2 mM glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin (Thermo Fisher Scientific). 16-HBE cells were grown at 37°C, 95% humidity and 5% CO2 condition. Prior to incubation with or without CSE, the cells were pretreated with 10 μM U0126 (Cell Signaling, Beverly, MA, USA) or 0.1 mM NAC (Sigma-Aldrich Co., St Louis, MO, USA) for 2 h.
CSE preparation
We prepared fresh CSE before each experiment as previously described16 with a slight modification. Briefly, one commercial filtered cigarette (Marlboro, each containing 1.3 mg of nicotine and 15 mg of tar according to the manufacturer’s report) was bubbled into a flask containing 10 mL of preheated Roswell Park Memorial Institute-1640 medium; this medium was defined as 100% CSE, adjusted to pH 7.4 and filtered through a 0.22-μm filter. The concentration of CSE was calculated by measuring the optical density (OD) at a wavelength of 320 nm using a DU800 Spectrophotometer (Beckman Coulter, Inc., Brea, CA, USA). The pattern of absorbance among a series of CSE solutions prepared in this manner revealed few differences, and the mean OD of the different batches was 1.42±0.13.
Cell viability
Cell viability was evaluated with Cell Counting Kit-8 (CCK-8; Dojindo, Tokyo, Japan) according to the manufacturer’s instructions. Briefly, 16-HBE cells were plated in the 96-well plates (5×103 cells per well) and incubated for 24 h. CSE was introduced to cells with different test concentrations (3%, 6%, 9%, 12% and 15%). Untreated cells served as the control. After 24 h, 48 h and 72 h treatments, the cells were washed twice with phosphate-buffered saline (PBS). In all, 20 μL of CCK-8 solution was added to each well of the plate and incubated for a further 1 h at 37°C. The OD of each well at 450 nm was recorded on a microplate reader (Thermo Fisher Scientific).
Real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was isolated with TRIzol (Thermo Fisher Scientific) according to the manufacturer’s protocol. Reverse transcription was performed using RT2 First Strand Kit (Qiagen NV, Venlo, the Netherlands) on the Eppendorf Mastercycler (Eppendorf AG, Hamburg, Germany). Quantitative real-time RT-PCR was performed to quantify the target genes expression using Ssoadvanced™ SYBR® Green Supermix (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and the Roche LightCycler 480 PCR system (Hoffman-La Roche Ltd., Basel, Switzerland). The following PCR conditions were used: 95°C for 2 min and then 40 cycles at 95°C for 10 sec and 60°C for 30 sec. β-Actin was used as the housekeeping gene. The qRT-PCR primer sequences are as follows:17,18 PlGF forward: 5′-TGTTCAGCCCATCCTGTGTC-3′ and reverse: 5′-AGGAGCTGCATGGTGACATT-3′, Egr-1 forward: 5′-CCGCAGAGTCTTTTCCTGAC-3′ and reverse: 5′-AGCGGCCAGTATAGGTGATG-3′, and β-actin forward: 5′-GCAAGAGATGGCCACGGCTG-3′ and reverse: 5′-CCACAGGACTCCATGCCCAG-3′.
Protein extraction and immunoblot analysis
Total protein from 16-HBE cells was extracted using ice-cold radioimmunoprecipitation assay buffer supplemented with 1 mM phenylmethanesulfonyl fluoride (PMSF) (Thermo Fisher Scientific) or phosphatase inhibitor cocktail (Calbiochem, La Jolla, CA, USA) for phosphoprotein. Cell lysates containing equal amounts of protein (50 μg) were separated by 10% sodium dodecyl sulfate polyacrylamide gels and transferred onto nitrocellulose membrane (Amersham, GE Healthcare, Milano, Italy) using a Mini-Protean 2 electrophoresis system (Bio-Rad Laboratories, Inc.). The membranes were blocked in phosphate-buffered saline plus 0.1% Tween 20 (PBST; Sigma-Aldrich Co.) containing 5% non-fat milk for 1 h and then incubated with respective primary antibodies at 4°C overnight. The antibodies used were rabbit monoclonal antibodies to Egr-1 (1:1,000; Abcam, San Diego, CA, USA), rabbit polyclonal antibodies to PlGF (1:1,000; Abcam), rabbit polyclonal antibodies to phospho- and total-p44/42 MAPK (ERK-1/2) (1:2,000; Cell Signaling Technologies [CST], Danvers, MA, USA) and rabbit polyclonal antibodies to glyceraldehyde 3-phosphate dehydrogenase (1:10,000; Sigma-Aldrich Co.), followed by incubation with horseradish peroxidase-conjugated anti-rabbit (1:2,000; CST). After washing again with Tris buffered saline with Tween (TBST), target bands were visualized using Immobilon Western Chemiluminescent horse radish peroxidase (HRP) Substrate (EMD Millipore, Billerica, MA, USA) and detected using the GeneGnome Imaging System (Syngene, Frederick, MD, USA). Band densities were quantified using densitometry (Bio-Rad Laboratories, Inc.). All immunoblots were repeated in triplicate.
Enzyme-linked immunosorbent assay (ELISA)
The cells were treated with CSE at the previously described doses for 2 h, followed by changing the media and a 4- to 24-h recovery. Cellular supernatants were analyzed using a Human PlGF Quantikine ELISA Kit (R&D Systems, Inc., Minneapolis, MN, USA) according to the manufacturer’s protocols.
Immunofluorescent staining and confocal microscopy
16-HBE cells were cultured on Lab-Tek-II chamber slides (four or eight well; Thermo Fisher Scientific) and treated with CSE, fixed with 4% paraformaldehyde in PBS for 20 min at 4°C and incubated with rabbit anti-Egr-1 or phospho-ERK-1/2 antibody (Abcam) at 4°C overnight, followed by Alexa Fluor 488 goat anti-rabbit IgG antibody (Thermo Fisher Scientific). Fluorescence was detected with a Leica-TCS-SP confocal microscope (Leica Microsystems, Wetzlar, Germany) and the accompanying software Leica v2.6.1 (Leica Microsystems).
RNA interference
Two independent siRNAs for knockdown of Egr-1 and a negative control siRNA (scRNA) were designed and synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China) and used in this experiment. Nucleotide sequences for siRNAs used were as follows: Egr-1 siRNA-1, 5′-CAGUAUCAUCUCCAUCAUA-3′; Egr-1 siRNA-2, 5′-GGUUACUACCUCUUAUCCA-3′ and negative control siRNA, 5′-CCUACGCCACCAAUUUCGU-3′. For transient transfection of siRNA, 16-HBE cells were seeded in six-well tissue culture plates and grown to 60%–70% confluence; either Egr-1 siRNA or scRNA was transfected to cells using lipofectamine RNAiMAX (Thermo Fisher Scientific), according to the manufacturer’s protocols with the following minor modifications. Briefly, scRNA or Egr-1 siRNA (150 nM) was incubated with 8 μL of lipofectamine RNAiMAX reagent in 200 μL Opti-MEM (Thermo Fisher Scientific) for 20 min. The siRNA mixtures were added to the 16-HBE cells in serum-free medium and cultured at 37°C in a CO2 incubator. After 24 h, the medium was replaced with growth medium and the cells were used for CSE exposure experiments.
Egr-1 transcriptional activity assay
16-HBE cells were treated with CSE for 4 h and then the NE-PER Nuclear and Cytoplasmic Extraction Reagent Kit (Thermo Fisher Scientific) was used to prepare nuclear extracts. The BD Mercury TransFactor Kit (BD Biosciences, San Jose, CA, USA) was used to measure Egr-1 transcriptional activity following manufacturer’s protocols. In brief, nuclear extracts (50 μg/mL) were added to incubation wells precoated with the PlGF DNA binding sequences. After incubating at room temperature for 1 h, blocking buffer was used to wash redundant samples; bound transcription factors in the wells were detected by rabbit anti-Egr-1 antibody and then a HRP conjugated secondary antibody was added to generate a color reaction. The enzymatic product was measured on a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Measurement of intracellular ROS
Intracellular ROS levels were measured in intact 16-HBE cells by the 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) assay using acetyl-ester (Molecular Probes, Carlsbad, CA, USA) according to the manufacturer’s instructions. In brief, cells were treated with NAC (10 mmol/L) for 2 h, followed by addition of CSE (60 min); H2O2 served as a positive control. Then, cells were treated with 10 μmol/L CM-H2DCFDA for 30 min at 37°C in darkness. The excess probe was washed out. Fluorescence of chloromethyl-2′,7′-dichlorodihydrofluorescein (CM-DCF; excitation/emission: 495/530 nm) was recorded by a microplate reader (BioTek Instruments, Inc.). A superoxide dismutase (SOD) assay kit (Cell Biolabs, Inc., San Diego, CA, USA) was also used to detect the activity of SOD in intact 16-HBE cells according to the manufacturer’s instructions.
Statistical analysis
Data were presented as mean ± standard error of the mean from triplicate independent experiments. An unpaired Student’s t-test or Mann–Whitney U test was used for two-group comparisons and one-way analysis of variance (ANOVA) was used for multiple-group comparisons. Statistical analyses were performed using GraphPad Prism version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). A P-value <0.05 was considered as statistically significant.
Results
CSE induces PlGF mRNA, protein expression and release from human bronchial epithelial cells
Like other growth factors of the VEGF family, PlGF is synthesized and released to activate target cells.19 To explore the impact of CSE on PlGF expression, 16-HBE cells were stimulated by the indicated concentrations of CSE. Subsequently, the media samples and cells were collected, and the mRNA, protein and secretion of PlGF were assessed by quantitative polymerase chain reaction (qPCR), Western blot and ELISA, respectively (Figure 1A, C and E). The results from these experiments demonstrated that CSE increased PlGF expression in a dose-dependent manner. When cells were exposed to low concentration (6%) of CSE, PlGF upregulated slightly, while high concentration (12%) of CSE increased PlGF expression significantly compared to the control. A time-dependent effect of PlGF production was also observed after CSE stimulation (Figure 1B, D and F). ELISA assay showed that PlGF release significantly increased when 16-HBE cells were exposed to CSE (12%) for 8 h (Figure 1C). At this time, we examined the cell viability by CCK-8 assay, and the absorbance had no significant difference from 16-HBE cells after exposure for 8 h to 12% CSE (1.09±0.04), compared with untreated cells (1.19±0.06). However, we observed a significant decrease in absorbance at a higher concentration (15%) of CSE (0.71±0.06; Figure 1G).
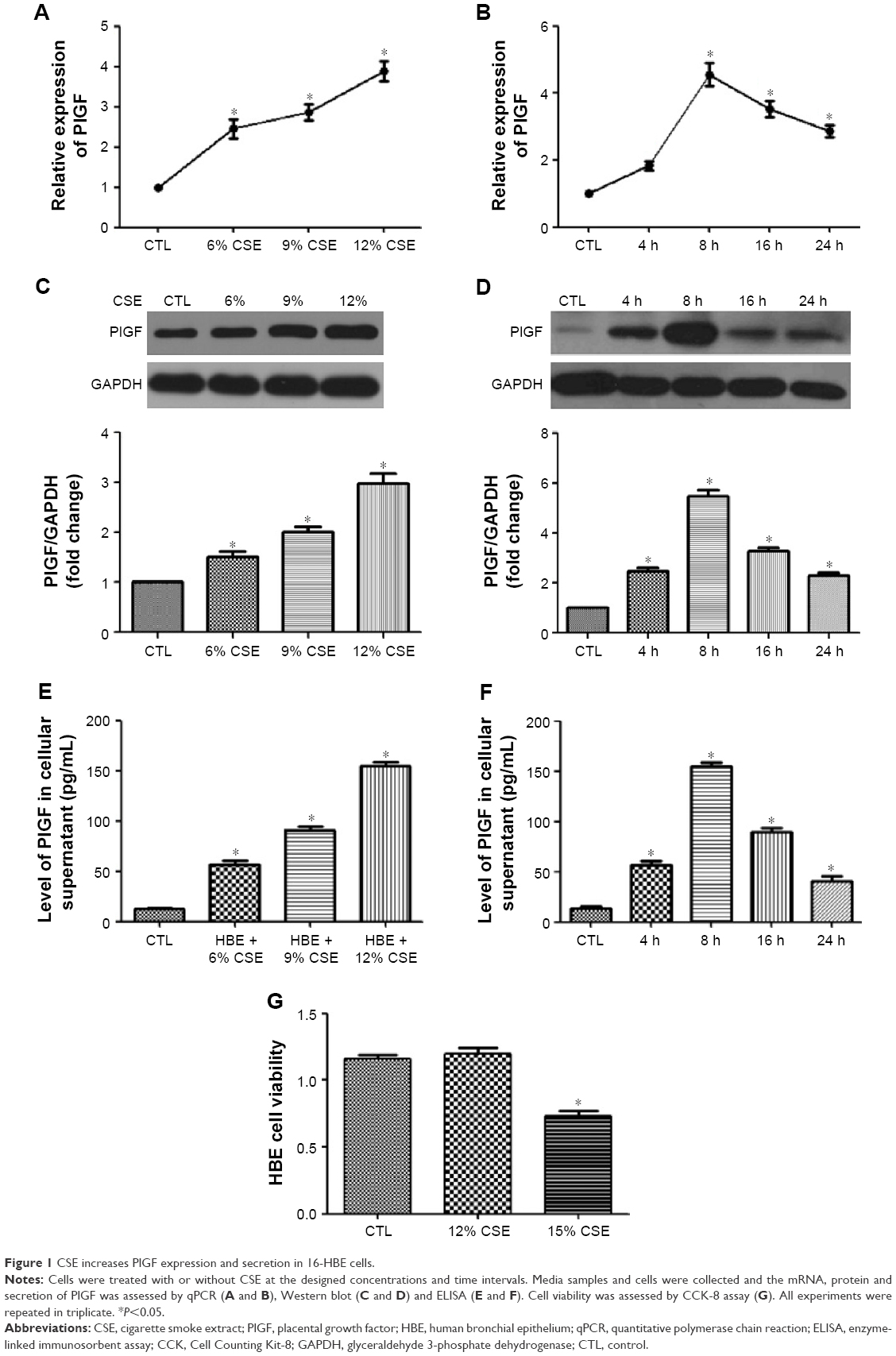 | Figure 1 CSE increases PlGF expression and secretion in 16-HBE cells. |
CSE increases Egr-1 expression, nuclear translocation and transcriptional activity in 16-HBE cells
Subconfluent, quiescent 16-HBE cells were first treated with increasing concentrations (3%–12%) of CSE. Exposure to CSE dose dependently increased Egr-1 mRNA and total protein levels (Figure 2A and C). Time-course analysis demonstrated that 12% CSE increases Egr-1 expression on both mRNA and protein levels (Figure 2B and D) within 4 h, which gradually declined to baseline after 8 h. Consistent with this, nuclear localization of Egr-1 was detectable within 15 min (Figure 1E) in CSE-treated cells. These results demonstrate that CSE induces Egr-1 upregulation and increases its nuclear translocation. At the same time, we measured the PlGF promoter binding activity of Egr-1 using the nuclear extracts prepared from CSE-treated 16-HBE cells. Compared with untreated control 16-HBE cells, CSE significantly increases Egr-1 binding to its consensus oligonucleotide sequence in a dose-dependent manner (Figure 2F).
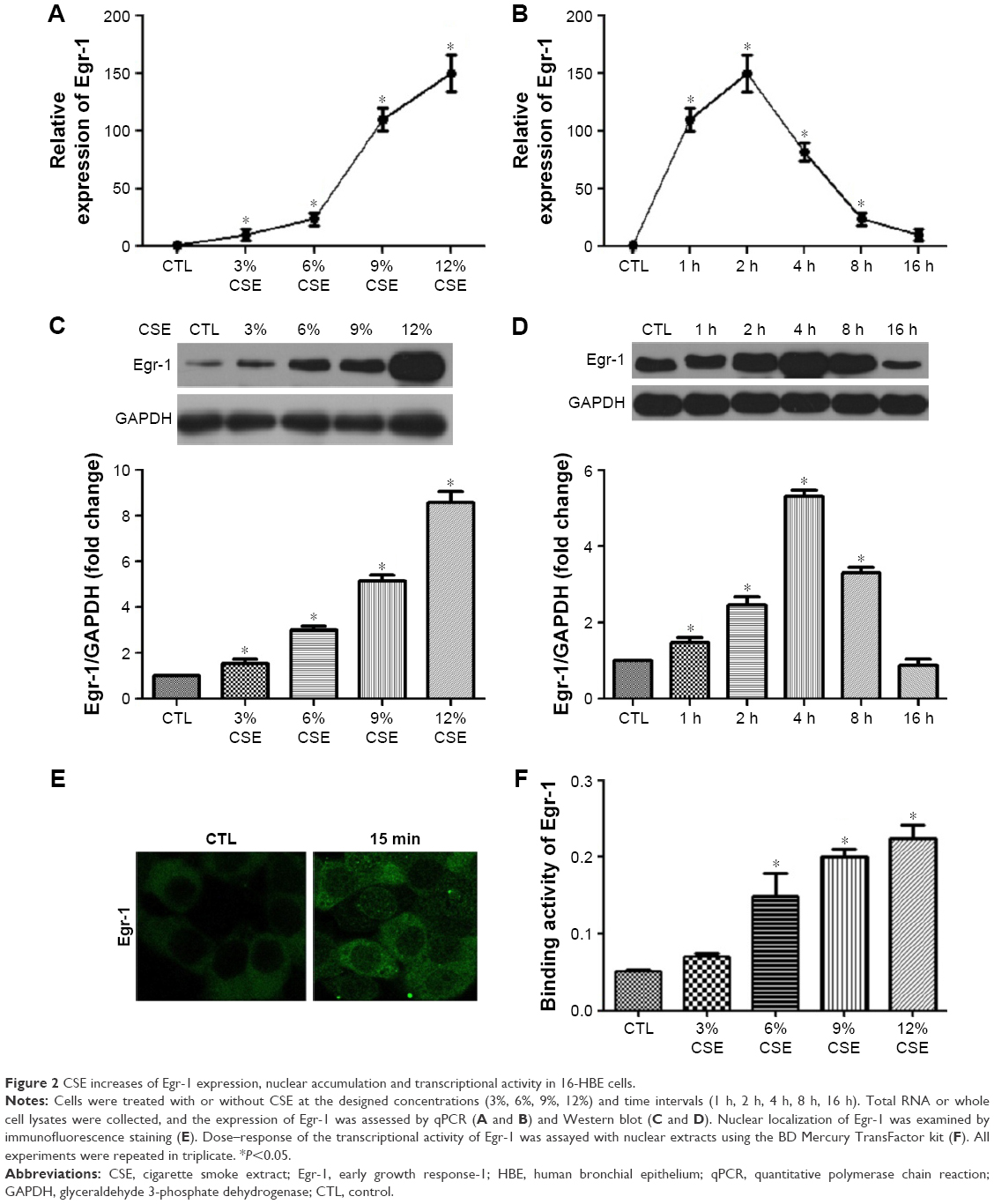 | Figure 2 CSE increases of Egr-1 expression, nuclear accumulation and transcriptional activity in 16-HBE cells. |
Egr-1 siRNA inhibits CS-stimulated PlGF production
Next, we use two independent small-interfering RNAs (Egr-1 siRNAs) to examine the role of Egr-1 in CS-mediated PlGF induction in 16-HBE cells. In our findings, Egr-1 siRNAs significantly silence Egr-1 expression at the mRNA level (Figure 3A) and protein level (Figure 3B) in 16-HBE cells, compared with scRNA, in response to the 12% CSE treatment. Furthermore, upon exposure to 12% CSE, 16-HBE cells transfected with Egr-1 siRNA exhibited reduced PlGF mRNA (Figure 3C) and protein (Figure 3D) expression. The maximum (60%) decline in PlGF levels was detected in 16-HBE cells transfected with 150 nM Egr-1 siRNA-2 (Figure 3C and D). Importantly, the inhibitory effect of scRNA was not been observed on CSE-induced PlGF expression.
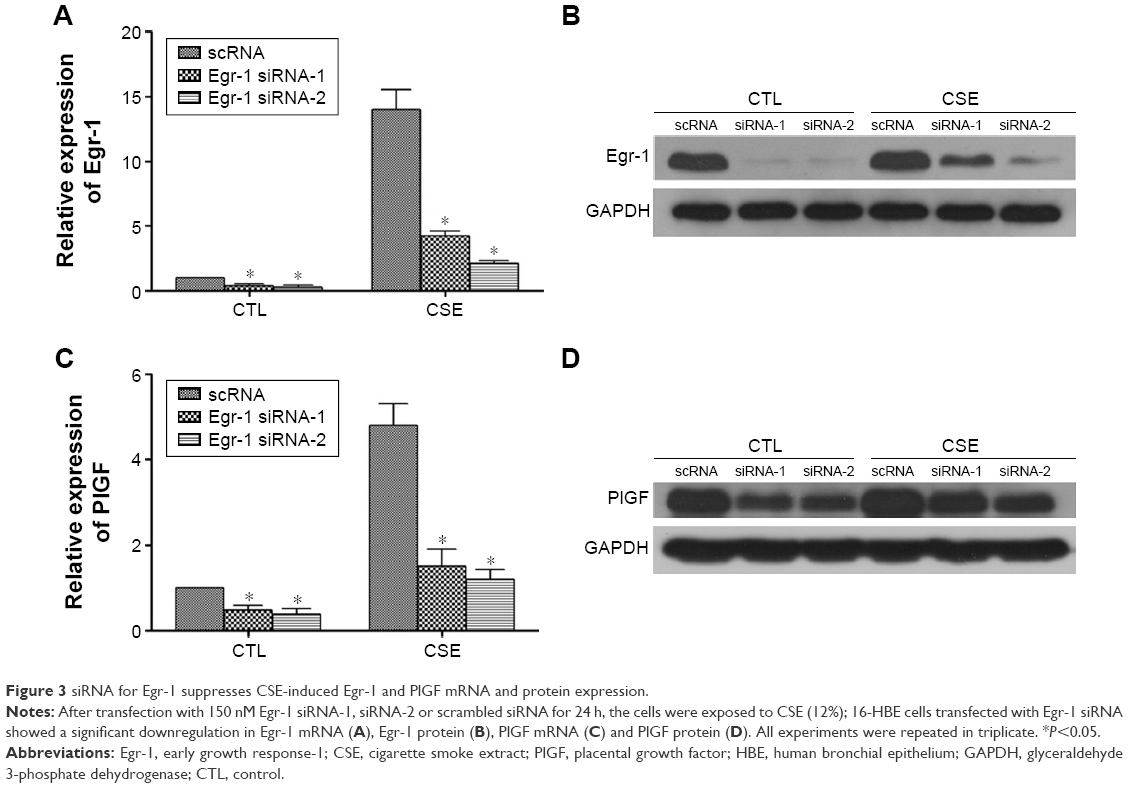 | Figure 3 siRNA for Egr-1 suppresses CSE-induced Egr-1 and PlGF mRNA and protein expression. |
ERK-1/2 contributes to CSE-induced Egr-1 and PlGF expression
MAPKs, such as ERK-1/2, are involved in growth and survival and can be activated by growth factors or oxidative stress.17,20 Therefore, the impact of CSE on phosphorylation of this protein was first measured. CSE rapidly induced phosphorylation of ERK-1/2 within 5 min, which declined to basal levels by 120 min (Figure 4A). Nucleus accumulation of phospho-ERK-1/2 was found within 5 min and sustained up to 120 min later (Figure 4B and C). U0126, an inhibitor of the ERK-1/2, inhibited not only CSE-induced Egr-1 upregulation but also PlGF expression (Figure 4D). Collectively, these data demonstrate that ERK-1/2 contributes to CSE-induced Egr-1 and PlGF expression.
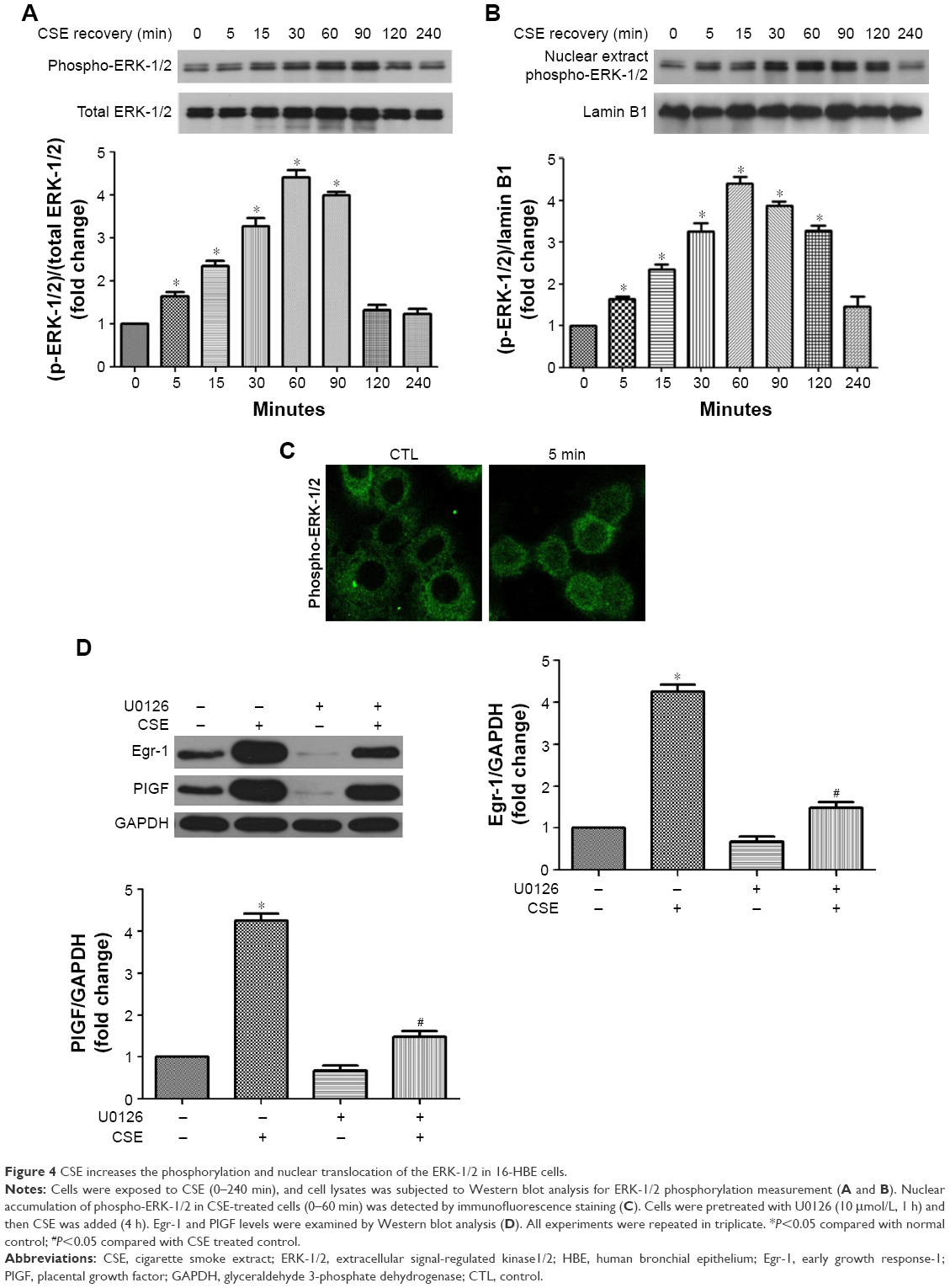 | Figure 4 CSE increases the phosphorylation and nuclear translocation of the ERK-1/2 in 16-HBE cells. |
CSE-induced cellular ROS causes ERK-1/2 activation and Egr-1 upregulation
CSE has been reported to be a potent inducer of oxidative stress.3 Therefore, a CM-H2DCFDA assay was used to examine the impact of CSE on cellular ROS contents in 16-HBE cells. CM-H2DCFDA is non-fluorescent, but in the presence of ROS, when this reagent is oxidized, it becomes green fluorescent and could act as an indicator of oxidative stress levels. When CSE was added to cells, the DCF fluorescence surged immediately and continued linearly for at least 1 h (Figure 5A). At 60 min, the DCF fluorescence induced by CSE had increased ~12-fold over baseline, whereas the fluorescence intensity was only 6.5-fold for cells treated with a known inducer of oxidative stress, H2O2 (100 μmol/L).21 Similar results were obtained from an SOD assay. Pretreatment of cells with the antioxidant NAC significantly reduced the CSE-induced increase in DCF fluorescence (Figure 5A). NAC also partly inhibited CSE-induced ERK-1/2 phosphorylation as well as the induction of Egr-1 and PlGF mRNA (Figure 5B and C). Taken together, our results demonstrate that CSE induces a surge in cellular ROS levels, and the ensuing oxidative stress increases Egr-1 and PlGF expression via activation of ERK-1/2.
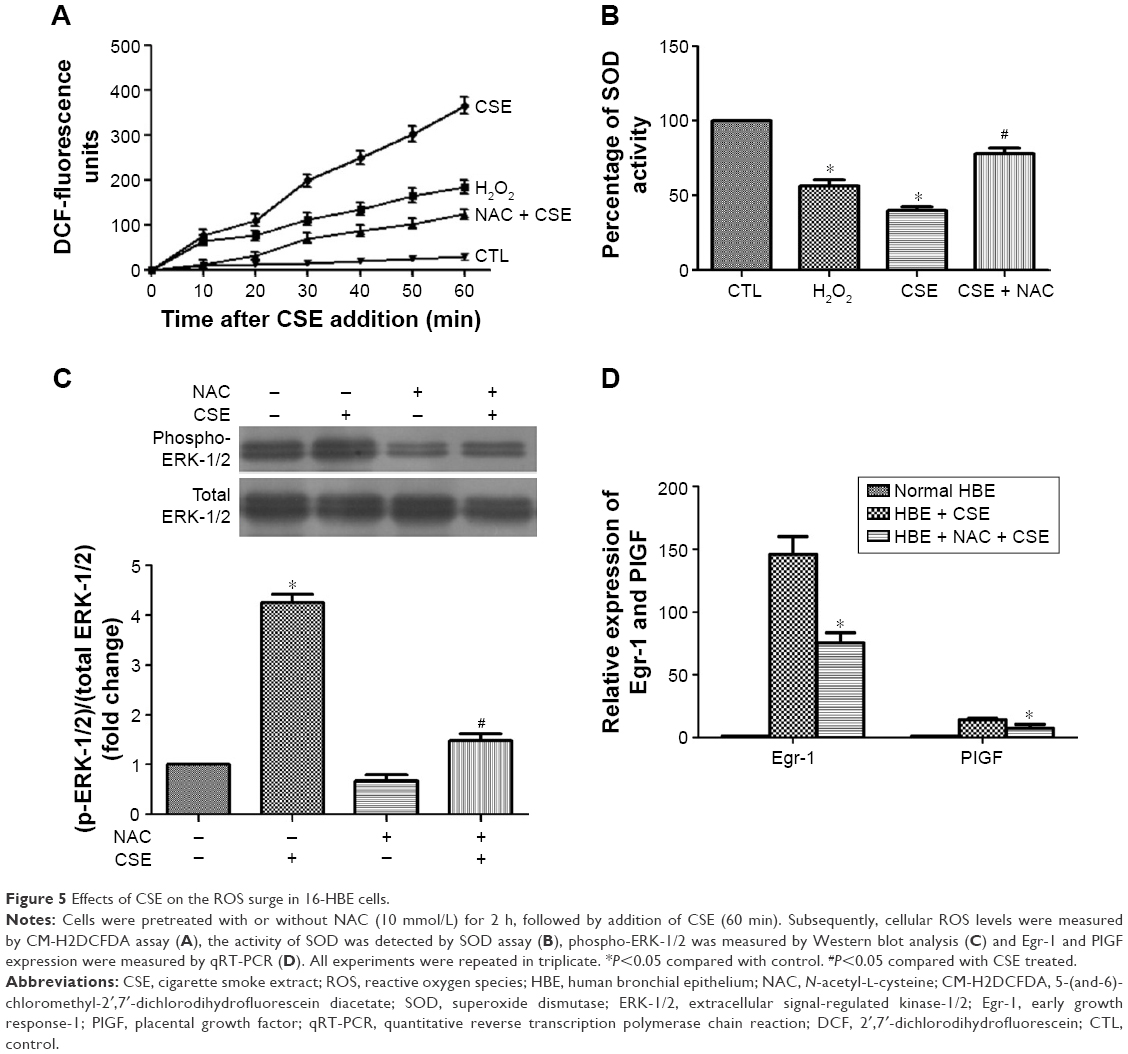 | Figure 5 Effects of CSE on the ROS surge in 16-HBE cells. |
Discussion
Our study is the first to demonstrate that ROS-dependent activation of the MAPK (ERK-1/2)/Egr-1 pathway positively regulates CSE-induced PlGF production and secretion in airway epithelia. We demonstrated that CSE specifically induced Egr-1 protein activation and nuclear translocation in 16-HBE cells and that by specifically inhibiting Egr-1, using siRNA or ERK, with the chemical inhibitor U0126, we could significantly inhibit CSE-induced PlGF mRNA and protein production. We also showed that the antioxidant NAC could partly abolish the ERK-1/2 and Egr-1 mediated activation of CSE-induced PlGF production.
CS contains >4,000 different chemical components, including oxidant compounds, and it is the main etiological factor contributing to COPD.22 Inhalation of CS is thought to lead to widespread oxidative damage that is capable of extending through the airways and lung parenchyma.7,23 Correlative studies have demonstrated that the expression of both VEGF and its receptor is decreased in lung tissue of patients with COPD.24 Moreover, CS is known to disrupt components of VEGF and its receptor VEGFR2/FLK1, which results in the expression levels of VEGF and VEGFR2/FLK1 decrease significantly in the lungs of rats and human beings.25
In this article, we investigated the role of PlGF in cigarette-related disease development. Previously, PlGF was implicated in tissue ischemia, inflammation, malignancy, eclampsia and multiple other diseases,26 but its role in cigarette-related diseases is not well known. For example, one study found that PlGF levels significantly elevate in serum and BALF of COPD patients and negatively relate with forced expiratory volume in 1 sec.13 In a separate study from the same group, it was discussed that the neutrophil’s elastic protease (pig pancreas) could increase the expression of PlGF and induce lung epithelial cell apoptosis.27 However, neither study addressed whether CS could directly stimulate PlGF overproduction in human airway epithelial cells. In our findings, CSE directly increased PlGF (both mRNA and protein levels) expression and secretion from bronchial epithelial cells in a dose- and time-dependent manner. This observation is consistent with a report from Li et al28 that CS induced oxidative stress, autophagy, apoptosis and emphysema-related PlGF in the rats. Our data suggest that overproduction of PlGF induced by CS may play a potential role in the development of cigarette smoking-related pulmonary diseases.
Egr-1 is a zinc finger containing transcription factor and is known as an immediate early response protein that is rapidly induced in multiple cells by a broad spectrum of extracellular stimuli and environmental signals.29 Once activated, Egr-1 will translocate to the nucleus and activate the expression of its downstream genes, which participate in all sorts of biological processes, such as proliferation, differentiation, apoptosis and survival.30 Notably, Egr-1 has been found to upregulate in the lung tissues of COPD patients who were smokers.31 However, the specific role of Egr-1 in smoking-related diseases is not well illuminated. A conserved Egr-1 binding site is found near the PlGF promoters of mouse and human genomes.32–34 Hou et al27 reported that neutrophil elastase induced PlGF expression through promoting Egr-1 transcriptional activity. Our results demonstrate that CSE can induce Egr-1 expression, nuclear translocation and transcriptional activity in 16-HBE cells. Moreover, PlGF expression was significantly inhibited by Egr-1 siRNA. Our results forcefully suggested that Egr-1 is activated by CS to promote the transcription of PlGF.
It has been reported that MAPK is activated in the pathological process of chronic airway inflammatory disease, such as inflammatory factor and matrix metalloproteinases expression, mucus overproduction and airway remodeling.35 The MAPK (ERK-1/2) pathway has also been implicated in Egr-1-mediated transcriptional events that are associated with tumor angiogenesis.36 The present study reveals that CSE-induced PlGF expression is mainly mediated by the MAPK (ERK-1/2) pathway based on our following findings: first, CSE rapidly induced ERK-1/2 phosphorylation and increased its nuclear ingression (Figure 4A and B) and second, ERK-1/2 inhibitor U0126 inhibited CSE-induced Egr-1 and PlGF upregulation (Figure 4C).
CS can generate toxic ROS, and ROS-mediated oxidative stress and inflammation have been increasingly implicated in chronic airway inflammatory diseases, including COPD.37 In the present study, CSE induced intracellular ROS levels’ surge immediately, and the antioxidant NAC can inhibit this ROS surge (Figure 5A and B). Consistent with this, CSE increased ERK-1/2 phosphorylation as well as Egr-1 and PlGF expression, were also partly abolished by NAC (Figure 5C and D), indicating that ROS mediated the CSE-induced PlGF expression. In this context, our results suggest that chronic exposure to CS may destroy the intracellular balance of oxidation and antioxidation and result in upregulation of many mediators, including PlGF; increased airway inflammation; cell apoptosis and emphysema development.
Conclusion
We showed that CSE stimulates nuclear translocation and transcriptional activity of Egr-1 via ROS-dependent activation of the ERK-1/2 cascades, leading to an increase in PlGF production. Based on our findings and previous observations, we propose the following model for the molecular mechanisms underlying CSE-induced PlGF expression in 16-HBE cells (Figure 6). Collectively, these findings imply that ROS, ERK-1/2, Egr-1 and PlGF might play a potential role in amplifying airway inflammation and cigarette smoking-related diseases. Pharmacological approaches that target the ROS/MAPK (ERK-1/2)/Egr-1 system and their downstream signaling components could yield useful therapeutic targets for these diseases.
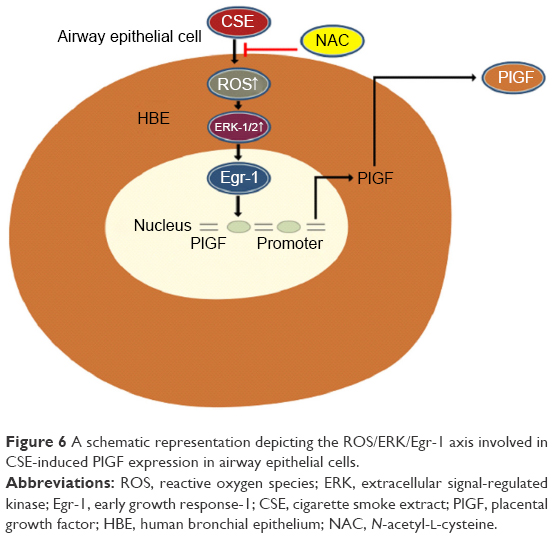 | Figure 6 A schematic representation depicting the ROS/ERK/Egr-1 axis involved in CSE-induced PlGF expression in airway epithelial cells. |
Acknowledgment
This work was supported in part by grants from the Science and Technology Project of Zhanjiang City (No 2016B01062) and the Doctoral Startup Foundation of Affiliated Hospital of Guangdong Medical University (No 20160005).
Disclosure
The authors report no conflicts of interest in this work.
References
Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
Buist AS, McBurnie MA, Vollmer WM, et al. International variation in the prevalence of COPD (the BOLD study): a population-based prevalence study. Lancet. 2007;370(9589):741–750. | ||
Csordas A, Bernhard D. The biology behind the atherothrombotic effects of cigarette smoke. Nat Rev Cardiol. 2013;10(4):219–230. | ||
Zuo L, He F, Sergakis GG, et al. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am J Physiol Lung Cell Mol Physiol. 2014;307(3):L205–L218. | ||
Caramori G, Kirkham P, Barczyk A, Di Stefano A, Adcock I. Molecular pathogenesis of cigarette smoking–induced stable COPD. Ann N Y Acad Sci. 2015;1340(1):55–64. | ||
Hoffmann RF, Zarrintan S, Brandenburg SM, et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res. 2013;14(1):1–13. | ||
Rahman I, MacNee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med. 1996;21(5):669–681. | ||
Holmes DI, Zachary I. The vascular endothelial growth factor (VEGF) family: angiogenic factors in health and disease. Genome Biol. 2005;6(2):1–10. | ||
Luttun A, Brusselmans K, Fukao H, et al. Loss of placental growth factor protects mice against vascular permeability in pathological conditions. Biochem Biophys Res Commun. 2002;295(2):428–434. | ||
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. | ||
Oura H, Bertoncini J, Velasco P, Brown LF, Carmeliet P, Detmar M. A critical role of placental growth factor in the induction of inflammation and edema formation. Blood. 2003;101(2):560–567. | ||
Luttun A, Tjwa M, Moons L, et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. 2002;8(8):831–840. | ||
Cheng SL, Wang HC, Yu CJ, Yang PC. Increased expression of placenta growth factor in COPD. Thorax. 2008;63(6):500–506. | ||
Tsao PN, Su YN, Li H, et al. Overexpression of placenta growth factor contributes to the pathogenesis of pulmonary emphysema. Am J Respir Crit Care Med. 2004;169(4):505–511. | ||
Cheng SL, Wang HC, Yu CJ, et al. Prevention of elastase-induced emphysema in placenta growth factor knock-out mice. Respir Res. 2009;10(1):115. | ||
Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds PN. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2007;37(6):748–755. | ||
Yoon YJ, Kim DK, Yoon CM, et al. Egr-1 activation by cancer-derived extracellular vesicles promotes endothelial cell migration via ERK1/2 and JNK signaling pathways. PLoS One. 2014;9(12):e115170. | ||
Zhou Y, Tu C, Zhao Y, Liu H, Zhang S. Placental growth factor enhances angiogenesis in human intestinal microvascular endothelial cells via PI3K/Akt pathway: potential implications of inflammation bowel disease. Biochem Biophys Res Commun. 2016;470(4):967–974. | ||
Dewerchin M, Carmeliet P. PlGF: a multitasking cytokine with disease-restricted activity. Cold Spring Harb Perspect Med. 2012;2(8):a011056. | ||
Dhingra S, Sharma AK, Singla DK, Singal PK. p38 and ERK1/2 MAPKs mediate the interplay of TNF-α and IL-10 in regulating oxidative stress and cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007;293(6):H3524–H3531. | ||
Jin N, Hatton ND, Harrington MA, Xia X, Larsen SH, Rhoades RA. H 2 O 2-induced egr-1, fra-1, and c-jun gene expression is mediated by tyrosine kinase in aortic smooth muscle cells. Free Radic Biol Med. 2000;29(8):736–746. | ||
van der Toorn M, Rezayat D, Kauffman HF, et al. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L109–L114. | ||
Pastukh VM, Zhang L, Ruchko MV, et al. Oxidative DNA damage in lung tissue from patients with COPD is clustered in functionally significant sequences. Int J Chron Obstruct Pulmon Dis. 2011;6:209–217. | ||
Sakao S, Tatsumi K, Hashimoto T, et al. Vascular endothelial growth factor and the risk of smoking-related COPD. Chest. 2003;124(1):323–327. | ||
Marwick JA, Stevenson CS, Giddings J, et al. Cigarette smoke disrupts VEGF165-VEGFR-2 receptor signaling complex in rat lungs and patients with COPD: morphological impact of VEGFR-2 inhibition. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L897–L908. | ||
Autiero M, Luttun A, Tjwa M, Carmeliet P. Placental growth factor and its receptor, vascular endothelial growth factor receptor-1: novel targets for stimulation of ischemic tissue revascularization and inhibition of angiogenic and inflammatory disorders. J Thromb Haemost. 2003;1(7):1356–1370. | ||
Hou HH, Cheng SL, Chung KP, et al. PlGF mediates neutrophil elastase-induced airway epithelial cell apoptosis and emphysema. Respir Res. 2014;15:106. | ||
Li PC, Tsai WH, Chien CT. Dietary Monascus adlay supplements facilitate suppression of cigarette smoke-induced pulmonary endoplasmic reticulum stress, autophagy, apoptosis and emphysema-related PLGF in the rat. Food Chem. 2013;136(2):765–774. | ||
Gashler A, Sukhatme VP. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 1995;50:191–224. | ||
Thiel G, Cibelli G. Regulation of life and death by the zinc finger transcription factor Egr-1. J Cell Physiol. 2002;193(3):287–292. | ||
Ning W, Li C-J, Kaminski N, et al. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2004;101(41):14895–14900. | ||
Patel N, Kalra VK. Placenta growth factor-induced early growth response 1 (Egr-1) regulates hypoxia-inducible factor-1α (HIF-1α) in endothelial cells. J Biol Chem. 2010;285(27):20570–20579. | ||
Green CJ, Lichtlen P, Huynh NT, et al. Placenta growth factor gene expression is induced by hypoxia in fibroblasts a central role for metal transcription factor-1. Cancer Res. 2001;61(6):2696–2703. | ||
Krämer B, Meichle A, Hensel G, Charnay P, Krönke M. Characterization of an Krox-24/Egr-1-responsive element in the human tumor necrosis factor promoter. Biochim Biophys Acta. 1994;1219(2):413–421. | ||
Mercer BA, Kolesnikova N, Sonett J, D’Armiento J. Extracellular regulated kinase/mitogen activated protein kinase is up-regulated in pulmonary emphysema and mediates matrix metalloproteinase-1 induction by cigarette smoke. J Biol Chem. 2004;279(17):17690–17696. | ||
Kim JH, Choi DS, Lee OH, Oh SH, Lippman SM, Lee HY. Antiangiogenic antitumor activities of IGFBP-3 are mediated by IGF-independent suppression of Erk1/2 activation and Egr-1-mediated transcriptional events. Blood. 2011;118(9):2622–2631. | ||
Rajendrasozhan S, Yang SR, Edirisinghe I, Yao H, Adenuga D, Rahman I. Deacetylases and NF-kappaB in redox regulation of cigarette smoke-induced lung inflammation: epigenetics in pathogenesis of COPD. Antioxid Redox Signal. 2008;10(4):799–811. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.