Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Cigarette smoke exposure reduces leukemia inhibitory factor levels during respiratory syncytial viral infection
Authors Poon J ![]() , Campos M, Foronjy RF, Nath S
, Campos M, Foronjy RF, Nath S ![]() , Gupta G, Railwah C
, Gupta G, Railwah C ![]() , Dabo AJ, Baumlin N
, Dabo AJ, Baumlin N ![]() , Salathe M
, Salathe M ![]() , Geraghty P
, Geraghty P ![]()
Received 1 December 2018
Accepted for publication 12 April 2019
Published 18 June 2019 Volume 2019:14 Pages 1305—1315
DOI https://doi.org/10.2147/COPD.S196658
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Justin Poon,1 Michael Campos,2 Robert F Foronjy,1 Sridesh Nath,1 Gayatri Gupta,1 Christopher Railwah,1 Abdoulaye J Dabo,1 Nathalie Baumlin,3 Matthias Salathe,3 Patrick Geraghty1
1Division of Pulmonary & Critical Care Medicine, Department of Medicine, State University of New York Downstate Medical Center, Brooklyn, NY, USA; 2Division of Pulmonary, Critical Care, and Sleep Medicine, University of Miami, Miami, FL, USA; 3Department of Internal Medicine, University of Kansas Medical Center, Kansas City, KS, USA
Background: Viral infections are considered a major driving factor of chronic obstructive pulmonary disease (COPD) exacerbations and thus contribute to disease morbidity and mortality. Respiratory syncytial virus (RSV) is a frequently detected pathogen in the respiratory tract of COPD patients during an exacerbation. We previously demonstrated in a murine model that leukemia inhibitory factor (LIF) expression was increased in the lungs during RSV infection. Subduing LIF signaling in this model enhanced lung injury and airway hypersensitivity. In this study, we investigated lung LIF levels in COPD patient samples to determine the impact of disease status and cigarette smoke exposure on LIF expression.
Materials and methods: Bronchoalveolar lavage fluid (BALF) was obtained from healthy never smokers, smokers, and COPD patients, by written informed consent. Human bronchial epithelial (HBE) cells were isolated from healthy never smokers and COPD patients, grown at the air–liquid interface and infected with RSV or stimulated with polyinosinic:polycytidylic acid (poly (i:c)). Mice were exposed to cigarette smoke daily for 6 months and were subsequently infected with RSV. LIF expression was profiled in all samples.
Results: In human BALF, LIF protein was significantly reduced in both smokers and COPD patients compared to healthy never smokers. HBE cells isolated from COPD patients produced less LIF compared to never smokers during RSV infection or poly (i:c) stimulation. Animals exposed to cigarette smoke had reduced lung levels of LIF and its corresponding receptor, LIFR. Smoke-exposed animals had reduced LIF expression during RSV infection. Two possible factors for reduced LIF levels were increased LIF mRNA instability in COPD epithelia and proteolytic degradation of LIF protein by serine proteases.
Conclusions: Cigarette smoke is an important modulator for LIF expression in the lungs. Loss of LIF expression in COPD could contribute to a higher degree of lung injury during virus-associated exacerbations.
Keywords: chronic obstructive pulmonary disease, leukemia inhibitory factor, respiratory syncytial virus, cigarette smoke
Plain language summary
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death worldwide. Little is known about leukemia inhibitory factor (LIF), an immune response protein that has an important role in preventing lung injury, in COPD. Our group has previously demonstrated that LIF protects the lung during a respiratory syncytial virus (RSV) infection, a common virus observed in the lungs of COPD patients. This current study demonstrates that cigarette smoke inhalation significantly reducs LIF levels in the lungs. Reduced LIF levels were observed in lung samples from COPD patients and active smokers. Equally, lung cells isolated from COPD patients or mice exposed to smoke have reduced LIF responses during an RSV infection, which could contribute to the enhanced lung damage observed in COPD patients. Finally, we identify two events that could contribute to reduced LIF levels in COPD. LIF is sensitive to the hostile environment induced by cigarette smoke inhalation, with LIF RNA and protein degradating quicker in the lungs of COPD patients. In summary loss of LIF in the lungs of smokers and COPD patients could worsen lung damage, especially during viral infections, and could contribute to disease progression.
Introduction
Chronic obstructive pulmonary disease (COPD) is a leading cause of morbidity and mortality, and is considered the third leading cause of death worldwide and in the USA.1 COPD is a preventable disease with airflow limitation that is not fully reversible.2 There are multiple risk factors for disease development, but cigarette smoking is the most common etiologic factor in the developed world.3 Acute exacerbations of COPD, defined as a sudden worsening of COPD that typically lasts for several days, are major contributors to disease morbidity and mortality.4,5 Rhinovirus, influenza, and respiratory syncytial virus (RSV) are frequently detected in the respiratory tract of COPD patients.6 RSV infections are frequently reported in infants, the elderly and immunocompromised patients but also in healthy adults.7,8 Studies show that RSV is a common cause of COPD exacerbations and is associated with the onset of severe airway symptoms.9,10 RSV typically infects the airway epithelium.11 Those infected with RSV develop mild to severe cough and dyspnea.12 Wheezing and asthma symptoms are observed following severe RSV lower respiratory tract infection.13 Multiple factors are associated with lung damage in COPD and during RSV infection. We and others have identified leukemia inhibitory factor (LIF), an interleukin 6 class cytokine, as an important regulator of inflammation, with LIF expression protecting against lung injury.14–17 However, little is known about LIF in COPD.
The primary function of LIF is to preserve the totipotency of embryonic stem cells18, but higher levels of LIF are detected in several disease states, including acute respiratory distress syndrome.19 However, several studies have demonstrated that LIF protects the lung from injury during pneumonia16 and during RSV infection.17 LIF reduces cytokine expression16 and alters alveolar neutrophil numbers.14,16 Lif deficient mice have altered immune responses during an experimental autoimmune encephalomyelitis model.20 We have previously demonstrated that neutralizing LIF signaling enhanced lung damage, airway hyperresponsiveness, chemokine (C-X-C motif) ligand (CXCL)1, CC chemokine ligands (CCL)5, CXCL10, CCL3, and CCL2 in mice during an RSV infection.17 Overexpression of LIF in airway epithelial cells protects the airways during hyperoxia in mice, with improved survival and decreased pulmonary edema.15 LIF is a prominent signal transducer and activator of transcription 3 (STAT3)-activating cytokine that facilitates tissue protection during pneumonia.16 Loss of STAT3 enhances smoke-induced inflammation in mice.21 LIF regulates apoptosis, with investigators suggesting that LIF acts as a pro-apoptotic mediator22,23 while others suggesting that LIF has anti-apoptotic potential.24,25 Enhanced Fas, Fap, interleukin (Il)24, and tumor necrosis factor superfamily member (Tnfsf)15 expression is observed following LIF depletion in animals with bacterial pneumonia,16 which could contribute to changes in cell fate.
In this study, we investigated whether smoke exposure or the disease status of COPD predisposes the lung to reduced LIF expression during an RSV infection. Human bronchoalveolar lavage fluid (BALF), fully differentiated human bronchial epithelial (HBE) cells and a mouse cigarette smoke and RSV infection model were utilized to examine LIF expression following RSV infection. Our findings indicate that smoke exposure and underlining pulmonary disease modulate LIF responses, which could not only worsen symptoms during a viral infection but also contribute to worsening lung damage.
Methods
RSV culture
Human RSV strain A2 (ATCC, Manassas, VA; #VR-1540) was used to infected cells and mice throughout this study and was maintained as previously described.17 Non-infected cells were processed in the same manner as RSV infected cells and the resulting sample collection was used as a mock control.
Human samples
BALF was collected from healthy never smokers, smokers, and COPD patients free from exacerbations for 6 months (see Table 1 for demographics). Written informed consent was obtained from all study participants and was approved by the institutional review board of the University of Miami. Primary HBE cells were obtained from organ donors whose lungs were rejected for transplant (see Table 2 for demographics). Consent for research was obtained by the Life Alliance Organ Recovery Agency of the University of Miami. All consents were IRB-approved and conformed to the Declaration of Helsinki. The diagnosis of COPD was provided by the clinician taking care of the patient. Since no confirmatory lung function was available, we only classified and used cells in the COPD category if the explanted lungs showed signs of emphysema.
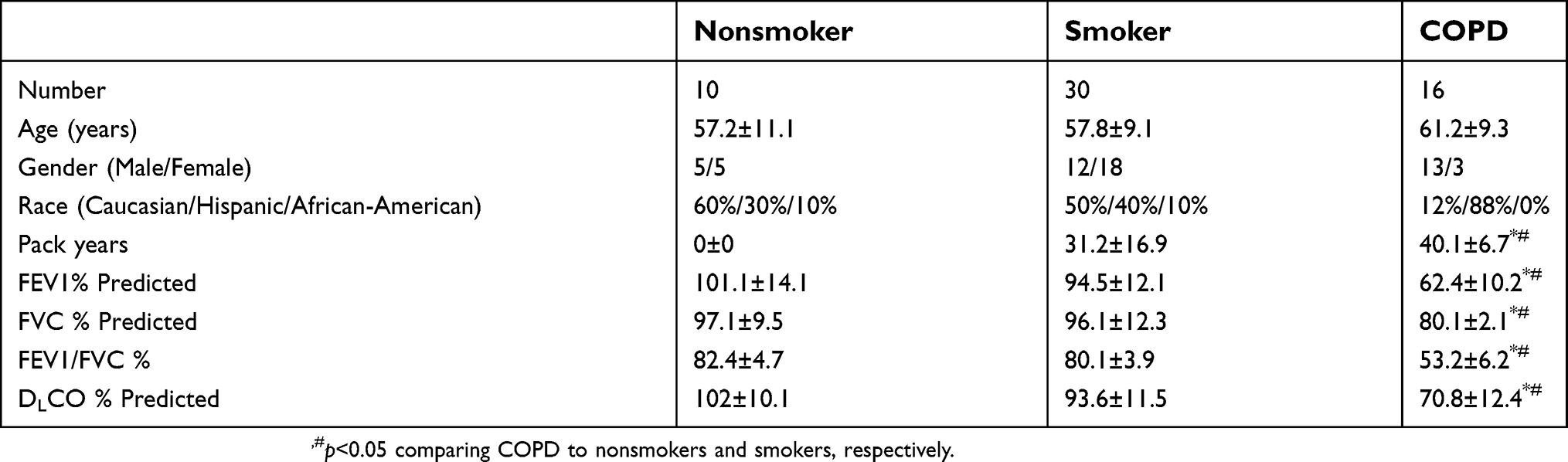 | Table 1 Patient demographics |
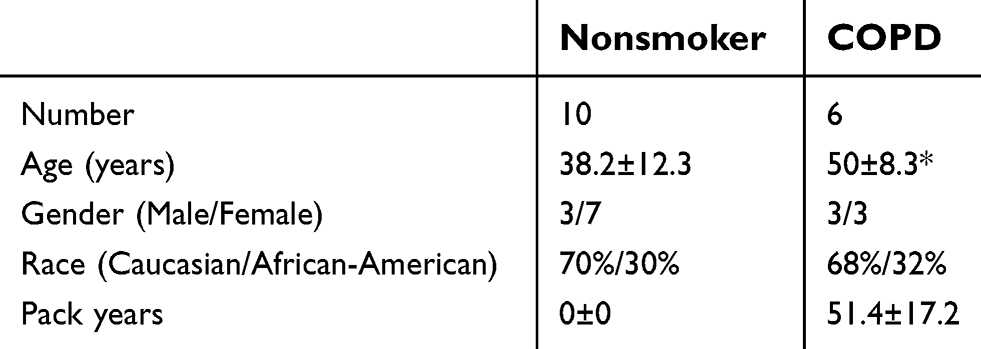 | Table 2 Patient demographics for epithelial cell donors |
Airway epithelial cells isolated from healthy, smokers and COPD lungs, were de-differentiated through expansion and re-differentiated at an air–liquid interface (ALI) on 24-mm T-clear filters (Costar Corning, Corning, NY) as previously described.26 RSV, mock, or 1 µg/mL polyinosinic:polycytidylic acid (poly (i:c)) were added to the apical surface of the cultures and incubated for 2 hrs at 37°C, 5% CO2. The multiplicity of infection for RSV was 0.3. Subsequently, the apical surface was washed five times with PBS and ALI conditions were restored. Twenty-four hours later the apical surface was rinsed with 600 μL PBS, and the rinse was harvested and investigated for LIF production. The cells were collected for RNA. Undifferentiated HBE cells were also transfected with either a scrambled control small interfering RNA (siRNA) (Qiagen, Gaithersburg, MD), human antigen R (HuR; encoded by the ELAVL1 gene), tristetraprolin (TTP; encoded by the ZFP36 gene), or Dcr-1 homolog (DICER1)-specific siRNA.
Animal model
C57BL/6J were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice were maintained in a specific pathogen-free facility at SUNY Downstate. Eight-week-old mice were used at the initiation point for all experiments. Mice were exposed to cigarette smoke in a specially designed chamber (Teague Enterprises, Davis, CA) for 3 hrs a day at a total particulate matter concentration of 80–120 mg/m3 as per our established protocol,27 using 3R4F reference cigarettes (University of Kentucky, Lexington, Kentucky, USA). Following 6 months of smoke exposure, mice were anesthetized by intraperitoneal (IP) injection of a mixture of ketamine and xylazine. Animals were intranasally administered 1×106 plaque-forming units RSV or an equal volume of mock. Animals were euthanized on day 7 post-RSV or mock administering. At the time of euthanasia, BALF isolation was performed on the mice in accordance with our previously published protocol.28 All animal experiments were performed with approval from SUNY Downstate’s Hospital’s Institutional Animal Care and Use Committee approval. This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and according to the Declaration of Helsinki conventions for the use and care of animals.
LIF detection
LIF was detected by real-time reverse transcription-PCR (RT-PCR), Luminex assays, and immunoblotting. Human LIF and mouse Lif gene expressions were performed by RT-PCR and corrected to ACTB or Actb using the following primers: human LIF forward 5ʹ-GAA GAA GCT GG CTG TCA A-3ʹ, human LIF reverse 5ʹ-ACA TCT GGA CCC AAC TCC T-3ʹ, human ACTB forward 5ʹ-GAT GAG ATT GGC ATG GCT-3ʹ, human ACTB reverse 5ʹ-CAC CTT CAC CGT TCC AGT-3ʹ, mouse Lif forward 5ʹ-TAG GAG TCA GGG AAG GAC-3ʹ, mouse Lif reverse 5ʹ-GAC AGC TGT GCT GGA TCA-3ʹ, mouse Actb forward 5ʹ-GTT GGA GCA AAC ATC CCC CA-3ʹ, mouse Actb reverse 5ʹ-CGC GAC CAT CCT CCT CTT AG-3ʹ (Life technologies/Applied Biosystems, Carlsbad, CA). Changes in LIF gene expression are presented as relative expression of LIF compared to controls and corrected to ACTB or Actb. LIF was examined in apical washes from the HBE cultures and mouse BALF using Luminex beads assays (Bio-Rad Magnetic Bio-Plex pro-human Cytokine Bead Panel and the Bio-Plex pro mouse cytokine panel) with the BioRad Bio-Plex 200 system (BioRad, Hercules, CA). Human BALF data were standardized to total BALF protein concentrations. Total protein was determined by bicinchoninic acid assays, according to the manufacturer’s instructions (Thermo Fisher). Human BALF data are now presented as picograms of LIF per microgram of total BALF protein. LIF was detection in BALF ranging from 30 to 100 pg/mL, and the limit of detection for the kit was 3.85 mg/mL. LIF data from cells and mice are presented as pg/mL of apical cell wash or BALF collected. Protein was collected from lung tissue by bead beater disruption (Minibeadbeater-16, BioSpec Products, Bartlesville, OK, USA). Tissue was placed in radio-immunoprecipitation assay buffer with 50 mg of 1 mm diameter Zirconia beads (BioSpec Products) and disrupted for 30 s in the bead beater. Soluble proteins were collected following a 10 mins centrifugation at 13,000× g at 4°C. Immunoblots were conducted to determine levels of LIF (Abcam, Cat # ab135629), LIFR (Abcam, Cat # ab101228), and β-actin (Cell Signaling Technologies, Cat #4967). All antibodies were polyclonal rabbit antibodies. Chemiluminescence detection was performed using the Bio-Rad Laboratories Molecular Imager ChemiDoc XRS+ imaging system. Densitometry was performed on each target and represented as a ratio of pixel intensity compared to β-actin, using Bio-Rad Laboratories Image Lab software (version 4.0, build 16).
Analysis of LIF mRNA degradation
The mRNA stability within HBE cells was measured indirectly by analyzing the mRNA half-life following transcription inhibition using actinomycin D, assuming changes in mRNA levels reflect mRNA degradation. HBE cells were treated with 2.5 μg/mL of actinomycin D (Sigma Aldrich) for 1 hr. RNA was extracted using Qiagen RNeasy kits according to the manufacturer’s instructions. LIF RT-PCR was then performed with ACTB used as a normalization control. Results were determined relative to time zero after actinomycin D treatment.
LIF degradation analysis
Recombinant LIF protein (250 ng; R&D Systems) was incubated with 10 μL of BALF (healthy nonsmoker or COPD) in PBS to a final volume of 20 μL for 24 hrs at 37°C. COPD BALF was also pretreated with 10 mM pefabloc for 30 mins prior to incubation with recombinant LIF. A series of concentrations of human sputum neutrophil elastase (NE) (Elastin products Inc, Owensville, Missouri) were incubated with recombinant LIF for 24 hrs at 37°C. LIF degradation was examined by immunoblotting samples and detection with an LIF antibody.
Statistical analyses
For statistical analysis, data from multiple animals or multiple separate cell experiments were pooled. Data are expressed as means ± SEM F-tests and normality testing (D’Agostino & Pearson omnibus normality test) were performed on all data sets. Differences between groups of mice were compared by two-way analysis of variance. Individual differences between groups were tested by multiple comparison and analysis using the Bonferroni post-test. Pairs of groups were compared by Student’s t-test (two-tailed). P-values for significance were set at 0.05. All analysis was performed using GraphPad Prism Software (Version 5 for Mac OS X).
Results
LIF protein is reduced in human BALF from both smokers and COPD patients
Enhanced LIF expression is observed in airway epithelial cells after RSV infection,17 and LIF is protective for lung injury.14–17 In this study, we measured LIF protein levels in human BALF fluid from age-matched healthy nonsmoker (NS) controls, smokers, and subjects with COPD with stable lung function 6 months prior to BALF collection. LIF protein was significantly reduced in both smokers and COPD patients compared to NS controls (Figure 1). There was a 31% decrease in LIF levels from the nonsmoking to the smoking group (from 2.2±0.21 to 1.5±0.13 pg/µg BALF total protein, respectively, p<0.05). Similarly, there was a 42% LIF reduction from healthy nonsmokers compared to COPD subjects (from 2.2±0.21 to 1.3±0.19 pg/µg BALF total protein, respectively, p<0.05). Therefore, cigarette smoke and the disease status play a role in suppressing LIF protein levels in human BALF samples in the absence of an infection.
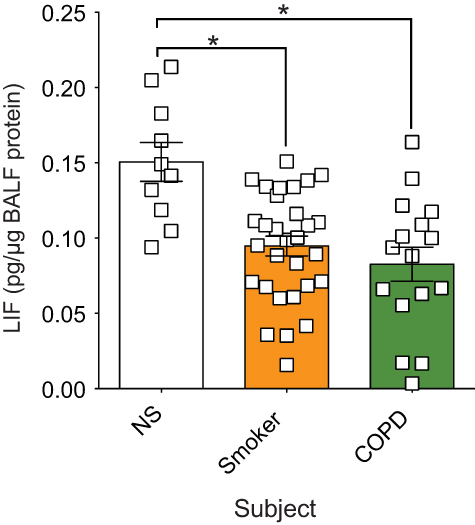 | Figure 1 In human BALF, leukemia inhibitory factor (LIF) protein is significantly reduced in both smokers and chronic obstructive pulmonary disease (COPD) patients compared to nonsmokers. LIF was measured in lung BALF from age-matched healthy control non-smoker subjects (NS; n=10), smokers (n=30), and subjects with COPD (n=16) by Luminex bead assays. LIF levels were standardized to BALF total protein concentration. Graphs are represented as mean LIF concentration (pg of LIF per µg BALF protein) ± standard error of the mean (SEM) *p<0.05 compared to nonsmoking controls. *p<0.05 comparing both groups connected by a line by Student’s t-tests and corrected with Bonferroni analysis. |
Proteolytic degradation of LIF protein in COPD BALF and increased LIF mRNA instability in COPD epithelia
To determine why LIF levels are reduced in COPD, we examine LIF protein and mRNA stability. Equal concentrations of LIF recombinant protein were incubated in the presence of BALF from either healthy nonsmokers or COPD subjects for 24 hrs; then, LIF immunoblots were performed. LIF recombinant protein underwent degradation in the presence of COPD BALF but not in the presence of BALF from nonsmokers (Figure 2A). BALF from COPD subjects was also pretreated with pefabloc, a serine protease inhibitor, prior to incubation with LIF recombinant protein. Inhibition of serine protease activity in BALF from COPD patients prevented LIF protein degradation (Figure 2A). Alternatively, incubation of LIF recombinant protein with various concentrations of NE demonstrated that NE can cleave LIF protein (Figure 2B).
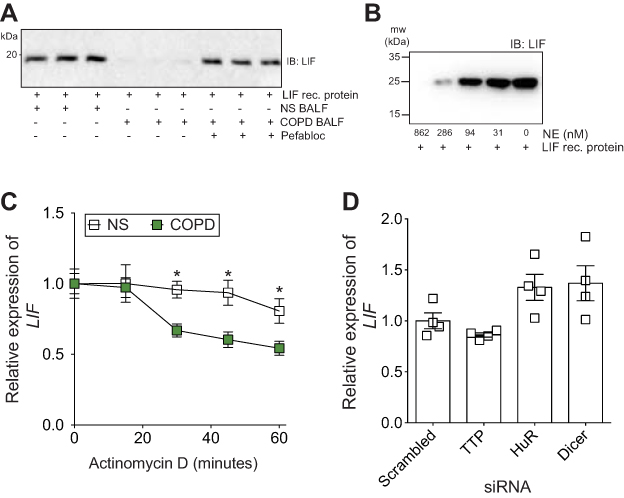 | Figure 2 Increased leukemia inhibitory factor (LIF) mRNA instability in chronic obstructive pulmonary disease (COPD) epithelia and proteolytic degradation of LIF protein. (A) Equal concentrations of LIF recombinant protein were incubated with BALF from nonsmokers or COPD patients for 24 hrs and LIF immunoblots were performed. COPD BALF was also pretreated with pefabloc before incubation with LIF recombinant protein. (B) Equal conentrations of LIF recombinant protein were inclubed with various concentrations of neutrophil elastase (NE) for 24 hours and LIF immunoblots were performed. (C) Human bronchial epithelial (HBE) cells from nonsmokers (NS) and COPD patients were treated with Actinomycin D. LIF expression was measured by qPCR. Relative gene expression of LIF was expressed as comparison to time zero of Actinomycin D treatment for NS and COPD samples. (D) Gene silencing was performed for human antigen R (HuR), tristetraprolin (TTP), and dicer in HBE cells from NS and LIF gene expression determined relative to scramble negative control transfected cells. Graph shows the mean ± standard error of the mean (SEM), with each measurement, performed three times on at least four subjects/group. *p<0.05 comparing NS to COPD at each corresponding time point by two-way analysis of variance (ANOVA) with Bonferroni post hoc analysis. |
The stability of LIF mRNA was examined in HBE cells from COPD subjects and nonsmokers, indirectly by analyzing the mRNA half-life following transcription inhibition using actinomycin D. In the presence of actinomycin D (Figure 2C), LIF mRNA degraded faster in fully differentiated HBE cells from COPD patients compared to cells from healthy nonsmokers. At 30-min post-Actinomycin D treatment, significant LIF degradation was observed in COPD epithelia compared to cells from nonsmokers with a 43% reduction in mRNA (Figure 2C). To determine whether LIF mRNA is regulated by common mRNA stabilizing or destabilizing proteins, gene silencing was performed for HuR, TTP, and dicer, and LIF expression was analyzed in HBE cells. LIF expression was unaltered by HuR, TTP, and dicer expression in HBE cells (Figure 2D).
HBE cells isolated from COPD patients produce less LIF during RSV infection
To determine whether LIF levels are lower in COPD samples during an RSV infection, we examined LIF levels in fully differentiated HBE cells that were isolated from nonsmokers and COPD subjects and subsequently infected with RSV. In both cells form nonsmokers and COPD patients, LIF gene expression was increased 24-hr post-RSV infection (Figure 3A). However, the level of LIF gene expression in COPD patients was significantly less than in nonsmokers, with a 72% difference observed (p<0.05). LIF protein secreted onto the apical surface of HBE cells was also reduced in cells isolated from COPD subjects (Figure 3A; p<0.05). Twenty-four-hours post-RSV infection, LIF secretion increased from 151.3 to 426.3 pg/mL in cells from nonsmokers. In COPD cells, LIF secretion increased from 75.3 to 180.0 pg/mL, which is 57% less compared to cells from nonsmokers. HBE cells from NS and COPD subjects were also exposed to poly (i:c), a common stimulus used to simulate viral infections, and LIF gene expression and LIF protein secretion were quantified. Similar to RSV infection, poly (i:c) stimuli induced a LIF response that was weaker in HBE cells isolated from COPD subjects (Figure 3B).
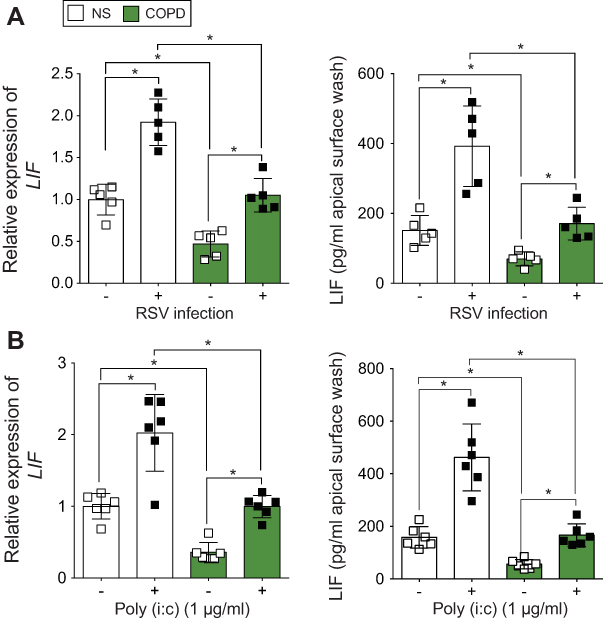 | Figure 3 Human bronchial epithelial (HBE) cells isolated from chronic obstructive pulmonary disease (COPD) patients produce less leukemia inhibitory factor (LIF) compared to cells from never smokers during respiratory synctial virus (RSV) infection. Fully differentiated HBE cells grown at the air liquid interface (ALI) from healthy and COPD individuals (n=5/group) were infected with (A) RSV or mock or treated with (B) polyinosinic:polycytidylic acid (poly (i:c)), and RNA and apical surface washes were taken 24 hrs later. (A and B) LIF gene expression was quantified by quantitative polymerase chain reaction (qPCR) and apically release LIF protein from apical surface washing with a Luminex bead assay. Graphs are represented as relative expression of LIF and LIF pg/mL of cell apical surface wash of the mean ± standard error of the mean (SEM), where each assay was performed in triplicate. *p<0.05 compared to mock treated mice by two-way analysis of variance (ANOVA) with Bonferroni post hoc analysis. |
Cigarette smoke exposure reduces LIF and LIFR levels in mouse lungs
To determine whether cigarette smoke exposure affects lung LIF levels, we examined LIF and LIFR expression in mouse lungs following smoke exposure, by performing immunoblots on total lung samples from room air and cigarette smoke-exposed mice. Lung LIF protein was lower in mice exposed to cigarette smoke (Figure 4; p<0.05). Similarly, LIFR protein was also lower in smoke-exposed mice (p<0.05). Cigarette smoke exposure reduced LIF and LIFR by 32% and 72%, respectively. Therefore, smoke exposure is an important modulator of LIF responses.
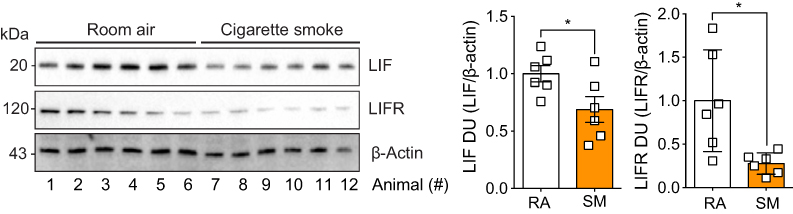 | Figure 4 Animals exposed to cigarette smoke had reduced expression of leukemia inhibitory factor (LIF) and its corresponding receptor, LIF receptor (LIFR). Animals were exposed to cigarette smoke for 6 months, immunoblots were performed on whole lung tissues proteins for LIF, LIFR, and β-actin. Densitometry analysis was performed. Graphs are represented as mean ± standard error of the mean (SEM), where each measurement was performed on six animals/group. *p<0.05 comparing both groups by Student’s t-tests. |
While COPD epithelia reveal reduced LIF responses during RSV infection, the impact of cigarette smoke exposure on LIF responses during RSV infection has yet to be determined. Relative LIF gene expression in the lung tissue and LIF protein concentration in BALF was measured in smoke-exposed mice, 7-day post-RSV infection (Figure 5). LIF expression increased 3.7-fold during an RSV infection in room air-exposed animals but only by 2.2-fold in smoke-exposed mice (p=0.02). This is a 41% reduction in LIF expression due to smoke exposure. Similarly, LIF levels in BALF post-RSV infection were reduced by 53% in the smoke-exposed group compared to the room air-exposed group (84.0±20.5 vs 177.3±20.5 pg/mL, p=0.01). Therefore, cigarette smoke subdues lung levels of LIF during RSV infection.
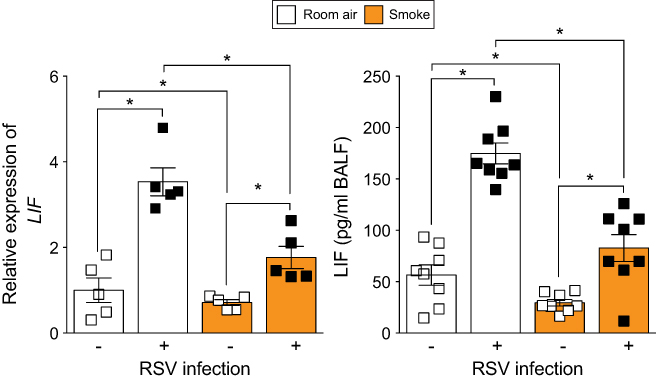 | Figure 5 Smoke-exposed animals have reduced lung leukemia inhibitory factor (LIF) expression during respiratory syncytial virus (RSV) infection. Animals were exposed to cigarette smoke for 6 months and then infected with RSV or mock. LIF expression was quantified in whole lung samples by qPCR and in BALF by Luminex bead assay 7-day post-RSV infection. Graphs are represented as mean ± standard error of the mean (SEM), where each measurement was performed on ≥5 animals/group. *p<0.05 comparing both groups connects by a line determined by analysis of variance (ANOVA) with Bonferroni post hoc analysis. |
Discussion
LIF protects the lung from injury14–17 and here, we demonstrate that smoke exposure and the disease state of COPD significantly reduces LIF baseline responses and upon infection with RSV or poly (i:c) stimulation. LIF protein is reduced in human BALF from smokers and COPD subjects compared to healthy never smokers, and the epithelia of COPD patients express less LIF, possibly due to reduced mRNA stability and increased cleavage of LIF protein by serine proteases. LIF protein is more susceptible to degradation in COPD lung microenvironment, with recombinant LIF protein undergoing degradation in the presence of COPD BALF which is prevented by serine protease inhibitors. NE can directly cleave LIF. Importantly, HBE cells from COPD subjects and mice pre-exposed to cigarette smoke produce less LIF during an RSV infection or following stimulation with poly (i:c) mimicking a viral infection. Therefore, this study demonstrates that cigarette smoke and the COPD microenvironment result in the inhibition of LIF production and enhanced LIF proteolytic degradation, which could significantly influence lung injury and repair.
In other cells and tissues, LIF expression is observed to play a role in minimizing tissue damage or aiding in replacing dead or stressed cells, with injured olfactory sensory neurons releasing LIF as a stimulus to initiate their own replacement.29 Equally, LIF promotes neurogenesis of fetal cerebrum in rats and is a critical step in brain development.30 Lower LIF and LIFR expression is also observed in pancreatic cancer and overexpression of LIFR significantly suppressed pancreatic cancer cell colony formation, migration, invasion, and wound healing ability. It is interesting that LIF counters the replication and metastasis of cancer cells but enhances the proliferation of cells involved in tissue repair.31,32 This unique feature of LIF underscores its importance in lung maintenance and suggests a potential mechanistic link between COPD and cancer, two smoking-related lung diseases. Here, we observed reduced LIF and LIFR expressions in the diseased lung, with reduced LIF responses upon RSV infection or poly (i:c) stimulation. Therefore, maintaining LIF expression could be a critical step to tissue repair and remodeling following stressful stimuli.
Several factors influence LIF expression. Interferon-gamma inhibits LIF expression33 while several other inflammatory mediators, such as IL1β,34 enhance it. LIF production is also influenced by estrogen35 and p53.36 E26 transformation-specific binding sites in the human LIF promoter are critical for its inducibility in response to T-cell activators.37 Insulin and epidermal growth factor downregulate LIFRα, which correlates with reduced cell responsiveness to LIF.38 Here we report that LIF mRNA appears to be less stable in COPD epithelia than cells from healthy never smokers. Others have reported that there are highly conserved AU-rich elements in the LIF 3ʹ UTR that are important for LIF regulation.39 Interleukin enhancer binding factor 3 (ILF3) plays a significant role in the redox-dependent Lif mRNA stabilization, with H2O2 enhancing mRNA stability of Lif in mouse Müller cells.39 ILF3 is highly expressed in samples from GOLD-2 smokers.40 But little is known about ILF3 in COPD and requires further testing. Here we examined the influence of HuR, TTP, and dicer on LIF expression in HBE cells, as all three proteins significantly influence mRNA fate especially inflammation responses. However, LIF expression was not influenced by these factors. Recently, HuR and TTP were reported to be unchanged in COPD samples compared to controls, determined by immunostaining41 but other methods are required to confirm their quantifiable expression. Ricciardi et al,41 also report that AU-rich element-binding factor 1 (AUF-1) is decreased in COPD samples. LIF mRNA also interacts with nucleolin and poly(rC)-binding protein 1 (PCBP1) at the proximal AU-rich region of its mRNA, which enhances LIF mRNA and protein levels.42 Nucleolin is regulated by HuR and miR-494 in Hela cells43 but little is known about its regulation in COPD. Interestingly, nucleolin is the receptor at the cell surface that interacts with the RSV fusion protein44 and investigating its role in COPD exacerbation would be of interest. Alternatively, PCBP1 is a negative regulator of epithelial-mesenchymal transition45, and PCBP1 plays an important role as an iron chaperone for ferritin that assists in the mineralization of ferritin.46 Disruption of iron homeostasis is a critical step in COPD pathogenesis47 but the role of PCBP1 and LIF on iron regulation in COPD is unknown. ILF3, PCBP1, nucleolin, and AUF-1 could possibly influence LIF and require investigation in COPD models and tissue samples.
We also report that LIF protein is sensitive to protein degradation in the presence of COPD BALF but not in age-matched BALF from healthy never smokers. Serine proteases appear to mediate this degradation, as the serine protease inhibitor pefabloc prevented COPD BALF degradation of LIF and NE directly degrades LIF. To our knowledge, this is the first reported evidence of LIF protein degradation in BALF. Secreted LIF protein has a short half-life due to serum protease.48 However, further studies are required to identify whether other serine proteases also cleave LIF and investigate the specific cleavage site targets for LIF protein degradation in the lungs.
LIF is a multifunctional protein with a broad range of functions in multiple tissues. Therefore, its role in COPD and in exacerbations could be multifaceted. LIF can regulate many responses due to its regulation of cell differentiation and fate. LIF binds to its specific receptor LIFR, then recruits gp130 to form a high-affinity receptor complex to induce the activation of the downstream signal pathways, including JAK/STAT3, AKT, EKR1/2, and mTOR signal pathways. LIF also can regulate signaling via influencing mRNA fate of other genes, with the removal of LIF from mouse embryonic stem cells resulting in increased decay rates of mRNA of multiple targets.49 LIF is known to regulate several signaling processes, including gene expression of IGF-binding protein 3, amphiregulin, and immune response gene-1.50 LIF could be a significant regulator of multiple signaling processes in smoke-associated diseases but further investigations are needed. It is also important to note that LIF regulates several important events during pregnancy,51 such as implantation, which could link smoke exposure to infertility if LIF inhibition by cigarette smoke is systemic.
Conclusions
This study demonstrates that lung levels of LIF are reduced in COPD patients. LIF levels are reduced by smoke exposure and the disease state, and are significantly subdued during an RSV infection or poly (i:c) stimulation. This reduction in expression could be due to LIF mRNA stability and degradation of LIF protein in the COPD airways by serine proteases. Therefore, reduction of LIF expression in the COPD airways could contribute to worsening of lung injury during viral exacerbations.
Abbreviations list
RSV, respiratory syncytial virus; BALF, bronchoalveolar lavage fluid; HBE, bronchial epithelial; LIF, leukemia inhibitory factor; ARDS, acute respiratory distress syndrome; CXCL, chemokine (C-X-C motif) ligand; CCL, CC chemokine ligands; STAT3, signal transducer and activator of transcription 3; Fap, fibroblast activation protein; IL, interleukin; Tnfs; ALI, air-liquid interface; MOI, multiplicity of infection; PFU, plaque forming units; ETS, E26 transformation-specific; AREs, AU-rich elements; ILF3, Interleukin enhancer binding factor 3; EMT, epithelial-mesenchymal transition; poly (i:c), polyinosinic:polycytidylic acid; siRNA, small interfering RNA; HuR, human antigen R; TTP, tristetraprolin; DICER1, Dcr-1 homolog; NE, neutrophil elastase; AUF-1, AU-rich element-binding factor 1.
Acknowledgments
This work was supported by grants made available to PG (Flight Attendant Medical Research Institute YCSA113380 and CIA160005 and the Alpha-1 Foundation 493373) and to MS (Flight Attendant Medical Research Institute, CIA160011and CIA13033, as well as James & Esther King Biomedical Program of the State of Florida, #5JK02).
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests.
References
1. Celli BR, Decramer M, Wedzicha JA, et al. An Official American Thoracic Society/European Respiratory Society Statement: research questions in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;191(7):e4–e27. doi:10.1164/rccm.201501-0044ST
2. Celli BR, MacNee W, Force AET. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932–946.
3. Rostron BL, Chang CM, Pechacek TF. Estimation of cigarette smoking-attributable morbidity in the United States. JAMA Intern Med. 2014;174(12):1922–1928. doi:10.1001/jamainternmed.2014.5219
4. Langsetmo L, Platt RW, Ernst P, Bourbeau J. Underreporting exacerbation of chronic obstructive pulmonary disease in a longitudinal cohort. Am J Respir Crit Care Med. 2008;177(4):396–401. doi:10.1164/rccm.200708-1290OC
5. Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57(10):847–852.
6. Sajjan US. Susceptibility to viral infections in chronic obstructive pulmonary disease: role of epithelial cells. Curr Opin Pulm Med. 2013;19(2):125–132. doi:10.1097/MCP.0b013e32835cef10
7. Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med. 2005;352(17):1749–1759. doi:10.1056/NEJMoa043951
8.
9. Falsey AR, Walsh EE, Esser MT, Shoemaker K, Yu L, Griffin MP. Respiratory syncytial virus-associated illness in adults with advanced chronic obstructive pulmonary disease and/or congestive heart failure. J Med Virol. 2019;91(1):65–71. doi:10.1002/jmv.25285
10. Kokturk N, Bozdayi G, Yilmaz S, et al. Detection of adenovirus and respiratory syncytial virus in patients with chronic obstructive pulmonary disease: exacerbation versus stable condition. Mol Med Rep. 2015;12(2):3039–3046. doi:10.3892/mmr.2015.3681
11. Guo-Parke H, Canning P, Douglas I, et al. Relative respiratory syncytial virus cytopathogenesis in upper and lower respiratory tract epithelium. Am J Respir Crit Care Med. 2013;188(7):842–851. doi:10.1164/rccm.201304-0750OC
12. Hacking D, Hull J. Respiratory syncytial virus–viral biology and the host response. J Infect. 2002;45(1):18–24.
13. Sigurs N, Gustafsson PM, Bjarnason R, et al. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med. 2005;171(2):137–141. doi:10.1164/rccm.200406-730OC
14. Ulich TR, Fann MJ, Patterson PH, et al. Intratracheal injection of LPS and cytokines. V. LPS induces expression of LIF and LIF inhibits acute inflammation. Am J Physiol. 1994;267(4 Pt 1):L442–L446. doi:10.1152/ajplung.1994.267.4.L442
15. Wang J, Chen Q, Corne J, et al. Pulmonary expression of leukemia inhibitory factor induces B cell hyperplasia and confers protection in hyperoxia. J Biol Chem. 2003;278(33):31226–31232. doi:10.1074/jbc.M301820200
16. Quinton LJ, Mizgerd JP, Hilliard KL, Jones MR, Kwon CY, Allen E. Leukemia inhibitory factor signaling is required for lung protection during pneumonia. J Immunol. 2012;188(12):6300–6308. doi:10.4049/jimmunol.1200256
17. Foronjy RF, Dabo AJ, Cummins N, Geraghty P. Leukemia inhibitory factor protects the lung during respiratory syncytial viral infection. BMC Immunol. 2014;15:41. doi:10.1186/s12865-014-0041-4
18. Williams RL, Hilton DJ, Pease S, et al. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature. 1988;336(6200):684–687. doi:10.1038/336684a0
19. Jorens PG, De Jongh R, Bossaert LL, et al. High levels of leukaemia inhibitory factor in ARDS. Cytokine. 1996;8(11):873–876. doi:10.1006/cyto.1996.9999
20. Linker RA, Kruse N, Israel S, et al. Leukemia inhibitory factor deficiency modulates the immune response and limits autoimmune demyelination: a new role for neurotrophic cytokines in neuroinflammation. J Immunol. 2008;180(4):2204–2213.
21. Geraghty P, Wyman AE, Garcia-Arcos I, Dabo AJ, Gadhvi S, Foronjy R. STAT3 modulates cigarette smoke-induced inflammation and protease expression. Front Physiol. 2013;4:267. doi:10.3389/fphys.2013.00267
22. Kamohara H, Sakamoto K, Ishiko T, Masuda Y, Abe T, Ogawa M. Leukemia inhibitory factor induces apoptosis and proliferation of human carcinoma cells through different oncogene pathways. Int J Cancer. 1997;72(4):687–695.
23. Schere-Levy C, Buggiano V, Quaglino A, et al. Leukemia inhibitory factor induces apoptosis of the mammary epithelial cells and participates in mouse mammary gland involution. Exp Cell Res. 2003;282(1):35–47.
24. Furue M, Okamoto T, Hayashi Y, et al. Leukemia inhibitory factor as an anti-apoptotic mitogen for pluripotent mouse embryonic stem cells in a serum-free medium without feeder cells. In Vitro Cell Dev Biol Anim. 2005;41(1–2):19–28. doi:10.1290/0502010.1
25. Lu Y, Fukuyama S, Yoshida R, et al. Loss of SOCS3 gene expression converts STAT3 function from anti-apoptotic to pro-apoptotic. J Biol Chem. 2006;281(48):36683–36690. doi:10.1074/jbc.M607374200
26. Nlend MC, Bookman RJ, Conner GE, Salathe M. Regulator of G-protein signaling protein 2 modulates purinergic calcium and ciliary beat frequency responses in airway epithelia. Am J Respir Cell Mol Biol. 2002;27(4):436–445. doi:10.1165/rcmb.2002-0012OC
27. Wallace AM, Hardigan A, Geraghty P, et al. Protein phosphatase 2A regulates innate immune and proteolytic responses to cigarette smoke exposure in the lung. Toxicol Sci. 2012;126(2):589–599. doi:10.1093/toxsci/kfr351
28. Foronjy RF, Mirochnitchenko O, Propokenko O, et al. Superoxide dismutase expression attenuates cigarette smoke- or elastase-generated emphysema in mice. Am J Respir Crit Care Med. 2006;173(6):623–631. doi:10.1164/rccm.200506-850OC
29. Bauer S, Rasika S, Han J, et al. Leukemia inhibitory factor is a key signal for injury-induced neurogenesis in the adult mouse olfactory epithelium. J Neurosci. 2003;23(5):1792–1803.
30. Tsukada T, Simamura E, Shimada H, et al. The suppression of maternal-fetal leukemia inhibitory factor signal relay pathway by maternal immune activation impairs brain development in mice. PLoS One. 2015;10(6):e0129011. doi:10.1371/journal.pone.0129011
31. Broholm C, Brandt C, Schultz NS, Nielsen AR, Pedersen BK, Scheele C. Deficient leukemia inhibitory factor signaling in muscle precursor cells from patients with type 2 diabetes. Am J Physiol Endocrinol Metab. 2012;303(2):E283–E292. doi:10.1152/ajpendo.00586.2011
32. Bauer S, Patterson PH. Leukemia inhibitory factor promotes neural stem cell self-renewal in the adult brain. J Neurosci. 2006;26(46):12089–12099. doi:10.1523/JNEUROSCI.3047-06.2006
33. Arici A, Engin O, Attar E, Olive DL. Modulation of leukemia inhibitory factor gene expression and protein biosynthesis in human endometrium. J Clin Endocrinol Metab. 1995;80(6):1908–1915. doi:10.1210/jcem.80.6.7775640
34. Carlson CD, Bai Y, Jonakait GM, Hart RP. Interleukin-1 beta increases leukemia inhibitory factor mRNA levels through transient stimulation of transcription rate. Glia. 1996;18(2):141–151. doi:10.1002/(SICI)1098-1136(199610)18:2<141::AID-GLIA6>3.0.CO;2-3
35. Ace CI, Okulicz WC. Differential gene regulation by estrogen and progesterone in the primate endometrium. Mol Cell Endocrinol. 1995;115(1):95–103.
36. Hu W, Feng Z, Teresky AK, Levine AJ. p53 regulates maternal reproduction through LIF. Nature. 2007;450(7170):721–724. doi:10.1038/nature05993
37. Bamberger AM, Jenatschke S, Schulte HM, Ellebrecht I, Beil FU, Bamberger CM. Regulation of the human leukemia inhibitory factor gene by ETS transcription factors. Neuroimmunomodulation. 2004;11(1):10–19. doi:10.1159/000072964
38. Blanchard F, Duplomb L, Wang Y, et al. Stimulation of leukemia inhibitory factor receptor degradation by extracellular signal-regulated kinase. J Biol Chem. 2000;275(37):28793–28801. doi:10.1074/jbc.M003986200
39. Agca C, Boldt K, Gubler A, et al. Expression of leukemia inhibitory factor in Muller glia cells is regulated by a redox-dependent mRNA stability mechanism. BMC Biol. 2015;13:30. doi:10.1186/s12915-015-0137-1
40. Ning W, Li CJ, Kaminski N, et al. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2004;101(41):14895–14900. doi:10.1073/pnas.0401168101
41. Ricciardi L, Col JD, Casolari P, et al. Differential expression of RNA-binding proteins in bronchial epithelium of stable COPD patients. Int J Chron Obstruct Pulmon Dis. 2018;13:3173–3190. doi:10.2147/COPD.S166284
42. Chakraborty A, Mukherjee S, Saha S, De S, Sengupta Bandyopadhyay S. Phorbol-12-myristate-13-acetate-mediated stabilization of leukemia inhibitory factor (lif) mRNA: involvement of Nucleolin and PCBP1. Biochem J. 2017;474(14):2349–2363. doi:10.1042/BCJ20170051
43. Tominaga K, Srikantan S, Lee EK, et al. Competitive regulation of nucleolin expression by HuR and miR-494. Mol Cell Biol. 2011;31(20):4219–4231. doi:10.1128/MCB.05955-11
44. Tayyari F, Marchant D, Moraes TJ, Duan W, Mastrangelo P, Hegele RG. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat Med. 2011;17(9):1132–1135. doi:10.1038/nm.2444
45. Liu Y, Gai L, Liu J, Cui Y, Zhang Y, Feng J. Expression of poly(C)-binding protein 1 (PCBP1) in NSCLC as a negative regulator of EMT and its clinical value. Int J Clin Exp Pathol. 2015;8(6):7165–7172.
46. Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science. 2008;320(5880):1207–1210. doi:10.1126/science.1157643
47. Zhang WZ, Butler JJ, Cloonan SM. Smoking-induced iron dysregulation in the lung. Free Radic Biol Med. 2018;133:238–247. doi:10.1016/j.freeradbiomed.2018.07.024
48. Yue X, Wu L, Hu W. The regulation of leukemia inhibitory factor. Cancer Cell Microenviron. 2015;2:3.
49. Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MS. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res. 2009;16(1):45–58. doi:10.1093/dnares/dsn030
50. Sherwin JR, Freeman TC, Stephens RJ, et al. Identification of genes regulated by leukemia-inhibitory factor in the mouse uterus at the time of implantation. Mol Endocrinol. 2004;18(9):2185–2195. doi:10.1210/me.2004-0110
51. Bhatt H, Brunet LJ, Stewart CL. Uterine expression of leukemia inhibitory factor coincides with the onset of blastocyst implantation. Proc Natl Acad Sci U S A. 1991;88(24):11408–11412.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.