")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Chrysin suppresses proliferation, migration, and invasion in glioblastoma cell lines via mediating the ERK/Nrf2 signaling pathway
Authors Wang J, Wang HD , Sun KJ , Wang XL, Pan H, Zhu JH, Ji XJ , Li X
Received 16 December 2017
Accepted for publication 16 February 2018
Published 3 April 2018 Volume 2018:12 Pages 721—733
DOI https://doi.org/10.2147/DDDT.S160020
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Juan Wang, Handong Wang, Kangjian Sun, Xiaoliang Wang, Hao Pan, Jianhong Zhu, Xiangjun Ji, Xiang Li
Department of Neurosurgery, Jinling Hospital, Medical School of Nanjing University, Nanjing, People’s Republic of China
Background: Chrysin, an active natural bioflavonoid, has been proven to protect against carcinogenesis. However, the role of chrysin in glioblastoma and the potential molecular mechanisms remain to be elucidated. In our previous study, we found that nuclear factor erythroid 2 (NF-E2)-related factor 2 (Nrf2) is highly expressed in a variety of glioblastoma cell lines associated with the mitogen-activated protein kinase (MAPK) pathway. The aim of this study was to evaluate the antitumor effects of chrysin in glioblastoma cells and how chrysin is related to the MAPK/Nrf2 signaling pathway.
Methods: A Cell Counting Kit-8 assay and a plate colony formation assay were performed to evaluate cell proliferation. Cell migration ability was tested by a wound-healing assay. Transwell migration and Matrigel invasion assay were used to test the migration and invasion potential of cells. Nrf2 was knocked down by shRNA transfection. Protein expression was determined by Western blotting and immunofluorescence staining. The in vivo anticancer effect was measured using tumor xenografts in nude mice.
Results: Chrysin inhibited the proliferation, migration, and invasion capacity of glioblastoma cells in dose- and time-dependent manners. Mechanistically, chrysin deactivated the Nrf2 signaling pathway by decreasing the translocation of Nrf2 into the nucleus and suppressing the expression of hemeoxygenase-1 (HO-1) and NAD(P)H quinine oxidoreductase-1, meanwhile, Nrf2 shRNA attenuated the anticancer activity of chrysin. Furthermore, chrysin downregulated the protein expression of p-extracellular signal-regulated kinase 1 and 2 (ERK1/2), but did not significantly affect p-JNK and p-P38 expression levels. However, the downregulated level of Nrf2 and the antitumor effect of chrysin in glioblastoma cell lines were partially abrogated by the ERK1/2 signaling inhibitor (U0126). Finally, chrysin inhibited tumor growth in U87 xenografts.
Conclusion: Our results show that chrysin exerts anticancer activity in glioblastoma cell lines possibly via the ERK/Nrf2 signaling pathway and indicate the potential application of chrysin as a natural sensitizer in chemotherapy.
Keywords: chrysin, glioblastoma, nuclear factor erythroid 2-related factor 2, Nrf2, extracellular signal-regulated kinase, ERK
Introduction
Glioma is the most common form of primary central nervous system tumors in adults. According to the classification of the World Health Organization (WHO), glioblastoma multiforme (GBM), described as grade IV glioma, is the most frequent and malignant histological subtype.1 Standard treatments for GBM for disease-free survival in randomized studies include reasonable surgical resection, radiotherapy, and chemotherapy with temozolomide.2 Despite decades of efforts and advances in therapeutics, the poor prognosis of patients with GBM has not improved, with a static median survival of ~15 months.3 Accordingly, new therapeutic strategies that can effectively suppress GBM are urgently required.
Chrysin (5,7-dihydroxyflavone) is a natural flavone found in many plant extracts, such as honey, propolis, and blue passion flowers (Passiflora caerulea, Passiflora incarnata, and Oroxylum indicum).4 Recently, several in vivo and in vitro studies have shown that chrysin suppresses the development and progression of cancer cells originating from prostate, skin, colon, breast, liver, and lung through selective mediating of multiple cell signaling pathways.5
Nuclear factor erythroid 2 (NF-E2)-related factor 2 (Nrf2), a transcription factor belonging to the Cap’n’Collar subfamily of leucine-zipper (b-ZIP) proteins, modulates cell responses to oxidative stress and physical/chemical insults through interaction with the antioxidant response element (ARE).6,7 The activity of Nrf2 is mainly regulated by its interaction with Kelch-like erythroid cell-derived protein with cap’n’collar homology-associated protein 1 (Keap1), various protein kinases, and epigenetic factors.8,9 Nrf2 target genes are mainly cytoprotective genes that can encode antioxidant enzymes and Phase II detoxifying enzymes, such as hemeoxygenase-1 (HO-1) and NAD(P)H quinine oxidoreductase 1 (NQO-1).10 Over the past few years, the dual roles of Nrf2 signaling in cancer onset and progression have become the subject of widespread interest and investigation.11 There are strong opinions that increased nuclear accumulation of Nrf2 in cancer cells prevents oxidative stress and inhibits chemotherapeutic agents and radiation, thus creating an advantageous environment for cell growth.12
Mitogen-activated protein kinases (MAPKs) are able to transduce signals from hormones, growth factors, cytokines, and environmental stresses and then elicit a wide range of cellular responses, such as cell growth, differentiation, survival, neuronal function, and immune responses.13,14 In addition, MAPKs take part in the concerted modulation of Nrf2 in inflammation and carcinogenesis.15 There are three major subfamilies of MAPKs: extracellular signal-regulated kinase 1 and 2 (ERK1/2), Jun N-terminal kinases (JNKs), and P38 MAPKs. In addition, a number of discussions highlight that chrysin is a promising dietary agent that can negatively regulate the Nrf2/ARE and the MAPK signaling pathway in cancer development, progression, and chemoresistance.4,5,16
Unfortunately, regarding the human glioblastoma multiform model, thus far no reports have elucidated the role of chrysin on Nrf2 and MAPKs. In our study, we aimed to investigate the anticancer effect of chrysin in human glioblastoma cells in vivo and in vitro and to determine how the chemical compound represses the Nrf2/ARE and MAPK signaling pathway.
Materials and methods
Reagents and antibodies
Chrysin and puromycin were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). ERK1/2 inhibitor (U0126), antibody against ERK1/2, p-ERK 1/2, p38, p-P38, JNK, p-JNK, β-actin, and goat anti-rabbit, anti-mouse immunoglobulin G (IgG) (H&L) secondary antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Nrf2, HO-1, NQO-1, and Keap1 antibody were from Abcam (Cambridge, MA, USA). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG and FITC-conjugated anti-rabbit IgG antibody were from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA).
Cell culture
T98, U251, U87 derived from human glioblastomas were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, People’s Republic of China). Normal human astrocyte (NHA) primary cultures were obtained from the Institute of Basic Medical Sciences (Beijing, People’s Republic of China) and maintained in their patented medium. The cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific) and 100 units/mL penicillin/streptomycin (HyClone, GE Healthcare Life Sciences, Logan, UT, USA) at 37°C in a humidified atmosphere of 95% air and 5% CO2. The medium was changed at 48–72 hour intervals. For each passage, cells were washed twice with phosphate-buffered saline (PBS; HyClone) and then incubated at 37°C with 0.25% trypsin-EDTA (Sigma-Aldrich Co.). In experimenting, monolayer cells were grown to ~70% confluence in culture medium before drug treatments.
Cell proliferation assay
Cell proliferation assay was performed using a Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan). According to the manufacturer’s protocol, NHAs and glioblastoma cells (T98, U251, U87) were seeded into 96-well plates at 5×103 cells in a total volume of 100 μL per well and cultivated for 24 hours to allow cell adherence. Subsequently, chrysin was dissolved in 1% DMSO and diluted in DMEM to final concentrations of 15, 30, 60, 120, and 240 μM. After separate treatment for 24, 48, and 72 hours, 10 μL CCK-8 solution was added into each well and cells were incubated for 2 hours at 37°C. Then, the 96-well plates were placed in an ELISA reader (Bio-Rad Laboratories Inc., Hercules, CA, USA), and the absorbance (value) at 450 nm wave length (OD450) was measured. Wells without cells but with medium were used as blank values and subtracted from all values. Experiments for each concentration were carried out four times. The proliferation rate of the cells was calculated by the following formula: cell viability = (the OD values of treated groups/the OD values of control group) × 100%.
Colony-forming assay
T98, U251, and U87 glioblastoma cells were seeded at a density of 1,000 cells/well into six-well plates and treated with chrysin (0, 30, 60, and 120 μM). The medium and treatment were changed every 3 days. After incubation at 37°C for 10 days, the colonies were washed with PBS, fixed with methanol for 20 minutes, stained with 0.1% crystal violet (Sigma-Aldrich Co) and visualized under a phase-contrast light microscope (Carl Zeiss Meditec AG, Jena, Germany).
Wound-healing assay
Migration of treated and untreated glioblastoma cells (U251 and U87) were assessed by a classical in vitro wound-healing assay. Monolayer cells were grown in normal growth media to 70% confluence in six-well tissue culture plates, followed by scratching with a 200 μL standard pipette tip through the culture dish. After washing three times with PBS to remove detached cells for scoring, cells were treated with chrysin (0, 10, 20, and 30 μM) in serum-free medium. At time intervals of 0, 12, 24, and 48 hours during treatment, cells migrating into the wound areas were observed and imaged using a microscope (Carl Zeiss Meditec AG, Jena, Germany). The acquired phase-contrast images were analyzed using GraphPad Prism 5 software (version 1.46; GraphPad Software, Inc., La Jolla, CA, USA).
Migration and invasion assay
Transwell assays were used to test the motility and invasion capacity of glioblastoma cells (U251 and U87) after the treatment with different concentrations of chrysin (0, 10, 20, and 30 μM). Briefly, Matrigel matrix (Corning Incorporated, Corning, NY, USA) was mixed with serum-free medium and added to the upper chamber of 6.5-mm transwells with 8.0-μm pore polycarbonate membrane inserts (Corning Incorporated) in a 37°C incubator for 30 minutes to form a thin gel layer. A total of 1×105 cells in 200 μL DMEM were placed on the upper chamber of a transwell insert; 600 μL of DMEM with 10% FBS was used as a chemoattractant in the bottom of the lower chamber. After incubation at 37°C in 5% CO2 for 24 hours, the cells on the interior of the upper chamber were removed carefully by a cotton swab; however, the cells adhered to the lower membrane surface were fixed by methanol and stained with 0.1% crystal violet. The migrating cells were reviewed, and four randomly selected ×200 microscopic fields were viewed and imaged under an inverted microscope.
shRNA interference and construction of Nrf2 knockdown cells
Nrf2 shRNA (Nrf2i) (target sequence, 5′GCA GTTCAATGAAGCTCAACT3′), the scramble shRNA (Sc, 5′UUCUCCGAACGUGUCACGUAA3′), and the polybrene solution were purchased from Hanbio Biotechnology (Shanghai, People’s Republic of China). Glioblastoma cells (U87) were seeded into six-well plates, polybrene was used according to the manufacturer’s protocol, and the cells were transfected with lentiviral particles containing either Nrf2i or Sc shRNA expression plasmid. After 24 hours, the medium containing virus was replaced with fresh complete medium. After an additional 48 hours of incubating, 2.0 μg/mL puromycin was added to the complete medium to obtain the stably transfected cells. Western blot verified the knockdown efficiency.
Immunofluorescence
Glioblastoma cells were pretreated on sterile glass cover slips in 12-well plates upon reaching ~40% confluence. The treated cells were fixed with 4% paraformaldehyde for 20 minutes, incubated with 0.3% Triton X-100 for 10 minutes, blocked with bovine serum albumin for 1 hour at room temperature and exposed to a primary anti-Nrf2 antibody at 4°C overnight. The next day, the cells were washed three times with PBS containing 0.1% Tween (PBST). Furthermore, the cover slips were incubated with the appropriate red-conjugated secondary antibody for 2 hours in a dark chamber at room temperature. Subsequently, the slides were washed three times with PBST, incubated with 4′,6′-diamidino-2-phenylindole (DAPI) at room temperature for 5 minutes and mounted with aqueous mounting medium. All images were obtained by ZEISS fluorescence microscope and analyzed using ImageJ software.
Western blot analysis
After different treatments, cells were harvested and lysed in Radio Immunoprecipitation Assay (Beyotime, Jiangsu, People’s Republic of China) buffer supplemented with 1 mM phenylmethylsulfonyl fluoride (Beyotime) to obtain whole protein lysate. The concentration of freshly extracted protein was determined by the BCA Protein Assay Kit (Beyotime). Equal amounts of protein from each sample were separated by electrophoresis on an 8%–12% SDS-PAGE gel and transferred to polyvinylidene fluoride (EMD Millipore, Billerica, MA, USA) membranes. Membranes were blocked with 5% nonfat dry milk powder in Tris-buffered saline containing 0.1% Tween (TBST) for 2 hours at room temperature and incubated with the indicated primary antibodies overnight at 4°C. After washing with TBST, membranes were incubated with the corresponding secondary HRP-conjugated antibodies. Finally, with an enhanced chemiluminescent HRP substrate (EMD Millipore), protein bands were visualized by chemiluminescence imaging system (Tanon 5200; Tanon, Shanghai, People’s Republic of China).
Tumor xenografts study
All studies involving mice were approved by the Animal Care and Use Committee of Nanjing University and conformed to Guide for the Care and Use of Laboratory Animals from National Institutes of Health. U87 cells (5×106) resuspended with 100 μL of PBS were injected subcutaneously in the right flank of male BALB/c athymic nude mice (Charles River Breeding Laboratories, Wilmington, MA, USA) at 4–6 weeks of age. The tumors were monitored every 3 days using the vernier caliper and tumor volume (V) was calculated by the following formula: V (mm3) = (major axis) × (minor axis)2 × 0.5236.17 The mice bearing an U87-derived tumor 70–100 mm3 in size were randomly divided into three groups. Two groups of nude mice were treated with chrysin (40 and 80 mg/kg/day) by oral gavage, and the other group underwent gastric lavage with refined olive oil as the control group once a day, 5 times per week because of its insoluble nature.18 Body weight was used to monitor the health of animals. At the end of the experiments, the tumors were removed, weighed, and photographed.
Statistical analysis
Data in at least three independent experiments were analyzed using SPSS software version 23.0 (IBM Corporation, Armonk, NY, USA) and presented as means ± standard deviation. Statistical differences between control and treated groups were assessed by unpaired Student’s t-test or one-way analysis of variance. In all the experiments, statistical significance was set at p<0.05.
Results
The anti-proliferation effects of chrysin on glioblastoma cells
To examine the effects of chrysin on cell proliferation, three human glioblastoma cell lines (T98, U251, and U87) and NHAs were treated with different concentrations of chrysin. A CCK-8 assay was used to assess cell viability in the cultures. Cells were treated with chrysin for 12, 24, and 48 hours, and 0.1% dimethyl sulfoxide (DMSO) was used as a vehicle control. The results showed that chrysin induced cell death in both concentration- and time-dependent manners in all tested glioblastoma cell lines, with the lowest concentration observed at 30 μM (Figure 1A–C). In contrast, the proliferation of NHAs was not affected by any concentration of chrysin (Figure 1A).
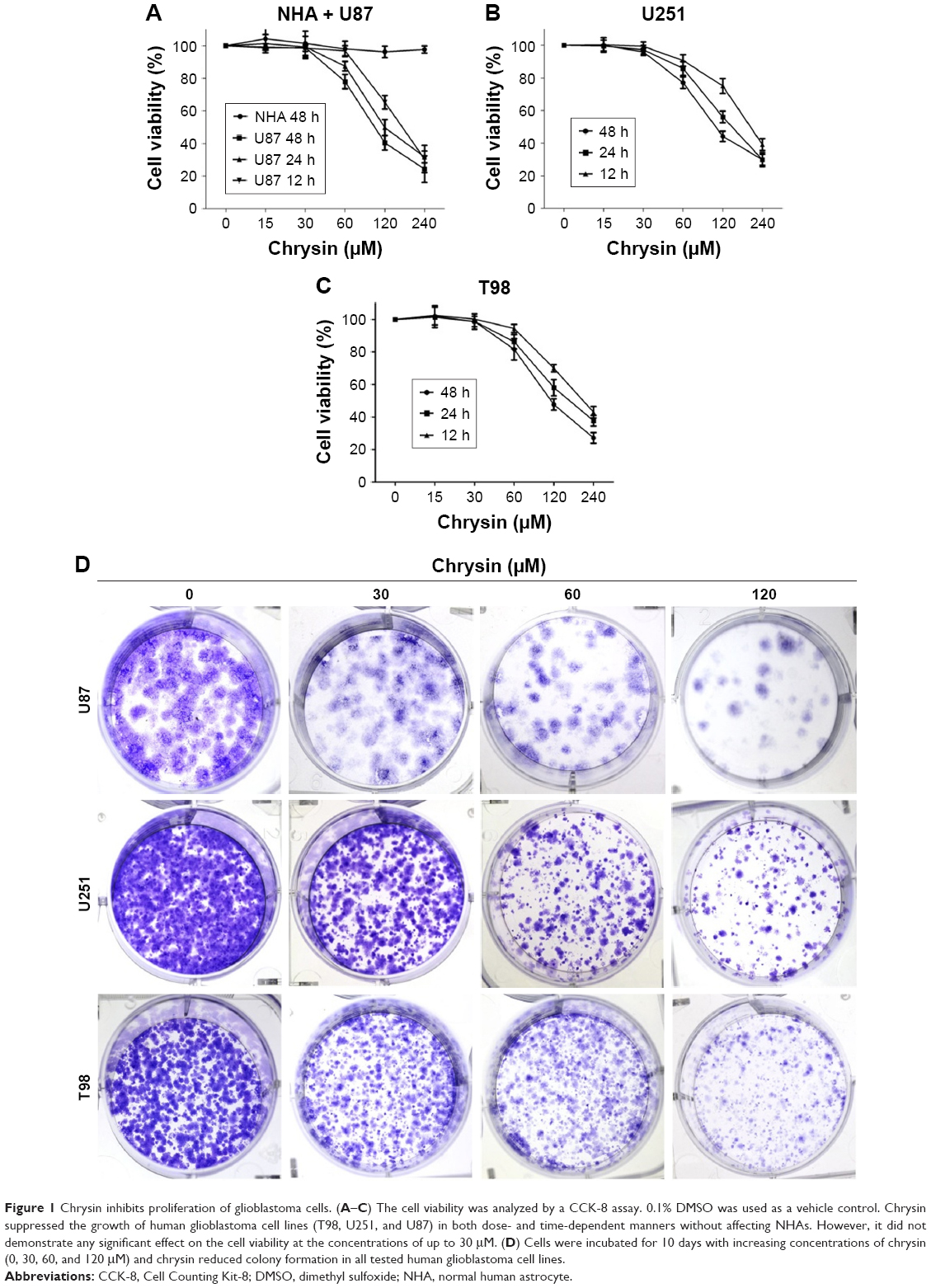 | Figure 1 Chrysin inhibits proliferation of glioblastoma cells. (A–C) The cell viability was analyzed by a CCK-8 assay. 0.1% DMSO was used as a vehicle control. Chrysin suppressed the growth of human glioblastoma cell lines (T98, U251, and U87) in both dose- and time-dependent manners without affecting NHAs. However, it did not demonstrate any significant effect on the cell viability at the concentrations of up to 30 μM. (D) Cells were incubated for 10 days with increasing concentrations of chrysin (0, 30, 60, and 120 μM) and chrysin reduced colony formation in all tested human glioblastoma cell lines. |
The long-term effects of chrysin on cell proliferation were determined by a colony-formation assay. As shown in Figure 1D, the size of independent colonies was much smaller in the chrysin-treated group than the vehicle-treated group, and the number of colonies was significantly reduced in the chrysin-treated group. The results demonstrated that chrysin suppresses the proliferation of glioblastoma cells in a dose-dependent manner without affecting NHAs.
Chrysin inhibits migration and invasion of glioblastoma cells
First, we used wound healing assays to measure the migration rate of glioblastoma cells (U251 and U87). The dosages and treatment duration are described in the “Materials and methods” section. Chrysin treatment significantly decreased the wound healing ability of glioblastoma cells, compared with the untreated control (Figure 2). Subsequently, we performed transwell migration and Matrigel invasion assays. In line with the wound healing assay, the migration and invasion capacity of glioblastoma cells was obviously reduced when pretreated with chrysin compared with vehicle-treated group (Figure 3). Taken together, these results suggest that chrysin inhibits the migration and invasion of glioblastoma cells in a concentration-dependent manner.
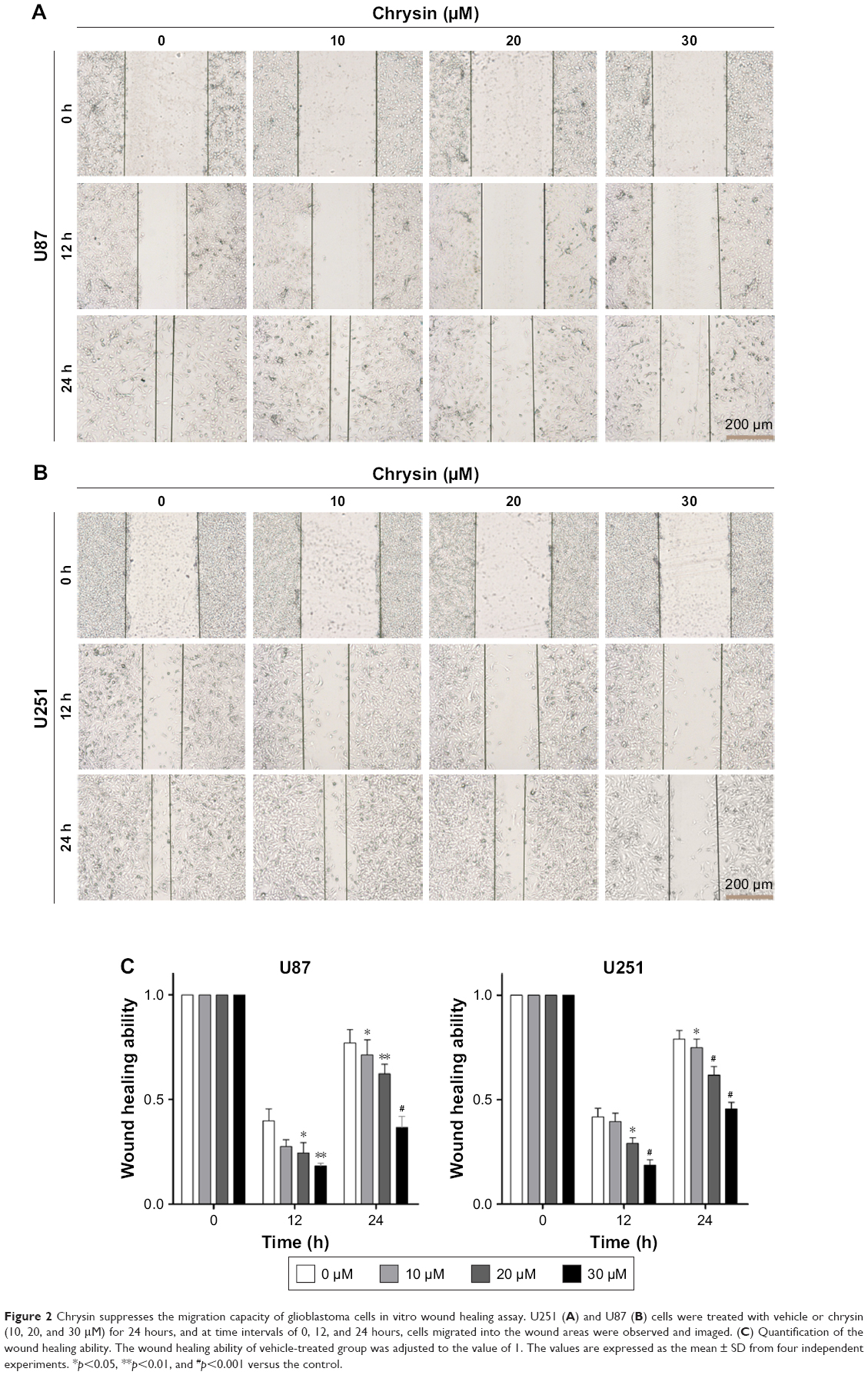 | Figure 2 Chrysin suppresses the migration capacity of glioblastoma cells in vitro wound healing assay. U251 (A) and U87 (B) cells were treated with vehicle or chrysin (10, 20, and 30 μM) for 24 hours, and at time intervals of 0, 12, and 24 hours, cells migrated into the wound areas were observed and imaged. (C) Quantification of the wound healing ability. The wound healing ability of vehicle-treated group was adjusted to the value of 1. The values are expressed as the mean ± SD from four independent experiments. *p<0.05, **p<0.01, and #p<0.001 versus the control. |
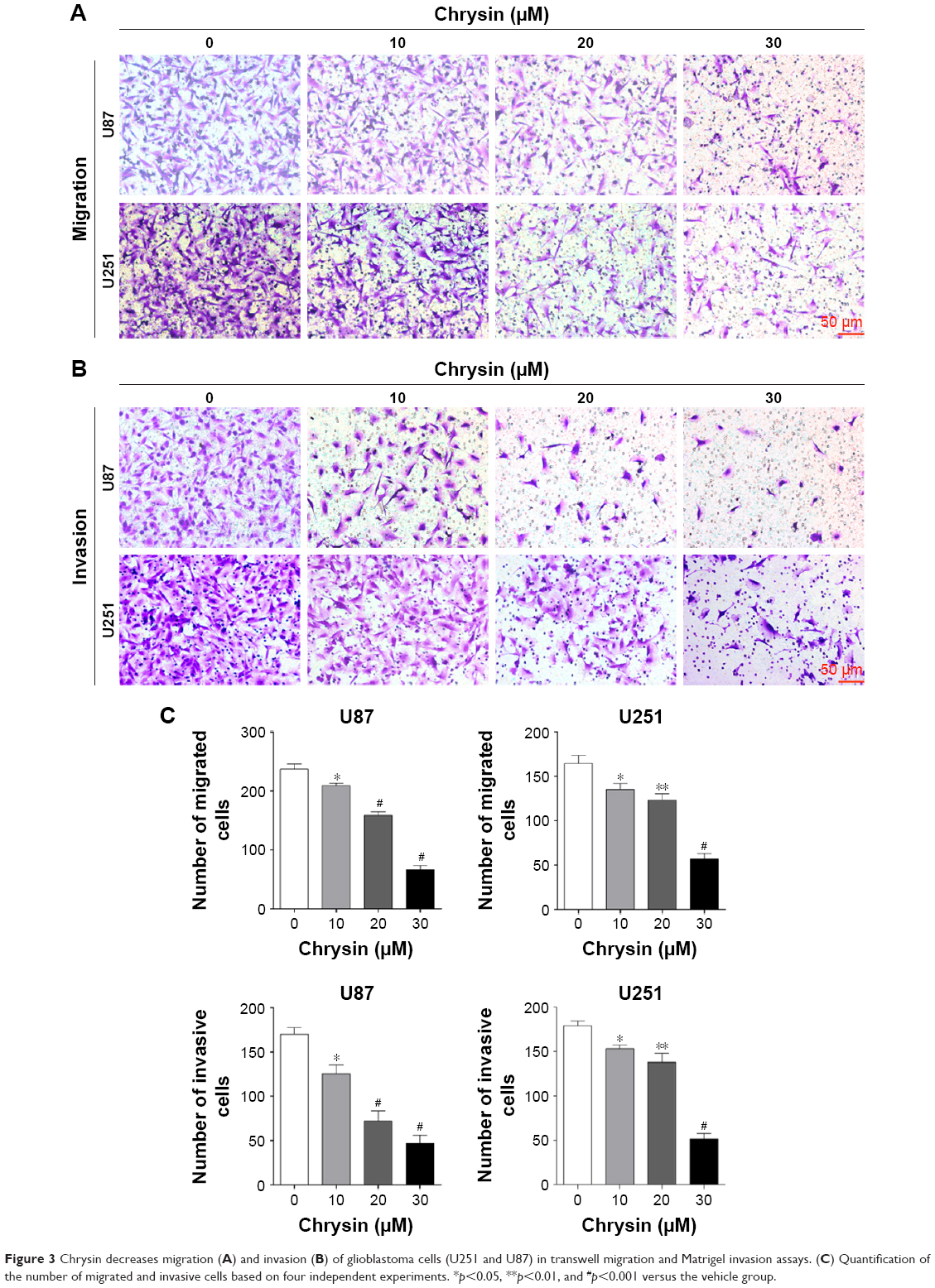 | Figure 3 Chrysin decreases migration (A) and invasion (B) of glioblastoma cells (U251 and U87) in transwell migration and Matrigel invasion assays. (C) Quantification of the number of migrated and invasive cells based on four independent experiments. *p<0.05, **p<0.01, and #p<0.001 versus the vehicle group. |
Chrysin inhibits glioblastoma cells by deactivating the Nrf2/ARE signaling pathway
Our previous studies indicate that the Nrf2/ARE signaling pathway is activated and plays an important role in the growth, migration, and invasion of glioblastoma.19,20
Furthermore, many studies reported that natural dietary flavonoids with well-defined anticancer activities were able to modulate the Nrf2/ARE signaling pathway. Therefore, we attempt to investigate whether chrysin can regulate the activity of Nrf2.21 As U87 was the most sensitive cell line in response to chrysin (Figure 1), this cell line was chosen for the subsequent experiments. First, cells were treated with 10, 30, and 60 μM chrysin for 24 hours; 0.1% DMSO was used as a vehicle control. Western blot analysis revealed that chrysin treatment reduced the protein levels of Nrf2 and Nrf2-target genes, including HO-1 and NQO-1, in a dose-dependent manner (Figure 4A and B). Importantly, the protein level of Keap1 was not visibly affected after the administration of chrysin at 24 hours (p>0.05). Second, we used lentivirus-mediated shRNA to establish the stable Nrf2 knockdown cells, as discussed previously.22 Compared with the shRNA control, transfection with shRNA-Nrf2 significantly abated Nrf2 protein expression according to Western-blot analysis (p<0.001, Figure 4C). Finally, cells preprocessed with shRNA-Nrf2 were treated with chrysin for 24 hours. As shown in Figure 4E and F, chrysin was unable to change the levels of Nrf2 protein with Nrf2 knockdown. The cell proliferation assay also revealed that silencing Nrf2 impaired glioblastoma cell proliferation and that chrysin was unable to change Nrf2i-mediated inhibition (Figure 4D). Collectively, these data further demonstrate that chrysin has anti-glioblastoma effects via inhibition of Nrf2.
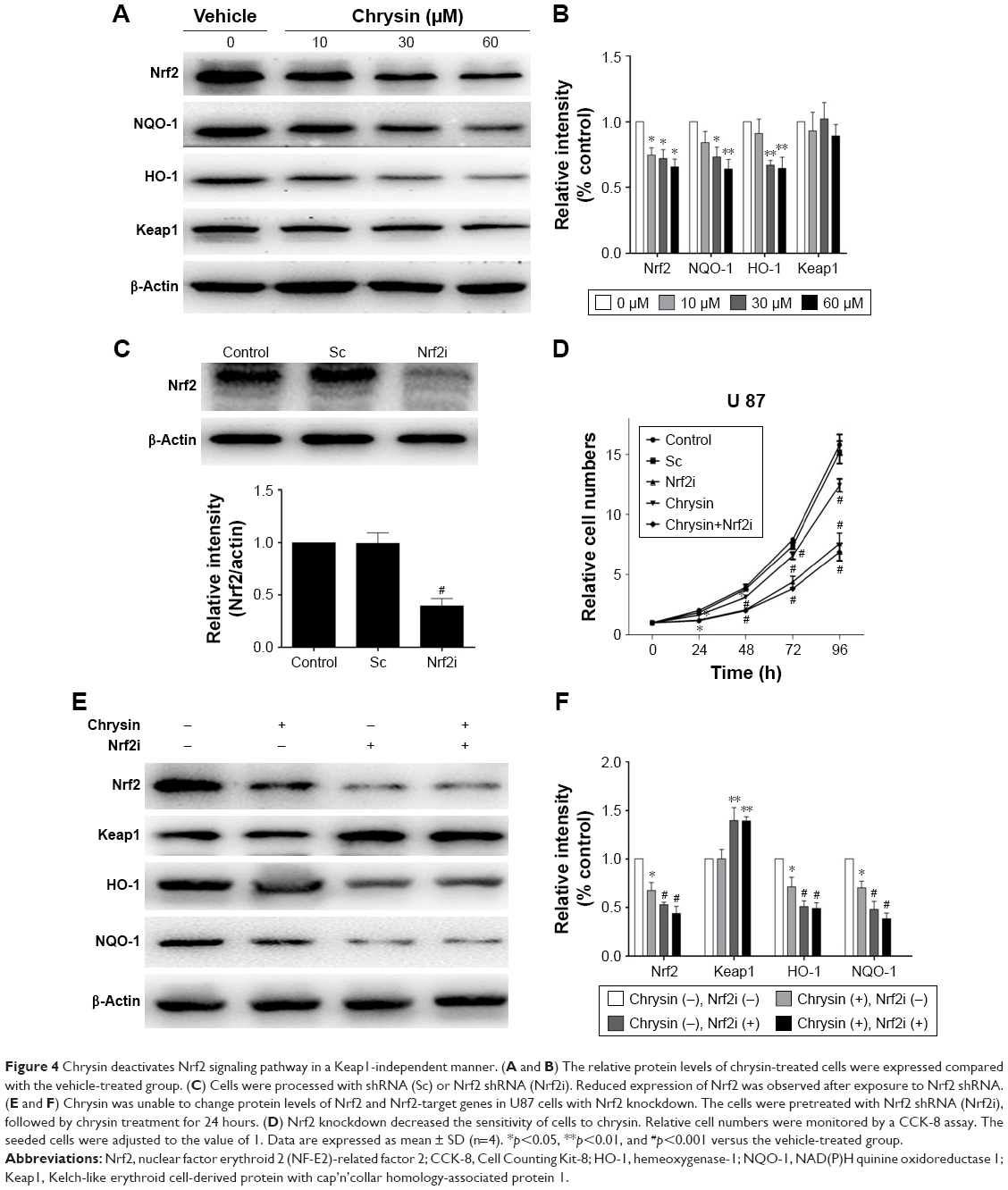 | Figure 4 Chrysin deactivates Nrf2 signaling pathway in a Keap1-independent manner. (A and B) The relative protein levels of chrysin-treated cells were expressed compared with the vehicle-treated group. (C) Cells were processed with shRNA (Sc) or Nrf2 shRNA (Nrf2i). Reduced expression of Nrf2 was observed after exposure to Nrf2 shRNA. (E and F) Chrysin was unable to change protein levels of Nrf2 and Nrf2-target genes in U87 cells with Nrf2 knockdown. The cells were pretreated with Nrf2 shRNA (Nrf2i), followed by chrysin treatment for 24 hours. (D) Nrf2 knockdown decreased the sensitivity of cells to chrysin. Relative cell numbers were monitored by a CCK-8 assay. The seeded cells were adjusted to the value of 1. Data are expressed as mean ± SD (n=4). *p<0.05, **p<0.01, and #p<0.001 versus the vehicle-treated group. |
Chrysin treatment downregulates the Nrf2 pathway via inhibition of ERK signaling
In malignant tumor cell models and specimens, the loss of regulation of MAPK signaling is strongly correlated with the disorder of Nrf2 expression.23,24 Accordingly, we further investigated whether chrysin affects MAPK activation and whether MAPKs play an important role in regulating Nrf2 expression. The results showed that chrysin treatment decreased the phosphorylation of ERK1/2 in a dose-dependent manner and had no noticeable effect on the expression of P 38, p-P 38, JNK, and p-JNK (Figure 5A and B). Thus, we hypothesized that ERK1/2 may participate in regulating Nrf2 expression in the presence of chrysin. To test this hypothesis, we assessed the effects of the ERK1/2 kinase inhibitor (U0126) on the protein expression of Nrf2. Our data confirmed that inhibition of ERK1/2 signaling by the ERK1/2 inhibitor (U0126) reduced the protein levels of Nrf2 (p<0.01, Figure 5C and D). To further investigate the effect of chrysin on ERK/Nrf2 expression, cells pretreated with U0126 were treated with chrysin for 24 hours. Western blot analysis (p>0.05, Figure 5C and D) demonstrated that chrysin was unable to alter U0126-mediated transcriptional inhibition. By immunofluorescence staining, we similarly found that chrysin and U0126 inhibited Nrf2 translocation into the nucleus in comparison with the control group; however, there was no statistical difference between the U0126 and the combined treatment group (p>0.05, Figure 5E). These data suggest that chrysin downregulates the Nrf2 pathway via inhibition of ERK signaling.
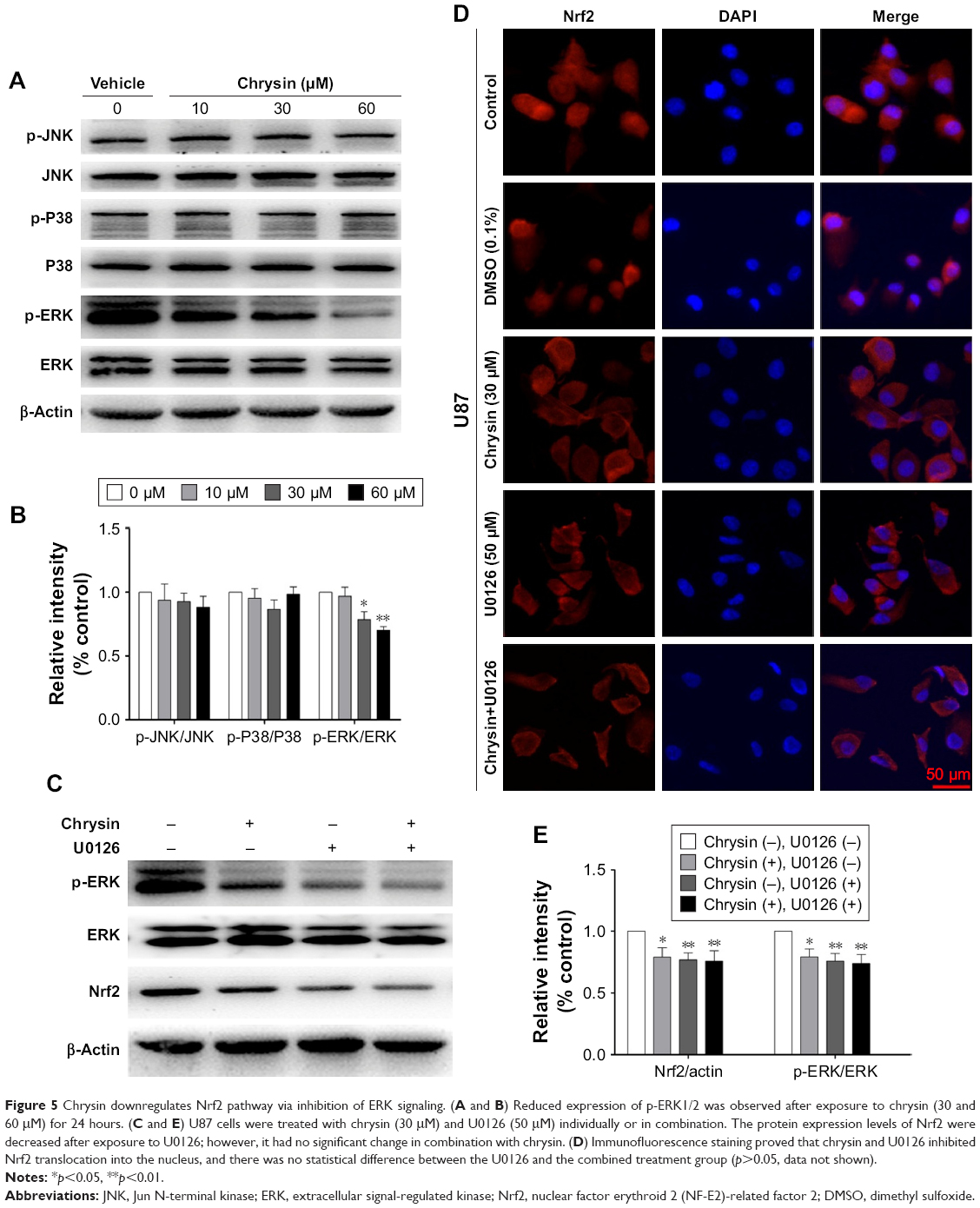 | Figure 5 Chrysin downregulates Nrf2 pathway via inhibition of ERK signaling. (A and B) Reduced expression of p-ERK1/2 was observed after exposure to chrysin (30 and 60 μM) for 24 hours. (C and E) U87 cells were treated with chrysin (30 μM) and U0126 (50 μM) individually or in combination. The protein expression levels of Nrf2 were decreased after exposure to U0126; however, it had no significant change in combination with chrysin. (D) Immunofluorescence staining proved that chrysin and U0126 inhibited Nrf2 translocation into the nucleus, and there was no statistical difference between the U0126 and the combined treatment group (p>0.05, data not shown). |
Chrysin inhibited in vivo tumor growth in tumorigenicity experiments
To assess whether the compound can suppress tumor growth in vivo, we employed immune-deficient BALB/c nude mice bearing U87 tumor xenografts. Chrysin treatment was administered when the tumor volume reached 70–100 mm3, as described in the “Materials and methods” section. After 3 weeks of treatment, tumor weight in mice administered 40 and 80 mg/kg chrysin was less than that in mice administered the refined olive oil as a control, and the mean tumor volume in the vehicle-treated group increased faster than that in the chrysin-treated group (Figure 6A, B, and D). None of the mice in the chrysin-treated group exhibited any weight loss or overt toxicity compared with the vehicle group (Figure 6C). Western blot analysis revealed that chrysin treatment inhibited tumor xenografts by downregulating the ERK/Nrf2 signaling pathway (Figure 6E and F).
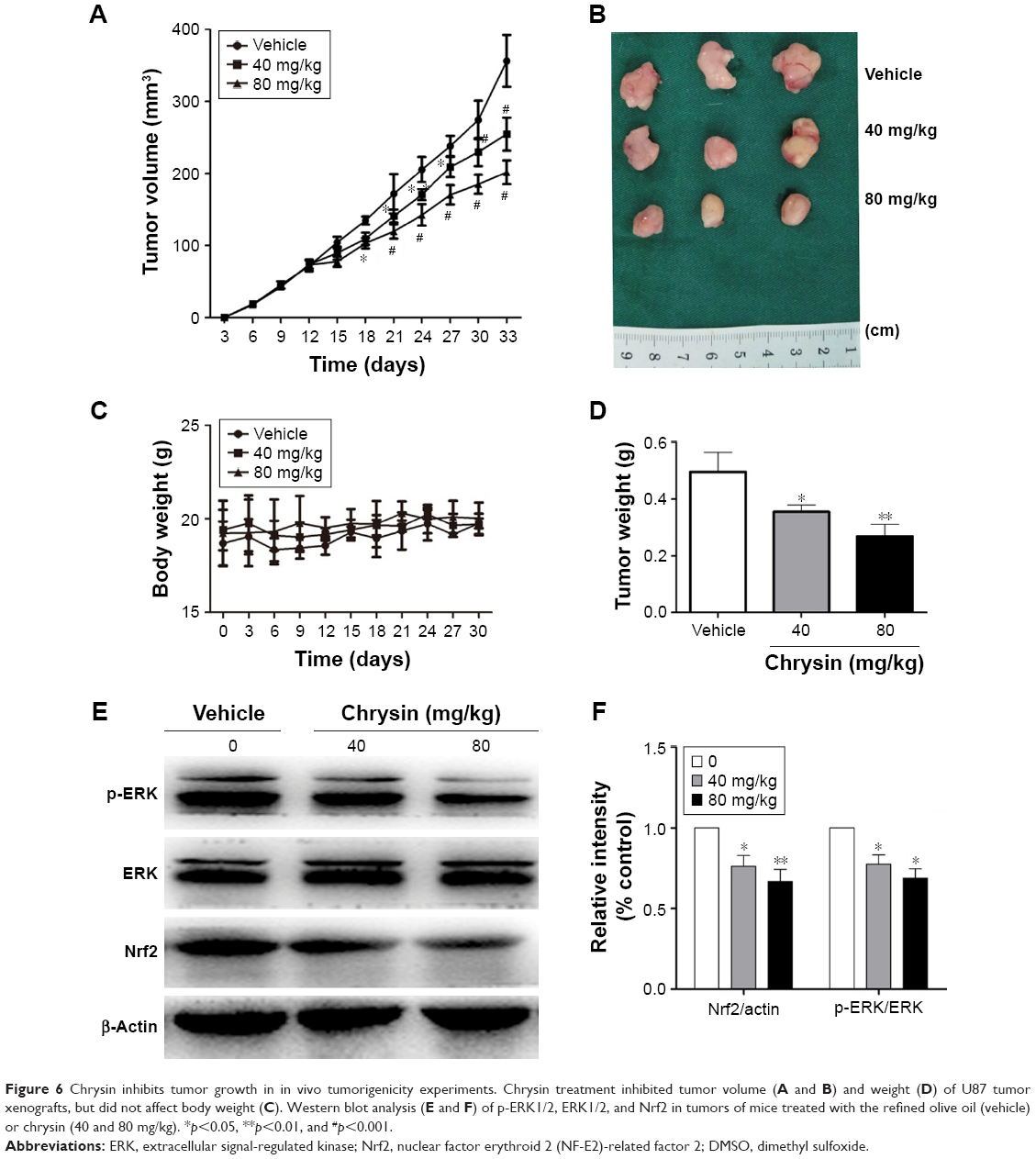 | Figure 6 Chrysin inhibits tumor growth in in vivo tumorigenicity experiments. Chrysin treatment inhibited tumor volume (A and B) and weight (D) of U87 tumor xenografts, but did not affect body weight (C). Western blot analysis (E and F) of p-ERK1/2, ERK1/2, and Nrf2 in tumors of mice treated with the refined olive oil (vehicle) or chrysin (40 and 80 mg/kg). *p<0.05, **p<0.01, and #p<0.001. |
Discussion
Despite years of basic research and clinical trials, GBM is still one of the deadliest primary tumors in the central nervous system.25 Several studies have contributed to the high morbidity and mortality of GBM, including multiple oncogenic signaling aberrations associated with excessive cell proliferation and migration and high cell invasiveness. In this work, we present that chrysin inhibited the proliferation, migration, and invasion of glioblastoma cells in vivo and in vitro in dose-dependent manners. In addition, we confirmed that anti-glioblastoma effects of chrysin are related to the downregulation of the Nrf2/ARE pathway in Keap1-independent cascades. Mechanistically, our data showed that the suppression induced by chrysin on ERK1/2 signaling pathway may lead to the inhibition of Nrf2/ARE signaling.
Chrysin, which is a natural dietary flavonoid, has been shown to display chemopreventive and therapeutic potential in the development of cancer, although the pivotal molecules targeted by its activity are not clear.26–28 For example, chrysin suppressed the expression of nuclear factor-kappaB and sensitized tumor necrosis factor-α-induced apoptosis in a panel of cancer cell lines, including human liver cancer cells (HCT-116), colorectal cancer cells (HCT-116), and human nasopharyngeal carcinoma cells (CNE-1).29 Chrysin was also reported to induce cell cycle arrest either through activating P38-MAPK leading to the accumulation of P21Waf1/Cip1 protein or by mediating the inhibition of proteasome activity.30 Moreover, professor Gao demonstrated that chrysin partially reversed doxorubicin resistance of human hepatocellular carcinoma cells by downregulating the PI3K-Akt and ERK pathway.31 However, it is interesting to note that chrysin has been reported to significantly inhibit the phosphorylation of JNK in murine microglia cells (BV-2), while having no influence on the phosphorylation of P38 and ERK.32 This disputation proved that the biological effects of chrysin are cell-type- and context-dependent. In addition, further studies are required to compare the molecular effects of chrysin on the MAPKs cascades in diverse disease. The results of our study suggest that chrysin had inhibitory effect on hyper-proliferation, migration, and invasiveness of the GBM cell models in vitro and in vivo and displayed dose-dependent effects, although chrysin seemed to have little impact on the proliferation of NHAs.
Nrf2, an essential cytoprotective transcription factor, plays a significant role in major stages of carcinogenesis such as initiation, promotion, and progression and has increasingly been considered as a prognostic molecular marker for determining the status of malignant tumor progression, specifically in anaplastic glioma.33,34 Previous research in our laboratory also determined that high expression of Nrf2 in glioblastoma was linked to survival, growth, angiogenesis, invasion, metastasis, and drug-resistance of glioblastoma cells and that blocking Nrf2 could inhibit glioblastoma growth.22,35 Therefore, the alternative inhibition of Nrf2 signal pathways should be regarded as a significant pharmacological target.21 Various natural flavonoids produce different biological effects on Nrf2, mainly depending on the cellular microenvironment. For example, low-dose resveratrol promoted Nrf2 activation, while higher concentrations of resveratrol antagonize Nrf2.36 In rat primary hepatocytes, chrysin, apigenin, and luteolin were reported to upregulate Nrf2/ARE signaling pathways to respond more robustly to oxidative assaults;37 however, others have reported that chrysin, apigenin, and luteolin reduced Nrf2 expression and increased the chemosensitivity of cells to anticancer drugs in cancer cell lines.4 These data suggested that dietary flavonoids exhibited agonistic as well as antagonistic activity of Nrf2 depending on the cell microenvironment and different disease states. Our present research revealed that anti-glioblastoma effect of chrysin was relevant to its inhibition on Nrf2 signal. These results are supported by previously published findings, which indicated that by downregulating the Nrf2 signaling pathway, chrysin could serve as an effective adjuvant sensitizer and enhance the sensitivity of drug-resistant hepatocellular carcinoma cells (BEL-7402) to chemotherapeutic drugs.31
As has been previously discussed, we briefly introduced the current knowledge on regulation of the Nrf2/ARE pathway involving Keap1-dependent cascades, Keap1-independent cascades, and epigenetic factors. Notably, we unexpectedly found that chrysin repressed the protein expression of Nrf2 and its major target genes without obvious induction of the protein levels of Keap1, which is primarily a negative modulator of Nrf2 under basal and stressed conditions. Based on this, we hypothesized that the nuclear inhibition of Nrf2 preceded the upregulation of Keap1 or that the binding of Keap1 to Nrf2 was enhanced; thus, other mechanisms need to be taken into account. The most recent research showed that flavonoid compounds regulated the expression of Nrf2 target antioxidative gene and had no effect on mRNA expression of Keap1.38 In addition, chrysin induces apoptosis of cancer cells through stimulating MAPKs and that MAPKs were involved in the regulation of Nrf2 activity in a Keap1-independent manner.39,40 For this reason, we performed further experiments to observe the mechanism engaged in the adjustment of Nrf2 activity. Finally, we provide evidence that chrysin suppressed the phosphorylation of ERK1/2 and has no observable effect on the expression of Nrf2 after treatment with an inhibitor of ERK1/2 (U0126). Accumulating evidence has shown that phosphorylation by MAPKs has a strong association with Nrf2 transcriptional activity, and the effects of MAPKs on Nrf2 are controversial. Several researchers have demonstrated that both ERK1/2 and JNK signaling pathway enhance Nrf2 signaling, while P38 pathway plays a negative role.24 In particular, our study confirmed that phosphorylation of Nrf2 by ERK1/2 may contribute to the downregulation of Nrf2 expression. However, we did not further explore the specificity and precise mechanism between the Keap1-dependent cascade and phosphorylation of ERK1/2.
Taken together, we present a novel function of chrysin by demonstrating that chrysin possesses anti-glioblastoma and chemopreventive properties, which can disturb the ERK/Nrf2 signaling pathway. This finding provides strong evidence that chrysin, a natural dietary flavonoid, is a promising candidate for the prevention and management of glioblastoma and other cancers.
Acknowledgments
This work was supported by grants from The National Natural Science Foundation of China (NSFC): grant numbers 81672503 (HD Wang) and 81401026 (XL Wang), and the China Postdoctoral Science Foundation: grant number 2015M572716 (H Pan).
Disclosure
The authors report no conflicts of interest in this work.
References
Wesseling P, Jacques TS. Taxonomy of CNS tumours; a series of three short reviews on the WHO 2016 classification and beyond. Neuropathol Appl Neurobiol. 2018;44(2):137–138. | ||
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | ||
Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005;109(1):93–108. | ||
Stepanic V, Gasparovic AC, Troselj KG, Amic D, Zarkovic N. Selected attributes of polyphenols in targeting oxidative stress in cancer. Curr Top Med Chem. 2015;15(5):496–509. | ||
Ji S, Li R, Wang Q, et al. Anti-H1N1 virus, cytotoxic and Nrf2 activation activities of chemical constituents from Scutellaria baicalensis. J Ethnopharmacol. 2015;176:475–484. | ||
McMahon M, Itoh K, Yamamoto M, et al. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61(8):3299–3307. | ||
Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7(3–4):385–394. | ||
Pandey P, Singh AK, Singh M, Tewari M, Shukla HS, Gambhir IS. The see-saw of Keap1-Nrf2 pathway in cancer. Crit Rev Oncol Hematol. 2017;116:89–98. | ||
Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol. 2013;85(6):705–717. | ||
Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48(2):91–104. | ||
Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med. 2016;22(7):578–593. | ||
Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27(20):2179–2191. | ||
Yang SH, Sharrocks AD, Whitmarsh AJ. MAP kinase signalling cascades and transcriptional regulation. Gene. 2013;513(1):1–13. | ||
Farooq A, Zhou MM. Structure and regulation of MAPK phosphatases. Cell Signal. 2004;16(7):769–779. | ||
Hu R, Saw CL, Yu R, Kong AN. Regulation of NF-E2-related factor 2 signaling for cancer chemoprevention: antioxidant coupled with antiinflammatory. Antioxid Redox Signal. 2010;13(11):1679–1698. | ||
Mantawy EM, Esmat A, El-Bakly WM, Salah ElDin RA, El-Demerdash E. Mechanistic clues to the protective effect of chrysin against doxorubicin-induced cardiomyopathy: plausible roles of p53, MAPK and AKT pathways. Sci Rep. 2017;7(1):4795. | ||
Wang Q, Wang H, Jia Y, Pan H, Ding H. Luteolin induces apoptosis by ROS/ER stress and mitochondrial dysfunction in gliomablastoma. Cancer Chemother Pharmacol. 2017;79(5):1031–1041. | ||
Kim KM, Lim HK, Shim SH, Jung J. Improved chemotherapeutic efficacy of injectable chrysin encapsulated by copolymer nanoparticles. Int J Nanomedicine. 2017;12:1917–1925. | ||
Pan H, Wang H, Zhu L, et al. The involvement of Nrf2-ARE pathway in regulation of apoptosis in human glioblastoma cell U251. Neurol Res. 2013;35(1):71–78. | ||
Ji X, Wang H, Zhu J, et al. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Int J Cancer. 2014;135(3):574–584. | ||
Lu MC, Ji JA, Jiang ZY, You QD. The Keap1-Nrf2-ARE pathway as a potential preventive and therapeutic target: an update. Med Res Rev. 2016;36(5):924–963. | ||
Jia Y, Wang H, Wang Q, Ding H, Wu H, Pan H. Silencing Nrf2 impairs glioma cell proliferation via AMPK-activated mTOR inhibition. Biochem Biophys Res Commun. 2016;469(3):665–671. | ||
Du ZX, Yan Y, Zhang HY, et al. Proteasome inhibition induces a p38 MAPK pathway-dependent antiapoptotic program via Nrf2 in thyroid cancer cells. J Clin Endocrinol Metab. 2011;96(5):E763–E771. | ||
Peluso I, Yarla NS, Ambra R, Pastore G, Perry G. MAPK signalling pathway in cancers: olive products as cancer preventive and therapeutic agents. Semin Cancer Biol. Epub 2017 Sep 11. | ||
Chen R, Cohen AL, Colman H. Targeted therapeutics in patients with high-grade gliomas: past, present, and future. Curr Treat Options Oncol. 2016;17(8):42. | ||
Davatgaran-Taghipour Y, Masoomzadeh S, Farzaei MH, et al. Polyphenol nanoformulations for cancer therapy: experimental evidence and clinical perspective. Int J Nanomedicine. 2017;12:2689–2702. | ||
Deldar Y, Pilehvar-Soltanahmadi Y, Dadashpour M, Montazer Saheb S, Rahmati-Yamchi M, Zarghami N. An in vitro examination of the antioxidant, cytoprotective and anti-inflammatory properties of chrysin-loaded nanofibrous mats for potential wound healing applications. Artif Cells Nanomed Biotechnol. Epub 2017 Jun 9. | ||
Javan Maasomi Z, Pilehvar Soltanahmadi Y, Dadashpour M, Alipour S, Abolhasani S, Zarghami N. Synergistic anticancer effects of silibinin and chrysin in T47D breast cancer cells. Asian Pac J Cancer Prev. 2017;18(5):1283–1287. | ||
Li X, Huang Q, Ong CN, Yang XF, Shen HM. Chrysin sensitizes tumor necrosis factor-alpha-induced apoptosis in human tumor cells via suppression of nuclear factor-kappaB. Cancer Lett. 2010;293(1):109–116. | ||
Weng MS, Ho YS, Lin JK. Chrysin induces G1 phase cell cycle arrest in C6 glioma cells through inducing p21Waf1/Cip1 expression: involvement of p38 mitogen-activated protein kinase. Biochem Pharmacol. 2005;69(12):1815–1827. | ||
Gao AM, Ke ZP, Shi F, Sun GC, Chen H. Chrysin enhances sensitivity of BEL-7402/ADM cells to doxorubicin by suppressing PI3K/Akt/Nrf2 and ERK/Nrf2 pathway. Chem Biol Interact. 2013;206(1):100–108. | ||
Ha SK, Moon E, Kim SY. Chrysin suppresses LPS-stimulated proinflammatory responses by blocking NF-kappaB and JNK activations in microglia cells. Neurosci Lett. 2010;485(3):143–147. | ||
Kanamori M, Higa T, Sonoda Y, et al. Activation of the NRF2 pathway and its impact on the prognosis of anaplastic glioma patients. Neuro Oncol. 2015;17(4):555–565. | ||
DeNicola GM, Karreth FA, Humpton TJ, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–109. | ||
Zhu J, Wang H, Fan Y, et al. Targeting the NF-E2-related factor 2 pathway: a novel strategy for glioblastoma (review). Oncol Rep. 2014;32(2):443–450. | ||
Plauth A, Geikowski A, Cichon S, et al. Hormetic shifting of redox environment by pro-oxidative resveratrol protects cells against stress. Free Radic Biol Med. 2016;99:608–622. | ||
Huang CS, Lii CK, Lin AH, et al. Protection by chrysin, apigenin, and luteolin against oxidative stress is mediated by the Nrf2-dependent up-regulation of heme oxygenase 1 and glutamate cysteine ligase in rat primary hepatocytes. Arch Toxicol. 2013;87(1):167–178. | ||
Ji LL, Sheng YC, Zheng ZY, Shi L, Wang ZT. The involvement of p62-Keap1-Nrf2 antioxidative signaling pathway and JNK in the protection of natural flavonoid quercetin against hepatotoxicity. Free Radic Biol Med. 2015;85:12–23. | ||
Yu R, Lei W, Mandlekar S, et al. Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J Biol Chem. 1999;274(39):27545–27552. | ||
Wang J, Huang X, Zhang K, Mao X. Vanadate oxidative and apoptotic effects are mediated by the MAPK-Nrf2 pathway in layer oviduct magnum epithelial cells. Metallomics. 2017;9(11):1562–1575. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.