Back to Journals » Veterinary Medicine: Research and Reports » Volume 10
Chronic Wasting Disease In Cervids: Prevalence, Impact And Management Strategies
Authors Rivera NA ![]() , Brandt AL
, Brandt AL ![]() , Novakofski JE
, Novakofski JE ![]() , Mateus-Pinilla NE
, Mateus-Pinilla NE ![]()
Received 18 June 2019
Accepted for publication 10 September 2019
Published 2 October 2019 Volume 2019:10 Pages 123—139
DOI https://doi.org/10.2147/VMRR.S197404
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Young Lyoo
Nelda A Rivera,1 Adam L Brandt,2 Jan E Novakofski,3 Nohra E Mateus-Pinilla1
1Illinois Natural History Survey-Prairie Research Institute, University of Illinois Urbana-Champaign, Champaign, IL, USA; 2Division of Natural Sciences, St. Norbert College, De Pere, WI, USA; 3Department of Animal Sciences, University of Illinois Urbana-Champaign, Urbana, IL, USA
Correspondence: Nohra E Mateus-Pinilla
Illinois Natural History Survey-Prairie Research Institute, University of Illinois Urbana-Champaign, 1816 South Oak Street, Champaign, IL 61820, USA
Tel +12173336856
Fax +12172440802
Email [email protected]
Abstract: Chronic wasting disease (CWD) is a transmissible spongiform encephalopathy (TSE) that affects members of the cervidae family. The infectious agent is a misfolded isoform (PrPSC) of the host prion protein (PrPC). The replication of PrPSC initiates a cascade of developmental changes that spread from cell to cell, individual to individual, and that for some TSEs, has crossed the species barrier. CWD can be transmitted horizontally and vertically, and it is the only TSE that affects free-ranging wildlife. While other TSEs are under control and even declining, infection rates of CWD continue to grow and the disease distribution continues to expand in North America and around the world. Since the first reported case in 1967, CWD has spread infecting captive and free-ranging cervids in 26 states in the US, 3 Canadian provinces, 3 European countries and has been found in captive cervids in South Korea. CWD causes considerable ecologic, economic and sociologic impact, as this is a 100% fatal highly contagious infectious disease, with no treatment or cure available. Because some TSEs have crossed the species barrier, the zoonotic potential of CWD is a concern for human health and continues to be investigated. Here we review the characteristics of the CWD prion protein, mechanisms of transmission and the role of genetics. We discuss the characteristics that contribute to prevalence and distribution. We also discuss the impact of CWD and review the management strategies that have been used to prevent and control the spread of CWD.
Keywords: CWD, prion, PRNP, PrPC, PrPSC, TSE
Introduction
Background
Chronic wasting disease (CWD) is the prion disease of the cervidae family.1 Prion diseases—or transmissible spongiform encephalopathies (TSEs)—are a group of progressive neurodegenerative disorders that affect animals and humans. The first TSE was discovered in the 18th century; at the time it was a strange disease that affected sheep, causing behavioral changes inducing excessive licking, scratching and altered gait.2 After Scrapie was first described in 1732, other diseases with similar neurological characteristic, such as Creutzfeldt-Jakob disease in 1920 (CJD)3,4 and Kuru5 in 1957 were identified in humans.
The agent causing these diseases was not clearly defined but was presumed to be a viral infection of the central nervous system.6–9 By the year 1959, researchers had linked Scrapie, Kuru and CJD by suggesting that they were related neuropathies.10,11 Eight more years passed before researchers considered that Scrapie was caused by a proteinase agent.12–14 By the same year, 1967, a new disease named CWD was discovered in a farmed mule deer (Odocoileus hemionus hemionus) in Colorado and later in mule deer and black-tailed deer (Odocoileus hemionus columbianus) in Colorado and Wyoming, USA;1 yet, the term TSE was far from being used as a disease category.2
It was not until 1982 when Prusiner used the term “prion”—derived from the words proteinaceous and infectious—to describe the causative infectious agent of Scrapie.15 The same year Prusiner and collaborators proved that the causative agent of Scrapie was a protein. In 1997, Prusiner and collaborators won the Nobel Prize for the discovery of “Prions – a new biological principle of infection” and for their contribution on prion research development. While the consensus is that prion proteins (denoted PrPSC from Scrapie) are the causative agents of prion diseases, and further evidence support that PRNP—a host gene that regulates the expression of the prion protein PrPC—plays a crucial role in the development of TSEs,16 some researchers proposed bacterias17,18 and viruses19 as causative agents of TSEs. However, these theories were soon dismissed.20–22
Even though the characteristics of the TSEs disease group are clear (Figure 1), they may present as inherited, infectious or sporadic disorders in a variety of hosts depending on the TSE (Table 1).23,24 Most TSEs are under control or declining. However, CWD is on the rise and is the only prion disease of wild free-ranging animals;25 CWD continues to affect several cervidae host species across the world. This review will introduce the unique characteristics of CWD and the influence of genetics. We will focus on prevalence and distribution, and examine the impact of CWD and suggested management strategies.
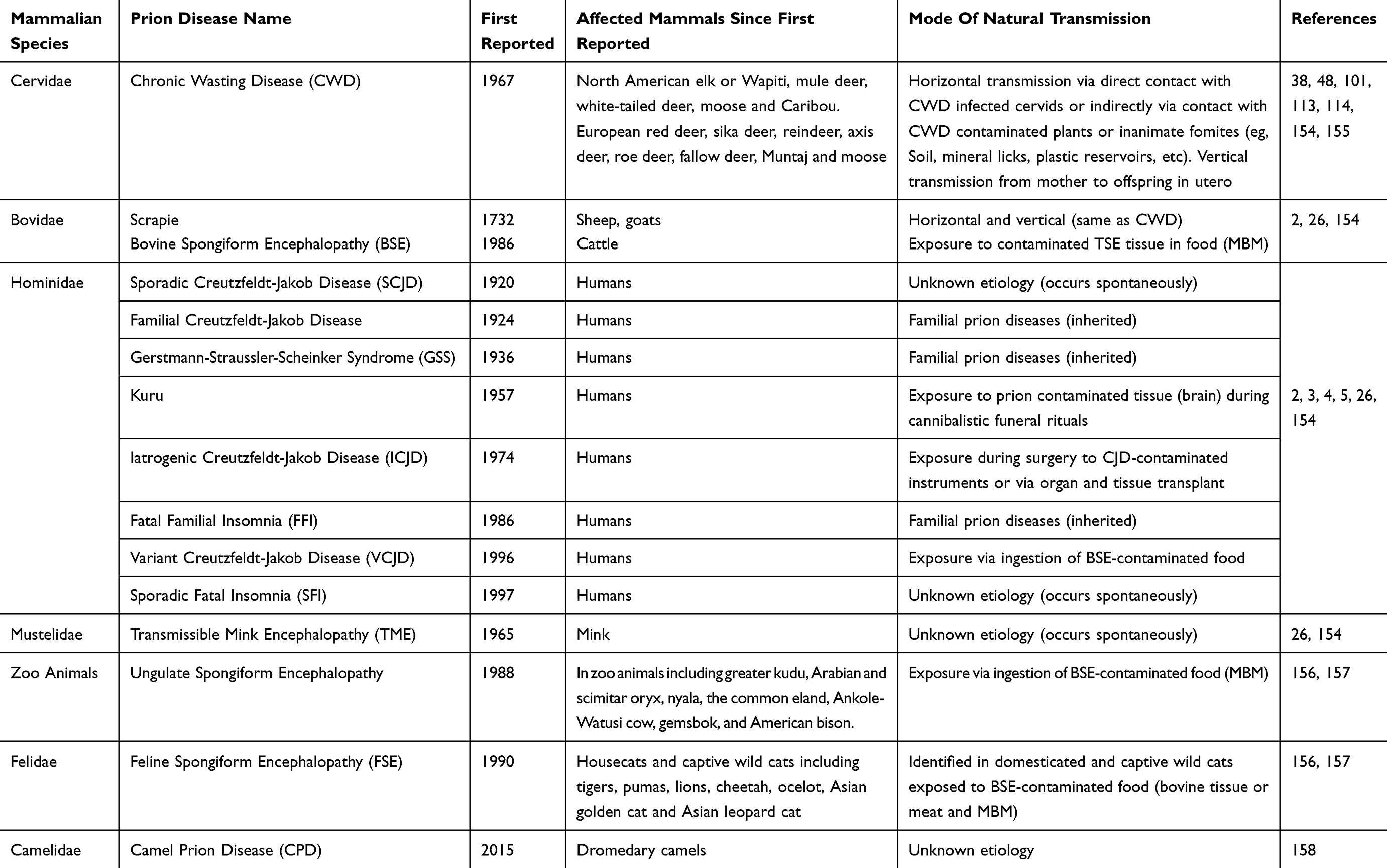 |
Table 1 Human And Animal Prion Diseases |
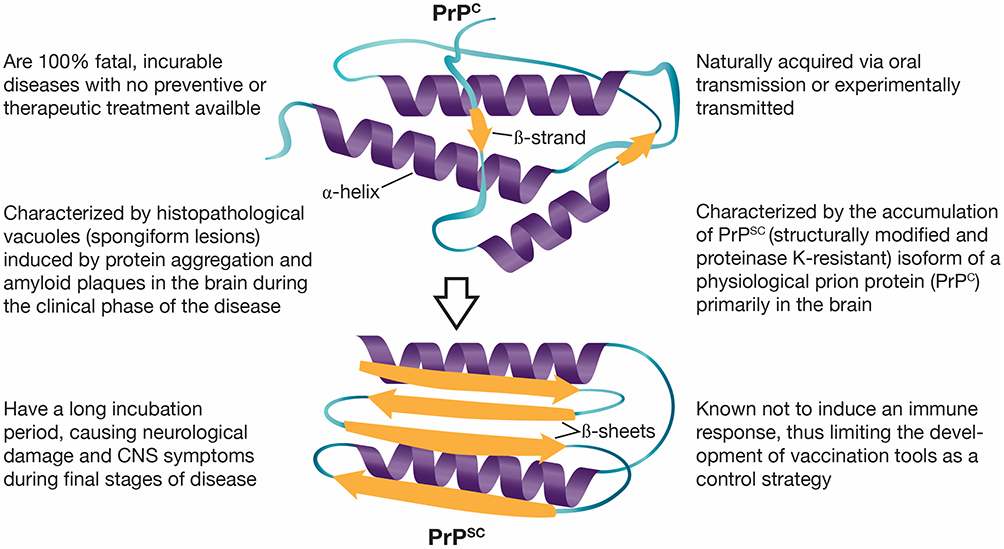 |
Figure 1 Characteristics of transmissible spongiform encephalopathies (TSEs) or prion diseases. Notes: Conformational changes of the host prion protein structure, from α-helices in the normal cell-surface glycoprotein (PrPC) to β-sheets in the misfolded isoform (PrPSC). Data from Doherr (2007),23 Prusiner (1998),26 Novakofski et al (2005),34 Image credit to Kerry L. Helms, Scientific Illustrator (Public domain).159Abbreviations: PrPC, the host prion protein; PrPSC, the misfolded isoform of the host prion protein. |
CWD Characteristics
Prion proteins (PrPC) are cell-surface glycoproteins with predominantly α-helical conformations. PrPC is encoded by the prion protein gene (PRNP), which is present in almost, if not all, mammalian species. The PrPC are expressed in several tissues and cell types,26 including epithelial, endothelial and immune cells.27–30 Above all, PrPC is highly expressed in neurons and neuroglial cells of the peripheral nervous system (PNS) and central nervous system (CNS).31,32 The infectious prion protein is the misfolded isoform (PrPSC) of the cellular PrPC. The posttranslational process that causes conformational changes from a predominantly α-helical isoform and a coil structure to a refolded β-pleated sheet33 confers resistance to proteases (eg, environmental, intestinal and intracellular) that would otherwise destroy the protein.34 Besides resistance, the β-sheet structure of the pathogenic PrPSC is prone to aggregation. Aggregation of PrPSC leads to the conversion of more PrPC to PrPSC, formation of amyloid plaques and vacuolization that cause progressive neurodegeneration.35 Because the modification from PrPC to PrPSC is posttranslational, the amino acid sequence of both, PrPC and PrPSC (209 residues), is identical within an individual.24 Hence, there is not a substantial immune response and inflammatory reaction to the infection.36,37 However, chronic inflammation may contribute to natural CWD transmission, as chronic inflammation may upregulate cytokines enabling PrPSC accumulation and propagation to other tissues.34 For example, follicular dendritic cells and mast cells express high levels of PrPC.28,36 Expression and release of PrPC from migratory cells, such as mast cells, may facilitate quick progress of the infectious prion from lymphoid tissues associated with the gastrointestinal track to the PNS, CNS and brain.28
Prions are infectious pathogens that, in the case of CWD, can be transmitted horizontally or vertically. Horizontal transmission is the most effective CWD transmission method, with reported incidence of disease in captive mule deer of 89%,38 and early infection detected in lymphoid tissue along the oral and digestive system 42 days post-oral inoculation in mule deer fawns.39 Horizontal transmission includes direct contact of an infected and susceptible animal or contact of susceptible animals with infected saliva, feces and urine. Indirect horizontal transmission involves environmental components and includes oral infection by ingestion of contaminated grass and/or soil during grazing and dust inhalation of infectious particles bound to soil.40,41 Based on social networks and contact patterns among free-ranging white-tailed deer (Odocoileus virginianus), direct contact is the primary contributor of CWD transmission among deer.42
Scientists considered vertical transmission (in utero) an unexpected or rare process. Researchers believed that high neonatal mortality in deer and elk populations coupled with the solitary nature of cervids during parturition reduced the importance of maternal transmission in sustaining CWD.43 However, TSEs vertical transmission has been confirmed in sheep, cattle, felids, humans and in transgenic mouse models.44–47 Recent studies based on experimental models of CWD demonstrated the transmission of CWD from doe (clinical and sub-clinical mothers) to fawns (full-term viable, full-term non-viable and in utero harvested offspring).48 These studies found 80% of the fetuses from CWD-positive muntjac deer dams PrPSC positive, suggesting previous underestimation of the transmission from mother to offspring for all TSEs.48 Vertical transmission contributes to CWD infection in naturally exposed elk populations.49
The development of clinical CWD can take months to years. The incubation time—the period between exposure to the pathogenic CWD prion to the development of clinical signs and symptoms—in both, naturally and experimentally infected cervids may vary from 2 to 4 years.43 Differences in incubation periods could relate to infectious dose, route of exposure, cervid species and/or genotype. For example, incubation periods in orally inoculated mule deer ranged from 3 months to 2 years, with differences in CNS accumulation timing associated to genotype profiles.50 Similar findings were reported for other cervid species; CWD-positive muntjac deer developed terminal disease in 18–24 months post-oral inoculation.69 Interestingly, viable offsprings from those CWD-positive muntjac dams surpassed the time usually seen for terminal disease in cervid species (18–24 months).48 Although the maximum incubation period in free-ranging cervids is unknown, most CWD cases have been reported in 3- to 7-year-old animals,43,51 which is similar to the age groups of captive elk and mule deer that succumb to CWD.1 Because the way prions spread throughout the body, pathological changes and distribution of PrPSC might be first identified in the lymphoreticular system and later in the CNS, similar to what has been described for sheep infected with scrapie.52 Retropharyngeal lymph node (RLN) and medulla oblongata at the level of the obex are early sites of PrPSC accumulation50 and considered gold standard tissues for postmortem CWD detection using immunohistochemistry (IHC). Peripheral accumulation and the excretion of the infective prion protein have been thought to occur only after central nervous system replication and was associated with the time of clinical disease manifestation.53 However, recent findings identified shedding in excreta concurrent with peripheral lymphoid accumulation.54 Newer antemortem detection methods such as serial protein misfolding cyclic amplification (sPMCA) and real-time quaking-induced conversion (RT-QuIC) are emerging as potential tools to detect low levels of PrPSC in excreta and identify early accumulation of PrPSC in peripheral tissue of sub-clinical CWD cases.49,55
A slow wasting process that leads to death characterizes CWD. Clinical signs—objective evidence of disease—include polydipsia and polyuria (excessive thirst or urination), sialorrhea (drooling or excessive salivation) and wasting (drastic weight loss). Behavioral changes include listlessness, aggression, lack of fear of people and depression.53 Signs of neurological damage at later stages are characterized by a lack of coordination, difficulty moving and ataxia (losing balance while walking). Other distinctive characteristics associated with CWD are drooping head and ears. The development of signs and symptoms—subjective evidence of disease—is progressive, with some of them such as polyuria and sialorrhea, appearing at later stages of disease and contributing to the shedding of the pathogenic PrPSC. Diagnosis based on clinical signs and behavioral changes is not possible, as these can be characteristics of other diseases.
Genetics
Eliminating or controlling the spread of TSEs has relied heavily on a solid understanding of the molecular mechanisms of the disease. Earlier work demonstrated that a pathogenic prion protein (PrPSc) is responsible for post-translational conversion of the host encoded cellular prion protein (PrPC) in several TSEs.16,24,56 The PRNP gene which encodes the PrP protein is well conserved among mammals,57 including cervids, and has implications for CWD.58 Though other loci were examined for their involvement (such as “Sinc”59 and Pid-160), variations in the prion protein (PRNP) gene have been shown to affect TSE progression61 and susceptibility.62–64 Due to this association, much of the research on the genetics of CWD has focused on this locus within affected cervids.
Complete genetic resistance has not yet been found for cervids, though examination of PRNP sequences has identified variable sites that may influence an individual animal’s susceptibility to or the rate of progression of CWD. The inferred amino acid sequence was described in Rocky Mountain elk (Cervus elaphus nelsoni), finding only a single mutation of methionine (M) to leucine (L) at cervid codon 132 with elk homozygous (M/M) overrepresented among those infected with CWD.65 Later studies demonstrated that the L mutation causes an increase in the incubation times.66 Few studies have been published involving moose (Alces alces) and reindeer (Rangifer tarandus) due to the rarity of naturally occurring cases of CWD in the wild.67 Among moose variable sites have been identified at codon 36 (N-asparagine or T-threonine),68 109 (K-lysine or Q-glutamine), 90 and 209 (M-methionine or I-isoleucine);69,70 however, it is unclear what protective qualities these mutations may or may not have for CWD infection. Mule deer and white-tailed deer have been studied extensively, likely due to the higher prevalence of CWD among these species.
Studies of the PRNP gene in mule deer and white-tailed deer have identified a number of mutations both in the amino acid and nucleotide sequences. Initial studies were complicated by an unexpressed process pseudogene in some but not all individuals.71 Further study of the pseudogene did not reveal any effects on CWD infection in deer and the presence of an asparagine mutation at codon 138 is absent in the functional gene, thereby easily distinguishing the two.72 The functional PRNP gene has been studied extensively, notably two coding mutations have been identified in the inferred amino acid sequence that has been linked to reduced CWD susceptibility. Examination of allele frequencies found few CWD-infected individuals with a substitution of histidine (H) for glutamine (Q) at codon 95 (aaQ95H) or a substitution of serine (S) for guanine (G) at codon 96 (aaG96S).72,73 The effects of these mutations were examined experimentally by orally infecting captive white-tailed deer with known genotypes finding that these mutations delay onset of CWD. Deer genotypes in this study included wild type (aa95QQ/aa96GG, N=6), heterozygous at aa95 only (aa95QH/aa96GG, N=1), aa96 only (aa95QQ/aa96GS N=4) or heterozygous for both positions (aa95QH/aa96GS, N=1).74 All deer presented with clinical signs of CWD (with the exception of two deer euthanized due to intercurrent disease); wild type genotypes having an average incubation period of 693 (± 27) days, those with only the aa96 mutations lasting 956 (± 107) days, and those with only the aa95 or both aa95 and 96 mutation genotypes succumbing to the disease after 1508 and 1596 days (respectively).74
Examination of complete PRNP nucleotide sequence corroborates these previous findings of CWD susceptibility and further the understanding of the role of this gene in disease management. Kelly et al75 found ten polymorphic sites in the PRNP gene from free roaming deer in Illinois (N = 196 deer, 76 CWD-positives and 120 CWD-negative). This study identified both of the nonsynonymous mutations (aa95 or nt285, and aa96 or nt286) confirming previous findings and identified three additional synonymous mutations which were determined to be more common among deer testing negative for CWD. Similarly, Wilson et al76 examined both white-tailed deer and mule deer in Canada finding fifteen variable sites among white-tailed deer and two variable sites in mule deer. Of these variable sites, only one nonsynonymous mutation (nt286) and four synonymous mutations were determined to be associated with CWD susceptibility in white-tailed deer,76 and one nonsynonymous (nt59) and one synonymous (nt393) mutation was identified in mule deer, each found significantly more often in mule deer testing negative for CWD.76
When the combined effects of synonymous and nonsynonymous mutations were considered from white-tailed deer in Illinois and Wisconsin twenty-six unique haplotypes were identified consisting of fourteen polymorphic sites (ten previously reported75,76 and four novel).77,78 Two haplotypes designated C and F were found less frequently among deer testing positive for CWD. Each haplotype contains one of the nonsynonymous mutations reported to reduce CWD susceptibility as well as one synonymous mutation. Haplotype C includes the nonsynonymous mutation nt286A (aa96S) and one synonymous at nt555T, and haplotype F contains the nonsynonymous mutation nt285C (aa95H) and one synonymous at nt60T. Unlike scrapie, no study has identified mutations that confer complete genetic resistance to CWD. Brandt et al77 found that deer with either the C or F haplotypes were less likely to be infected with CWD but still detected positive deer possessing these haplotypes.77,78 No studies have examined the protective effects of these mutations with regard to infectious dose of the prion protein.
The absence of complete genetic resistance to CWD does not preclude the use of genetics as a tool to manage the disease. Several studies analyzing white-tailed deer landscape and population genetics in Wisconsin and Illinois have led to a better understanding of deer movement patterns and other dynamics that may influence the spread of CWD. Deer were genotyped using microsatellite loci finding that population structure was largely influenced by female philopatry79 and landscape features can promote or inhibit movement thus influencing disease spread.80–82 The frequency of protective PRNP haplotypes may contribute to population level susceptibility and shape the way CWD spreads across the landscape. In Illinois where populations have higher frequencies of the protective C or F haplotypes, the geographic progression of the disease was slowed and confined to a smaller area.78 Control of CWD may require a multifactorial approach where genetic profiles can assist in the management of CWD.
Prevalence And Distribution
The origin of TSEs, and thus CWD, is not clear. Speculative theories suggest that TSEs might have a spontaneous origin, however, these theories are not proven.83,84 Prevalence of CWD varies across North America, reaching 30% for free-ranging populations in endemic areas,53 but can be, in unusual circumstances, as high as 80–90% in captive populations.54 Initial endemic zones were limited to northeastern Colorado and southeastern Wyoming, with eventual growth to southeastern Wisconsin, extending east to New York and West Virginia, and southward to New Mexico.85,86 Since the first report in captive mule deer in Colorado nearly 50 years ago,1,54 CWD in North America has spread to 26 US states and three provinces in Canada (Saskatchewan, Alberta and Quebec; Figure 2).87 The first cases of CWD outside North America were reported by the year 2000 in South Korea, after import of subclinical CWD infected farmed elk from Canada.88,89 Recently, CWD cases have been found in free-ranging reindeer and moose in Norway, Finland and Sweden.67,87,90 CWD’s expanding geographic distribution has been attributed to both natural movements of free-ranging cervids, as well as anthropogenic movement of infected farmed elk and deer. Movement of animal carcasses and other animal byproducts that are known to be infectious under experimental conditions—including natural cervid urine lures and antler velvet—may be involved in facilitating the spread of CWD.25,91
 |
Figure 2 Reported distribution of Chronic Wasting Disease (CWD) in North America. By 2019, 26 states and 3 Canadian provinces have reported CWD cases in captive and free-ranging cervid populations. Notes: Credit to Bryan Richards, USGS National Wildlife Health Center (Public domain).160 |
Since the initial report of CWD in mule deer (Odocoileus hemionus) in a Colorado research facility in the late 1960s,1 many captive and free-ranging cervid populations have been affected, and, by the time of writing this paper, the known host of CWD has grown to include moose (Alces alces), North American elk (Cervus canadensis and Cervus elaphus elaphus, also known as wapiti), white-tailed deer (Odocoileus Virginianus), red deer (Cervus elaphus), sika deer (Cervus nipon), reindeer (Rangifer tarandus), European moose (E. alces alces, also known as Eurasian Elk), mule deer (Odocoileus hemionus) and subspecies black-tailed deer (O. hemionus columbianus; Figure 3).92 Although the spread of CWD is well understood, no conclusive evidence to demonstrate a link between CWD in North America and CWD in European cervids has been established.67 This is mainly due to the lack of understanding of the origins of CWD coupled with no evidence of CWD in the cervid population in the European Union prior to 2016,93 when the first case of CWD in Europe was discovered in a free-ranging Norwegian reindeer.91 Still, prevalence of CWD in North America, Europe and possibly other parts of the world is unknown.
 |
Figure 3 Chronological identification of CWD in cervid species. Notes: Data from Haley and Hoover (2015),54 Benestad and Telling (2018),52 Ricci et al (2017),67 Chronic Wasting disease Alliance.92 |
Prevalence estimates are susceptible to the number of deer tested, representing the occurrence of disease in the tested population in a geographic region at a particular time. Based on hunter-harvested animal surveillance programs (1996–1999), CWD prevalence in an endemic area in Colorado was estimated at approximately 5% in mule deer, 2% in white-tailed deer and <1% in elk.52 By 2018, CWD rates of infection were estimated to occur in about one-third of Colorado’s elk population and about half of the state’s deer population.94 In Wisconsin, CWD prevalence in white-tailed deer doubled in some areas during a period of 6 years (2011–2016), with approximately 40–50% adult males and 20–30% adult females infected.95 Despite reports of increasing rates of CWD in specific locations in the US, surveillance data from other endemic areas indicate that CWD prevalence rates have remained low and changed little over long periods of time.96 This is the case in Illinois, where surveillance and management strategies were implemented and sustained since the first case of CWD was detected in 2002. Although prevalence rates found in Illinois in 2018 were lower than previous years, it was recognized that it is too early to suggest that this trend will continue. Thus, long-term surveillance and CWD management strategies need to continue to slowdown the spread of disease and the increase in prevalence rates to parts of the state that remain CWD free.96
Prevalence is influenced by biotic factors, such as sex and age, as well as abiotic factors associated to geographic location (eg, soil and pH characteristic). Trends in prevalence in endemic areas in Wisconsin have increased during the last 17 years, showing a rise in the prevalence from 8–10% to over 35% in adult males and from 3–4% to over 15% in adult females at the western monitoring area; during the same period of time the trends in prevalence increased from 2% to 13% in male yearlings and from 2% to 10% in yearling females.97 In Illinois, age and sex have been found to be associated with differences in prevalence. The mean prevalence rates during 2003–2018 have been 75% higher in males than in females;96 with higher rates in adult deer (1.93%) than in yearlings (0.89%) and fawns (0.45%); and higher rates of CWD in males than females, although, in this study, sex difference was not significant (P=0.079).98 More recently, the overall CWD prevalence in adult deer was estimated at 0.84% and was twice as high in males (1.07%) compared to females (0.54%).96
Horizontal transmission is the most important route of CWD infection52 and the most important contributing factor for CWD prevalence and incidence. Miller et al41 demonstrated that naïve animals could contract CWD when using sites where previous CWD-infected animals were housed. Prions enter the environment via decaying carcasses and excretion of bodily fluids that have been identified as containing high levels of PrPSC. Blood and saliva are the biological fluids with highest PrPSC levels, followed by fecal matter and urine, thus carrying high levels of infectivity. Other peripheral organs that accumulate large numbers of PrPSC include adrenal glands, thyroid glands, lungs, liver, kidneys, bladder, pancreas, gastrointestinal tract, retina, antler velvet, heart, tongue and skeletal muscle.52 Prions from all these tissues can enter the environment and remain infectious for long periods of time.41,99,100 Soils and other fomites acting as environmental reservoirs (eg, mineral licks) contribute to horizontal transmission.101
Because of cervid grazing behaviors, infection can be acquired via soil ingestion or soil inhalation, and by contact with bioavailable PrPSC from biological material in soil. It is not surprising that, because of this, much research in recent years has focused on soil properties (eg, organic matter, clay content, soil metals and soil pH) and its contribution to PrPSC persistence in the environment.98,100,102–109 For instance, attachment of the prion proteins to minerals in clay may limit migration of the infectious CWD protein through the soil column, maintaining infectious PrPSC at the soil surface, contributing to CWD dissemination.103 While some of these soil characteristics may influence PrPSC stability, persistence in the environment and infectivity,100,104–106 others—such as natural oxidants and soil humic acids—may interfere with conversion of PrPC to the pathogenic PrPSC,107 or degrade PrPSC and reduce CWD infectivity.108
Modeling studies based on CWD cases from surveillance programs have evaluated landscape features related to deer habitat and soil characteristics that could be environmental determinants for CWD risk, influencing prion availability and persistence.98,109–111 Evaluation of spatio-temporal patterns of CWD reported cases in white-tailed deer at the border of Wisconsin and Illinois—one of the hot spot areas for CWD in the US—found that landscape features such as larger and more compact forest, as well as lower elevation areas closer to rivers, were associated with higher risk of CWD; yet, other study found areas with small forest patches increased the risk of CWD occurrence.110 A geographical model focused on soil characteristics and its contribution to CWD in free-ranging deer found percent of clay and soil pH as the two most important predictors of the persistent presence of CWD in endemic areas.109 This is in agreement with findings by O’Hara-Ruiz et al98 that indicated that less clay and more sand enhance CWD persistence and transmission. Still, while some studies agree that more clay is associated with less CWD, others have found the opposite106 or no association to CWD incidence.112
Beside soil, other common environmental materials including wood, rocks, plastic, glass, cement, stainless steel, aluminum and grass plants have been proven to “bind, retain and release” prions.113,114 In the case of plants, these can act as carriers of infection by binding infectious prions from contaminated secretions, as well as by uptake of prions from contaminated soils, and mobilizing them to aerial parts of the plants including steam and leaves.113 After five decades of CWD research, many factors that influence CWD prevalence have been identified and several lines of evidence have expanded our understanding of how CWD spreads in nature. Nonetheless, many questions remain and significant challenges need to be addressed in order to effectively control CWD prevalence and reduce incidence at different geographic locations.
Impact
Chronic wasting disease has an ecologic, economic and social effect, with deep impact on the viability of cervid populations. An experimental study found a 60% decline in full-term viable offspring born to CWD-positive muntjac dams.48 Modeling studies have shown an annual population decline of 10.4% in white-tailed deer and 21% in sympatric mule deer populations in southeastern Wyoming, corroborating the population-limiting impact of CWD.115,116 Survival estimates indicate that CWD-infected mule deer were 4.5 more likely to die annually compared to CWD-negative deer116 and were more susceptible to predation than uninfected deer.117 The impact on elk populations in endemic areas in Colorado and South Dakota has also shown declines in survival118 and decrease in population growth rates.119 Conversely, the impact of CWD in low-density deer populations differs from places with high-density populations. Mule deer living in arid San Andres Mountains—part of the Chihuahuan Desert-range in southern New Mexico—showed weak population effects based on CWD prevalence and mortality data.120 Models reveal mixed results in long-term survival of cervid populations based on observed epidemics in endemic areas of Wisconsin, Colorado and Wyoming. Outcomes ranged from small host declines to moderate epidemics, and in some cases, to complete host extinction.121 Captive cervid facilities have been impacted by CWD, with over 175 herd facilities affected across the US and reported infection rates as high as 80% at some of these facilities.122 The extent of disease impact in other parts of the world is less understood. Because of the limited surveillance across Europe—especially in remote areas—it is not possible to exclude the possibility that CWD has been affecting cervids across Europe for decades.91,93
Another consideration of the potential impact of CWD is the risk to human health. Even though the only demonstrated zoonotic TSE is variant Creutzfeldt–Jakob disease (vCJD), which resulted from non-experimental transmission of classic bovine spongiform encephalopathy (BSE) from cattle to humans,123 no absolute molecular barrier to conversion of the human prion protein by the CWD prion protein has been found.124 Experimental studies and epidemiological investigations, coupled with careful surveillance, established a link between vCJD and BSE. Nevertheless, ongoing surveillance and epidemiological studies of humans living in CWD-endemic areas in North America and Canada have not shown any increases in human TSE cases,123,125 and have not been able to find associations between CWD and prion diseases in humans. Laboratory and epidemiological data support the role of a species barrier protecting humans from CWD.126–131
Experimental studies using humanized transgenic mice did not result in CWD transmission,127 and Raymond et al126 demonstrated a barrier at the molecular level that appears to limit the susceptibility of humans, cattle and sheep to CWD. Yet, susceptibility to CWD has been shown in cattle, cats, sheep and goats under experimental conditions following intracerebral inoculation.132 Oral inoculations, on the other hand, have been inefficient at inducing disease, suggesting a high species barrier under oral exposure. Only recently oral inoculation was successful at inducing prion disease in squirrel monkeys and swine.130,131,133 In swine, the species barrier was relatively high, as only low amount of prions was found in brain and lymphoid tissue.133 Interestingly, CWD has not been successfully transmitted to Cynomolgus macaques, which are genetically closer to humans than squirrel monkeys.129,131,134
More recently, the zoonotic potential of scrapie prions was demonstrated after serial transmission of different Scrapie isolates to humanized transgenic mice,135 coupled with the reported transmission of Scrapie prions to primates after long incubation periods of 10 years.136 Taking into account the long incubation periods—of a minimum of 5 years—that was required before clinical disease was observed after oral inoculation of Cynomolgus macaques with BSE, surveillance and research should continue and allow for long incubation periods to elucidate long-term effect of CWD in nonhuman primates and potential consequences to humans.
Beyond the direct impact of CWD on free-ranging cervid populations and potential effect on human health, there is an economic impact associated with management of CWD, and the effect of CWD on hunters and farmed cervid industry. For example, after the discovery and spread of CWD in North America, an estimated $32.3 million was spent by Wisconsin for CWD surveillance and management between 2001 and 2006.137 The potential economic losses per farm have been estimated at $290,000,138 and reached $53–$79 million in 2002 and $45–$72 million in 2003 for hunters in Wisconsin.139 Total depopulation was required at some captive cervid facilities, with costly government expenses associated with compensation.95 According to the 2016 National Survey of Fishing, Hunting, and Wildlife-Associated Recreation,140 an estimated 103 million Americans participated in fishing, hunting or other wildlife-associated recreational activity, spending $156.9 billion on equipment, travel, licenses and fees. Approximately 11.5 million were hunters, meaning that 4% of the Americans 16 years of age or older hunted in 2016. Revenues from hunting, fishing and wildlife-associated activities help to support wildlife and habitat conservation efforts. However, concerns about the potential and long-term impact of CWD to the cervid captive and wildlife populations, compounded with unknown risks of CWD transmission to humans, and evidence of risk of transmission to swine, could impact these revenues. CWD may reduce hunting and related activities in endemic areas, affecting the cost of management disease in areas where CWD becomes established. These, in turn, could affect jobs and communities that depend on the support of hunting and related activities across the nation.
Management Of CWD
Management guidelines for infectious diseases like CWD are difficult to develop and implement, as they need to account for factors that influence prevalence, incidence, transmission and geographic spread. Some of these factors include population dynamics, genetics, animal movement and dispersal, type of population (eg, captive or free-ranging cervids) and landscape characteristics (eg, forest areas or arid environments). Furthermore, the goals of proposed management and control strategies of CWD should be defined according to disease status in different regions; only then, strategies for control and/or prevention might be implemented. While depopulation of an infected herd followed by restocking after a period of 2 years is used for farmed deer,141 management intervention strategies for free-ranging populations are different; they consist of population reduction—to minimize disease transmission—and selective culling of deer in CWD endemic areas—to control CWD prevalence.142 These efforts require support from hunters and landowners so that the management can be applied. Eradication of CWD might not be realistic, but control is; once CWD has become established, management strategies should focus on limiting the growth of the number of infected individuals and therefore limit the increase in prevalence.
Farmed cervid CWD management programs in the US have been developed with the goal of creating a national approach to control CWD incidence and prevent spread between states. This is a collaborative effort among state regulatory agencies (eg, wildlife and animal health agencies), Animal and Plant Health Inspection Services (APHIS) and owners of farmed cervid facilities. The USDA-APHIS CWD management program includes a herd certification program that facilitates surveillance and interstate movement of non-infected animals. The program provides guidance on fence design, sampling strategies and response protocols if CWD is detected in a facility (eg, quarantine and carcass disposal, decontamination procedures and management of a herd during the epidemiological investigation).143 Depending on the epidemiological investigation, a herd could be classified as (a) CWD-positive (if an animal tested positive for CWD), (b) CWD-exposed (“if a CWD-positive animal resided in another herd (or multiple herds) within the previous 5 years”) or (c) epidemiologically linked herds (all herds with animals that were in contact with other animals that previously resided with a CWD-positive animal). If a herd is classified as CWD-positive or CWD-exposed, a quarantine of 5 years should be issued—based on the date the herd was last exposed to a CWD-positive animal—unless the herd is depopulated.143
The four CWD management strategies used in North American free-ranging wild deer include 1) general, non-selective population harvest (spatially targeted); 2) selective or targeted removal of clinical suspects (infected deer); 3) seasonal harvest (eg, summer) and 4) vaccination.144 Predictive models that evaluated these management strategies found that increased general hunting pressure with or without targeted sex group, the role of large predators, and seasonal hunting had some positive effect on CWD under specific conditions.145–147 Yet, analytical experimental studies that included vaccination (eg, intramuscular vaccination with two different prion peptide sequences) and oral administration of therapeutic compounds for prevention of CWD infection showed ineffective results.148,149 A study evaluating the use of mucosal immunization with an attenuated Salmonella vaccine expressing PrP found that the efficacy of the control measure was not clear.150 Two analytical observational studies based on planned culling as the intervention strategy found the control measure effective.144 Despite differences between intervention and control strategies, studies that evaluated differences between government culling and hunting, found that moderate but sustained intensity with continued and frequent culling is needed to reduce CWD prevalence.142 This effort minimizes the impact on recreational deer harvest.151 Furthermore, other studies suggested that management strategies focused on reducing population prevalence instead of deer abundance are more effective strategies in reducing CWD transmission.112
The objective of management and surveillance is to protect the health of captive and free-ranging herds from the spread of CWD, mitigate the negative consequences of reduced recreational hunting on the economy, decrease the geographic spread of CWD and reduce the potential of CWD prions to be transmitted to the environment, humans and any other species.98 Surveillance and monitoring of CWD provide essential data that help with the development of focused management strategies in endemic areas and guide direct management efforts. Moreover, they help with early detection of CWD, so timely dissemination of information and necessary action can be taken.67 For example, early detection of CWD in two captive herds and two wild deer in New York in 2005 prompted immediate actions that appear to have successfully mitigated CWD.152 However, for those regions where self-sustaining CWD epizootics continue to be a challenge, as in the state of Illinois, surveillance efforts have shown that continued intensive management, focused on specific areas infected with CWD, is a powerful management strategy that helps to keep disease prevalence low.98 There are two types of surveillance for CWD monitoring in free-ranging cervid populations: passive surveillance (which include the testing of road kills, dead, sick or suspect deer for CWD) and active surveillance (which include testing of hunter-harvested deer for CWD in target areas). Although knowledge gaps in the epidemiology of CWD still exist, only continued surveillance will inform CWD management and control strategies.
Science-based policies will help to develop effective management strategies that are relevant to the population monitored. The development of long-term sustainable management strategies is necessary in order to keep low prevalence and to avoid dissemination of CWD. Left unmanaged infection rates will affect the ability of cervidae herds to sustain themselves.94 CWD regulations to prevent further spread include restriction in translocation of captive cervids and movement of hunter-killed big game carcasses, high-risk tissues or bodily fluids that tend to concentrate high levels of PrPSC. Prion deposition in mineral licks was demonstrated in an enzootic area in Wisconsin, corroborating the participation of mineral licks as risk factors for CWD transmission, environmental reservoirs for CWD prions and as potential sources for cross-species contamination as they attract livestock and non-cervid wildlife species.101 The bans on baiting and feeding implemented in multiple states in North America and municipalities in Norway are crucial to reduce the congregation of animals and to reduce direct contact rates of susceptible with infected animals that in normal circumstances do not congregate and feed on the precise same small area.153 This regulatory ban helps to prevent direct transmission of infectious diseases like tuberculosis, brucellosis and CWD.
Conclusions
Chronic wasting disease (CWD) is a highly contagious prion disease that affects captive and free-ranging cervids. The infectious agent is the misfolded prion protein (PRPSC), which is primarily transmitted horizontally via direct contact between animals or indirectly through contact with infective secretions and contaminated fomites. CWD is epizootic in the US and continues to expand through North America; outbreaks in Europe are on the rise. Our current understanding of the long-term effect of CWD on free-ranging populations is still limited. However, we know the population-limiting impact of CWD, even at low prevalence rates it affects the possibility of a herd to thrive. CWD is a 100% fatal slowly progressive neurodegenerative disease, with long incubation periods wherein sub-clinical-infected animals contribute to the shedding of the pathogenic PrPSC. Given the longevity of infectious proteins in the environment and that the disease is in a free-roaming population, managers are unlikely to completely eliminate the disease, but they can control it. Some management approaches have helped sustain low CWD prevalence and slow the spread of the disease. Genetic tools identify animal movement patterns and population level susceptibility (ie, herd immunity), helping to contain or reduced areas affected by CWD. Yet, gaps in knowledge still exist. An improved understanding of population dynamics, deer behavior that influence CWD transmission among free-ranging cervids and prions in the environment is needed to facilitate CWD management. The effect of protective haplotypes that may be acting as a genetic barrier preventing the spread of CWD, potential therapeutic strategies that will help to protect and manage captive and free-ranging populations, as well as new tools for effective antemortem detection, environmental clean-up and prion protein degradation are all integral components of the future management of CWD. Regardless of the strategy, management of an infectious disease such as CWD is a joint responsibility that involves the government, state and local agencies, farmed cervid producers, hunters and the general public. The key to the success of CWD management in free-ranging deer involves public acceptance and a continual support and commitment to intervention. Only through ongoing scientific research and management based on scientific evidence can CWD be controlled. In the future, if a treatment or cure is identified, our chances to take advantage of those tools will be much better if CWD has been contained and prevalence rates are low.
Acknowledgments
We thank the landowners and the hunting communities for their efforts and willingness to participate in CWD surveillance, and their commitment to the scientific advances and management of CWD. The authors also thank Kerry L Helms for the prion protein illustration and Ramsés V Morales for reviewing the manuscript. This work was supported by the US Fish and Wildlife Service Federal Aid in Wildlife Restoration Project (W-146-R), the Illinois Natural History Survey-Prairie Research Institute and the University of Illinois Office of the Vice Chancellor for Research.
Disclosure
Nelda A Rivera and Nohra E Mateus-Pinilla report grants from the US Fish and Wildlife Service Federal Aid in Wildlife Restoration Project (W-146-R) during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Williams ES, Young S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis. 1980;16:89–98. doi:10.7589/0090-3558-16.1.89
2. Zabel MD, Reid C. A brief history of prions. Pathog Dis. 2015;73(9). doi:10.1093/femspd/ftv087
3. Creutzfeldt HG. Über eine eigenartige herdförmige Erkrankung des Zentralnervensystems (vorläufige Mitteilung). Z Ges Neurol Psychiat. 1920;57(1):1–18. doi:10.1007/BF02866081
4. Jakob A. Über eigenartige Erkrankungen des Zentralnervensystems mit bemerkenswertem anatomischem Befunde. Z Ges Neurol Psychiat. 1921;64:147–228. doi:10.1007/BF02870932
5. Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New Guinea: the endemic occurence of “kuru” in the native population. N Engl J Med. 1957;257(20):974–978. doi:10.1056/NEJM195711142572005
6. Eklund CM, Kennedy RC, Hadlow WJ. Pathogenesis of scrapie virus infection in the mouse. J Infect Dis. 1967;117:15–22. doi:10.1093/infdis/117.1.15
7. Gajdusek DC. Slow-virus infections of the nervous system. New Engl J Med. 1967;276:392–400. doi:10.1056/NEJM196702092760609
8. Gajdusek DC, Gibbs CJ. Slow, latent and temperate virus infections of the central nervous system. Res Publ Assoc Res Nerv Ment Dis. 1968;44:254–280.
9. Sigurdsson B. Rida, a chronic encephalitis of sheep: with general remarks on infections which develop slowly and some of their special characteristics. Brit Vet J. 1954;110:341–354. doi:10.1016/S0007-1935(17)50172-4
10. Hadlow WJ. Scrapie and kuru. Lancet. 1959;2:289–290. doi:10.1016/S0140-6736(59)92081-1
11. Klatzo I, Gajdusek DC, Zigas V. Pathology of Kuru. Lab Invest. 1959;4:799–847.
12. Alper T, Cramp WA, Haig DA, Clarke MC. Does the agent of scrapie replicate without nucleic acid? Nature. 1967;214(5090):764. doi:10.1038/214764a0
13. Griffith JS. Nature of the Scrapie agent: self-replication and scrapie. Nature. 1967;215:1043–1044. doi:10.1038/215714a0
14. Pattison IH, Jones KM. The possible nature of the transmissible agent of scrapie. Vet Rec. 1967;80:2–9. doi:10.1136/vr.80.1.2
15. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi:10.1126/science.216.4541.73
16. Büeler H, Aguzzi A, Sailer A, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73(7):1339–1347. doi:10.1016/0092-8674(93)90360-3
17. Bastian FO, Dash S, Garry RF. Linking chronic wasting disease to scrapie by comparison of Spiroplasma mirum ribosomal DNA sequences. Exp Mol Pathol. 2004;77(1):49–56. doi:10.1016/j.yexmp.2004.02.002
18. Broxmeyer L. Is mad cow disease caused by a bacteria? Med Hypotheses. 2004;63(4):731–739. doi:10.1016/j.mehy.2004.04.013
19. Manuelidis L. A 25 nm virion is the likely cause of transmissible spongiform encephalopathies. J Cell Biochem. 2007;100:897–915. doi:10.1002/jcb.21052
20. Alexeeva I, Elliott EJ, Rollins S, et al. Absence of Spiroplasma or other bacterial 16S rRNA genes in brain tissue of hamsters with scrapie. J Clin Microbiol. 2006;44(1):91–97. doi:10.1128/JCM.44.1.91-97.2006
21. French HM Characterization of Spiroplasma Mirum and Its Role in Transmissible Spongiform Encephalopathies [dissertation]. Louisiana State University and Agricultural and Mechanical College; 2011. Available from: https://digitalcommons.lsu.edu/cgi/viewcontent.cgi?referer=https://scholar.google.com/&httpsredir=1&article=4011&context=gradschool_dissertations.
22. Hamir AN, Greenlee JJ, Stanton TB, et al. Experimental inoculation of raccoons (Procyon lotor) with Spiroplasma mirum and transmissible mink encephalopathy (TME). Can J Vet Res. 2011;75(1):18–24.
23. Doherr MG. Brief review on the epidemiology of transmissible spongiform encephalopathies (TSE). Vaccine. 2007;25(30):5619–5624. doi:10.1016/j.vaccine.2006.10.059
24. Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95(23):13363–13383. doi:10.1073/pnas.95.23.13363
25. Angers RC, Seward TS, Napier D, et al. Chronic wasting disease prions in elk antler velvet. Emerg Infect Dis. 2009;15(5):696. doi:10.3201/eid1505.081458
26. Wulf MA, Senatore A, Aguzzi A. The biological function of the cellular prion protein: an update. BMC Biol. 2017;15(1):34. doi:10.1186/s12915-017-0375-5
27. Ford MJ, Burton LJ, Morris RJ, Hall SM. Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience. 2002;113(1):177–192. doi:10.1016/s0306-4522(02)00155-0
28. Haddon DJ, Hughes MR, Antignano F, Westaway D, Cashman NR, McNagny KM. Prion protein expression and release by mast cells after activation. J Infect Dis. 2009;200(5):827–831. doi:10.1086/605022
29. Peralta OA, Eyestone WH. Quantitative and qualitative analysis of cellular prion protein (PrPC) expression in bovine somatic tissues. Prion. 2009;3(3):161–170. doi:10.4161/pri.3.3.9772
30. Viegas P, Chaverot N, Enslen H, Perrière N, Couraud PO, Cazaubon S. Junctional expression of the prion protein PrPC by brain endothelial cells: a role in trans-endothelial migration of human monocytes. J Cell Sci. 2006;119(22):4634–4643. doi:10.1242/jcs.03222
31. Castle AR, Gill AC. Physiological functions of the cellular prion protein. Front Mol Biosci. 2017;4:19. doi:10.3389/fmolb.2017.00019
32. Moser M, Colello RJ, Pott U, Oesch B. Developmental expression of the prion protein gene in glial cells. Neuron. 1995;14(3):509–517. doi:10.1016/0896-6273(95)90307-0
33. Pan KM, Baldwin M, Nguyen J, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A. 1993;90(23):10962–10966. doi:10.1073/pnas.90.23.10962
34. Novakofski J, Brewer MS, Mateus-Pinilla N, Killefer J, McCusker RH. Prion biology relevant to bovine spongiform encephalopathy. J Anim Sci. 2005;83(6):1455–1476. doi:10.2527/2005.8361455x
35. Guiroy DC, Williams ES, Yanagihara R, Gajdusek DC. Topographic distribution of scrapie amyloid-immunoreactive plaques in chronic wasting disease in captive mule deer (Odocoileus hemionus hemionus). Acta Neuropathol. 1991;81(5):475–478. doi:10.1007/bf00310125
36. Aguzzi A. Prions and the immune system: a journey through gut, spleen, and nerves. Adv Immunol. 2003;81:123–172.
37. An introduction to prion biology and diseases. In Prusiner SB, editor. Cold Spring Harbor Monograph Archive. Vol. 41, second edition; 2004:1–87.
38. Miller MW, Williams ES. Prion disease: horizontal prion transmission in mule deer. Nature. 2003;425(6953):35. doi:10.1038/425035a
39. Sigurdson CJ, Williams ES, Miller MW, Spraker TR, O’Rourke KI, Hoover EA. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J Gen Virol. 1999;80(10):2757–2764. doi:10.1099/0022-1317-80-10-2757
40. Gough KC, Maddison BC. Prion transmission: prion excretion and occurrence in the environment. Prion. 2010;4(4):275–282. doi:10.4161/pri.4.4.13678
41. Miller MW, Williams ES, Hobbs NT, Wolfe LL. Environmental sources of prion transmission in mule deer. Emerg Infect Dis. 2004;10(6):1003. doi:10.3201/eid1006.040010
42. Schauber EM, Nielsen CK, Kjær LJ, Anderson CW, Storm DJ. Social affiliation and contact patterns among white-tailed deer in disparate landscapes: implications for disease transmission. J Mammal. 2015;96(1):16–28. doi:10.1093/jmammal/gyu027
43. Miller MW, Wild MA, Williams ES. Epidemiology of chronic wasting disease in captive Rocky Mountain elk. J Wildl Dis. 1998;34(3):532–538. doi:10.7589/0090-3558-34.3.532
44. Braun U, Tschuor A, Hässig M, et al. Protease-resistant prion protein (PrPres) in the blood of offspring of cows that developed BSE. Schweiz Arch Tierheilkd. 2009;151(9):433–436. doi:10.1024/0036-7281.151.9.433
45. Bencsik A, Debeer S, Petit T, Baron T. Possible case of maternal transmission of feline spongiform encephalopathy in a captive cheetah. PLoS One. 2009;4(9):e6929. doi:10.1371/journal.pone.0006929
46. Castilla J, Brun A, Díaz-San Segundo F, et al. Vertical transmission of bovine spongiform encephalopathy prions evaluated in a transgenic mouse model. J Virol. 2005;79(13):8665–8668. doi:10.1128/JVI.79.13.8665-8668.2005
47. Foster JD, Goldmann W, Hunter N. Evidence in sheep for pre-natal transmission of scrapie to lambs from infected mothers. PLoS One. 2013;8(11):e79433. doi:10.1371/journal.pone.0079433
48. Nalls AV, McNulty E, Powers J, et al. Mother to offspring transmission of chronic wasting disease in reeves’ muntjac deer. PLoS One. 2013;8(8):e71844. doi:10.1371/journal.pone.0071844
49. Selariu A, Powers JG, Nalls A, et al. In utero transmission and tissue distribution of chronic wasting disease-associated prions in free-ranging Rocky Mountain elk. J Gen Virol. 2015;96(Pt 11):3444. doi:10.1099/vir.0.069187-0
50. Fox KA, Jewell JE, Williams ES, Miller MW. Patterns of PrPCWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus). J Gen Virol. 2006;87(11):3451–3461. doi:10.1099/vir.0.81999-0
51. Spraker TR, Miller MW, Williams ES, et al. Spongiform encephalopathy in free-ranging mule deer (Odocoileus hemionus), white-tailed deer (Odocoileus virginianus) and Rocky Mountain elk (Cervus elaphus nelsoni) in northcentral Colorado. J Wildl Dis. 1997;33(1):1–6. doi:10.7589/0090-3558-33.1.1
52. Benestad SL, Telling GC. Chronic wasting disease: an evolving prion disease of cervids. In: Handbook of Clinical Neurology. Vol. 153; 2018:135–151. doi:10.1016/B978-0-444-63945-5.00008-8
53. Williams ES. Chronic wasting disease. Vet Pathol. 2005;42(5):530–549. doi:10.1354/vp.42-5-530
54. Haley NJ, Hoover EA. Chronic wasting disease of cervids: current knowledge and future perspectives. Annu Rev Anim Biosci. 2015;3(1):305–325. doi:10.1146/annurev-animal-022114-111001
55. Haley N, Richt J. Evolution of diagnostic tests for chronic wasting disease, a naturally occurring prion disease of cervids. Pathogens. 2017;6(3):35. doi:10.3390/pathogens6030035
56. Carlson GA, Kingsbury DT, Goodman PA, et al. Linkage of prion protein and scrapie incubation-time genes. Cell. 1986;46(4):503–511. doi:10.1016/0092-8674(86)90875-5
57. Van Rheede T, Smolenaars MM, Madsen O, de Jong WW. Molecular evolution of the mammalian prion protein. Mol Biol Evol. 2003;20(1):111–121. doi:10.1093/molbev/msg014
58. Robinson SJ, Samuel MD, O’Rourke KI, Johnson CJ. The role of genetics in chronic wasting disease of North American cervids. Prion. 2012;6(2):153–162. doi:10.4161/pri.19640
59. Dickinson AG, Meikle VMH, Fraser H. Identification of a gene which controls the incubation period of some strains of scrapie agent in mice. J Comp Pathol. 1968;78(3):293–299. doi:10.1016/0021-9975(68)90005-4
60. Kingsbury DT, Kasper KC, Stites DP, Watson JD, Hogan RN, Prusiner SB. Genetic control of scrapie and Creutzfeldt-Jakob disease in mice. J Immunol. 1983;131(1):491–496.
61. Carlson GA, Ebeling C, Yang S-L, et al. Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc Natl Acad Sci. 1994;91(12):5690–5694. doi:10.1073/pnas.91.12.5690
62. Baylis M, Goldmann W. The genetics of scrapie in sheep and goats. Curr Mol Med. 2004;4(4):385–396.
63. Roden JA, Nieuwhof GJ, Bishop SC, Jones DA, Haresign W, Gubbins S. Breeding programmes for TSE resistance in British sheep: I. Assessing the impact on prion protein (PrP) genotype frequencies. Prev Vet Med. 2006;73(1):1–16. doi:10.1016/j.prevetmed.2005.08.002
64. Collinge J, Palmer MS, Dryden AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob Disease. Lancet. 1991;337(8755):1441–1442. doi:10.1016/0140-6736(91)93128-v
65. O’Rourke KI, Besser TE, Miller MW, et al. PrP genotypes of captive and free-ranging Rocky Mountain elk (Cervus elaphus nelsoni) with chronic wasting disease. J Gen Virol. 1999;80(10):2679–2765. doi:10.1099/0022-1317-80-10-2765
66. O’Rourke KI, Spraker TR, Zhuang D, Greenlee JJ, Gidlewski TE, Hamir AN. Elk with a long incubation prion disease phenotype have a unique PrPd profile. Neuroreport. 2007;18(18):1935–1938. doi:10.1097/WNR.0b013e3282f1ca2f
67. Ricci A, Allende A, Bolton D, et al. Chronic wasting disease (CWD) in cervids. EFSA J. 2017;15(1).
68. Wik L, Mikko S, Klingeborn M, Stéen M, Simonsson M, Linné T. Polymorphisms and variants in the prion protein sequence of European moose (Alces alces), reindeer (Rangifer tarandus), roe deer (Capreolus capreolus) and fallow deer (Dama dama) in Scandinavia. Prion. 2012;6(3):256–260. doi:10.4161/pri.19641
69. Kreeger TJ, Montgomery DL, Jewell JE, Schultz W, Williams ES. Oral transmission of chronic wasting disease in captive Shira’s moose. J Wildl Dis. 2006;42(3):640–645. doi:10.7589/0090-3558-42.3.640
70. Baeten LA, Powers BE, Jewell JE, Spraker TR, Miller MW. A natural case of chronic wasting disease in a free-ranging moose (Alces alces shirasi). J Wildl Dis. 2007;43(2):309–314. doi:10.7589/0090-3558-43.2.309
71. Brayton KA, O’Rourke KI, Lyda AK, Miller MW, Knowles DP. A processed pseudogene contributes to apparent mule deer prion gene heterogeneity. Gene. 2004;326:167–173. doi:10.1016/j.gene.2003.10.022
72. O’Rourke KI, Spraker TR, Hamburg LK, Besser TE, Brayton KA, Knowles DP. Polymorphisms in the prion precursor functional gene but not the pseudogene are associated with susceptibility to chronic wasting disease in white-tailed deer. J Gen Virol. 2004;85(5):1339–1346. doi:10.1099/vir.0.79785-0
73. Johnson C, Johnson J, Vanderloo JP, Keane D, Aiken JM, McKenzie D. Prion protein polymorphisms in white-tailed deer influence susceptibility to chronic wasting disease. J Gen Virol. 2006;87:2109–2114. doi:10.1099/vir.0.81615-0
74. Johnson CJ, Herbst A, Duque-Velasquez C, et al. Prion protein polymorphisms affect chronic wasting disease progression. PLoS One. 2011;6(3):e17450. doi:10.1371/journal.pone.0017450
75. Kelly AC, Mateus-Pinilla NE, Diffendorfer J, et al. Prion sequence polymorphisms and chronic wasting disease resistance in Illinois white-tailed deer (Odocoileus virginianus). Prion. 2008;2(1):28–36. doi:10.4161/pri.2.1.6321
76. Wilson GA, Nakada SM, Bollinger TK, Pybus MJ, Merrill EH, Coltman DW. Polymorphisms at the PRNP gene influence susceptibility to chronic wasting disease in two species of deer (Odocoileus Spp.) in western Canada. J Toxicol Environ Health A. 2009;72(17–18):1025–1029. doi:10.1080/15287390903084264
77. Brandt AL, Kelly AC, Green ML, Shelton P, Novakofski J, Mateus-Pinilla NE. Prion protein gene sequence and chronic wasting disease susceptibility in white-tailed deer (Odocoileus virginianus). Prion. 2015;9(6):449–462. doi:10.1080/19336896.2015.1115179
78. Brandt AL, Green ML, Ishida Y, Roca AL, Novakofski J, Mateus-Pinilla NE. Influence of the geographic distribution of prion protein gene sequence variation on patterns of chronic wasting disease spread in white-tailed deer (Odocoileus virginianus). Prion. 2018;12(3–4):204–215. doi:10.1080/19336896.2018.1474671
79. Kelly AC, Mateus-Pinilla NE, Douglas M, et al. Utilizing disease surveillance to examine gene flow and dispersal in white-tailed deer. J Appl Ecol. 2010;47(6):1189–1198. doi:10.1111/j.1365-2664.2010.01868.x
80. Blanchong JA, Samuel MD, Scribner KT, Weckworth BV, Langenberg JA, Filcek KB. Landscape genetics and the spatial distribution of chronic wasting disease. Biol Lett. 2008;4(1):130–133. doi:10.1098/rsbl.2007.0523
81. Kelly AC, Mateus-Pinilla NE, Brown W, et al. Genetic assessment of environmental features that influence deer dispersal: implications for prion-infected populations. Popul Ecol. 2014;56(2):327–340. doi:10.1007/s10144-013-0427-9
82. Green ML, Manjerovic MB, Mateus-Pinilla N, Novakofski J. Genetic assignment tests reveal dispersal of white-tailed deer: implications for chronic wasting disease. J Mammal. 2014;95(3):646–654. doi:10.1644/13-MAMM-A-167
83. Baron T, Vulin J, Biacabe AG, et al. Emergence of classical BSE strain properties during serial passages of H-BSE in wild-type mice. PLoS One. 2011;6(1):e15839. doi:10.1371/journal.pone.0015839
84. Ortiz-Peláez A, Arnold ME, Vidal-Diez A. Epidemiological investigations on the potential transmissibility of a rare disease: the case of atypical scrapie in Great Britain. Epidemiol Infect. 2016;144(10):2107–2116. doi:10.1017/S0950268816000303
85. Sigurdson CJ, Aguzzi A. Chronic wasting disease. Biochim Biophys Acta. 2007;1772(6):610–618. doi:10.1016/j.bbadis.2006.10.010
86. Sigurdson CJ. A prion disease of cervids: chronic wasting disease. Vet Res. 2008;39(4):1–12. doi:10.1051/vetres:2007039
87. Expanding Distribution of Chronic Wasting Disease [USGS]. National Wildlife Health Center. 2019. Available from: https://www.usgs.gov/centers/nwhc/science/expanding-distribution-chronic-wasting-disease?qt-science_center_objects=0#qt-science_center_objects.
88. Kim TY, Shon HJ, Joo YS, Mun UK, Kang KS, Lee YS. Additional cases of chronic wasting disease in imported deer in Korea. J Vet Med Sci. 2005;67(8):753–759. doi:10.1292/jvms.67.753
89. Sohn HJ, Kim JH, Choi KS, et al. A case of chronic wasting disease in an elk imported to Korea from Canada. J Vet Med Sci. 2002;64(9):855–858. doi:10.1292/jvms.64.855
90. Pirisinu L, Tran L, Chiappini B, et al. Novel type of chronic wasting disease detected in Moose (Alces alces), Norway. Emerg Infect Dis. 2018;24(12):2210–2218. doi:10.3201/eid2412.180702
91. Benestad SL, Mitchell G, Simmons M, Ytrehus B, Vikøren T. First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet Res. 2016;47(1):88. doi:10.1186/s13567-016-0375-4
92. Chronic Wasting Disease Alliance [cwd-info.org]. Timeline. 2019. Available from: http://cwd-info.org/timeline/.
93. EFSA Panel on Biological Hazards (BIOHAZ). Scientific opinion on the results of the EU survey for Chronic Wasting Disease (CWD) in cervids. EFSA J. 2010;8(10):1861. doi:10.2903/j.efsa.2010.1861
94. Chronic Wasting Disease [Colorado Parks and wildlife]. Recent trends and implications in Colorado. 2018. Available from: https://cpw.state.co.us/Documents/Commission/2018/Feb/Item_17-CWP-Feb2018-PWCMtg.pdf.
95. Carlson CM, Hopkins MC, Nguyen NT, Richards BJ, Walsh DP, Walter WD. Chronic wasting disease—status, science, and management support by the U.S. Geological Survey: U.S. Geological Survey Open-File Report 2017–1138. 2018. doi:10.3133/ofr20171138
96. Dufford D, McDonald P. Illinois Chronic Wasting Disease: 2017–2018 Surveillance and Management Report. Springfield (IL): Illinois Department of Natural Resources; 2018. Available from: https://www.dnr.illinois.gov/programs/CWD/Documents/CWDAnnualReport20172018.pdf.
97. Prevalence and surveillance [Wisconsin Department of Natural Resources]. CWD prevalence in Wisconsin. Available from: https://dnr.wi.gov/topic/wildlifehabitat/prevalence.html.
98. O’Hara Ruiz M, Kelly AC, Brown M, Novakofski JE, Mateus-Pinilla NE. Influence of landscape factors and management decisions on spatial and temporal patterns of the transmission of chronic wasting disease in white-tailed deer. Geospat Health. 2013;8(1):215–227. doi:10.4081/gh.2013.68
99. Haley NJ, Seelig DM, Zabel MD, Telling GC, Hoover EA. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS One. 2009;4(3):e4848. doi:10.1371/journal.pone.0004848
100. Saunders SE, Bartz JC, Bartelt-Hunt SL. Soil-mediated prion transmission: is local soil-type a key determinant of prion disease incidence? Chemosphere. 2012;87(7):661–667. doi:10.1016/j.chemosphere.2011.12.076
101. Plummer IH, Johnson CJ, Chesney AR, Pedersen JA, Samuel MD. Mineral licks as environmental reservoirs of chronic wasting disease prions. PLoS One. 2018;13(5):e0196745. doi:10.1371/journal.pone.0196745
102. Rivera NA, Novakofski J, Weng HY, et al. Metals in obex and retropharyngeal lymph nodes of Illinois white-tailed deer and their variations associated with CWD status. Prion. 2015;9(1):48–58. doi:10.1080/19336896.2015.1019194
103. Johnson CJ, Pedersen JA, Chappell RJ, McKenzie D, Aiken JM. Oral transmissibility of prion disease is enhanced by binding to soil particles. PLoS Pathog. 2007;3(7):e93. doi:10.1371/journal.ppat.0030093
104. Davies P, Brown DR. Manganese enhances prion protein survival in model soils and increases prion infectivity to cells. PLoS One. 2009;4(10):e7518. doi:10.1371/journal.pone.0007518
105. Smith CB, Booth CJ, Pedersen JA. Fate of prions in soil: a review. J Environ Qual. 2011;40:449–461.
106. Walter WD, Walsh DP, Farnsworth ML, Winkelman DL, Miller MW. Soil clay content underlies prion infection odds. Nat Commun. 2011;2:200. doi:10.1038/ncomms1203
107. Russo F, Johnson CJ, Johnson CJ, McKenzie D, Aiken JM, Pedersen JA. Pathogenic prion protein is degraded by a manganese oxide mineral found in soils. J Gen Virol. 2009;90:275–280. doi:10.1099/vir.0.003251-0
108. Kuznetsova A, Cullingham C, McKenzie D, Aiken JM. Soil humic acids degrade CWD prions and reduce infectivity. PLoS Pathog. 2018;14(11):e1007414. doi:10.1371/journal.ppat.1007414
109. Dorak SJ, Green ML, Wander MM, et al. Clay content and pH: soil characteristic associations with the persistent presence of chronic wasting disease in northern Illinois. Sci Rep. 2017;7(1):18062. doi:10.1038/s41598-017-18321-x
110. Evans TS, Kirchgessner MS, Eyler B, Ryan CW, Walter WD. Habitat influences distribution of chronic wasting disease in white‐tailed deer. J Wildl Manage. 2016;80(2):284–291. doi:10.1002/jwmg.1004
111. Nobert BR, Merrill EH, Pybus MJ, Bollinger TK, Hwang YT. Landscape connectivity predicts chronic wasting disease risk in Canada. J Appl Ecol. 2016;53(5):1450–1459. doi:10.1111/1365-2664.12677
112. Storm DJ, Samuel MD, Rolley RE, et al. Deer density and disease prevalence influence transmission of chronic wasting disease in white‐tailed deer. Ecosphere. 2013;4(1):1–4. doi:10.1890/ES12-00141.1
113. Pritzkow S, Morales R, Moda F, et al. Grass plants bind, retain, uptake, and transport infectious prions. Cell Rep. 2015;11(8):1168–1175. doi:10.1016/j.celrep.2015.04.036
114. Pritzkow S, Morales R, Lyon A, Concha-Marambio L, Urayama A, Soto C. Efficient prion disease transmission through common environmental materials. J Biol Chem. 2018;293(9):3363–3373. doi:10.1074/jbc.M117.810747
115. DeVivo MT. Chronic Wasting Disease Ecology and Epidemiology of Mule Deer in Wyoming [dissertation]. Laramie, USA: University of Wyoming; 2015.
116. Edmunds DR, Kauffman MJ, Schumaker BA, et al. Chronic wasting disease drives population decline of white-tailed deer. PLoS One. 2016;11(8):e0161127. doi:10.1371/journal.pone.0161127
117. Miller MW, Swanson HM, Wolfe LL, et al. Lions and prions and deer demise. PLoS One. 2008;3(12):e4019. doi:10.1371/journal.pone.0004019
118. Monello RJ, Powers JG, Hobbs NT, Spraker TR, Watry MK, Wild MA. Survival and population growth of a free‐ranging elk population with a long history of exposure to chronic wasting disease. J Wildl Manage. 2014;78(2):214–223. doi:10.1002/jwmg.665
119. Sargeant GA, Weber DC, Roddy DE. Implications of chronic wasting disease, cougar predation, and reduced recruitment for elk management. J Wildl Manage. 2011;75(1):171–177. doi:10.1002/jwmg.27
120. Bender LC, Rodden CL, Mathis P, et al. Impacts of chronic wasting disease on a low density mule deer (Odocoileus hemionus) population in the San Andres Mountains, Chihuahuan Desert, New Mexico. Acta Zool Mex. 2019;35(1):1. doi:10.21829/azm.2019.3502203
121. Almberg ES, Cross PC, Johnson CJ, Heisey DM, Richards BJ. Modeling routes of chronic wasting disease transmission: environmental prion persistence promotes deer population decline and extinction. PLoS One. 2011;6(5):e19896. doi:10.1371/journal.pone.0019896
122. Keane DP, Barr DJ, Bochsler PN, et al. Chronic wasting disease in a Wisconsin white-tailed deer farm. J Vet Diag Invest. 2008;20(5):698–703. doi:10.1177/104063870802000534
123. Houston F, Andréoletti O. The zoonotic potential of animal prion diseases. Handb Clin Neurol. 2018;153:447–462. doi:10.1016/B978-0-444-63945-5.00025-8
124. Barria MA, Balachandran A, Morita M. Molecular barriers to zoonotic transmission of prions. Emerg Infect Dis. 2014;20(1):88. doi:10.3201/eid2001.130858
125. Waddell L, Greig J, Mascarenhas M, Otten A, Corrin T, Hierlihy K. Current evidence on the transmissibility of chronic wasting disease prions to humans—a systematic review. Transbound Emerg Dis. 2018;65(1):37–49. doi:10.1111/tbed.12612
126. Raymond GJ, Bossers A, Raymond LD, et al. Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J. 2000;19(17):4425–4430. doi:10.1093/emboj/19.17.4425
127. Kong Q, Huang S, Zou W. Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J Neurosci. 2005;25(35):7944–7949. doi:10.1523/JNEUROSCI.2467-05.2005
128. Mussil B, Motzkus D, Hesse G, et al. Transmission of CWD to non-human primates: interim results of a 6 year risk assessment study on the transmissibility to humans. Prion. 2015;9:S87–S87.
129. Race B, Williams K, Orrú CD, Hughson AG, Lubke L, Chesebro B. Lack of transmission of chronic wasting disease to cynomolgus macaques. J Virol. 2018;92(14):e00550–18. doi:10.1128/JVI.00550-18
130. Marsh RF, Kincaid AE, Bessen RA, Bartz JC. Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus). J Virol. 2005;79(21):13794–13796. doi:10.1128/JVI.79.21.13794-13796.2005
131. Race B, Meade-White KD, Phillips K, Striebel J, Race R, Chesebro B. Chronic wasting disease agents in nonhuman primates. Emerg Infect Dis. 2014;20(5):833. doi:10.3201/eid2005.130778
132. Gilch S, Chitoor N, Taguchi Y, Stuart M, Jewell JE, Schätzl HM. Chronic wasting disease. Top Curr Chem. 2011;305:51–77.
133. Moore SJ, Greenlee MH, Kondru N, et al. Experimental transmission of the chronic wasting disease agent to swine after oral or intracranial inoculation. J Virol. 2017;91(19):e00926–17. doi:10.1128/JVI.00926-17
134. Hayasaka K, Gojobori T, Horai S. Molecular phylogeny and evolution of primate mitochondrial DNA. Mol Biol Evol. 1988;5(6):626–644. doi:10.1093/oxfordjournals.molbev.a040524
135. Cassard H, Torres JM, Lacroux C, et al. Evidence for zoonotic potential of ovine scrapie prions. Nat Commun. 2014;5:5821. doi:10.1038/ncomms5972
136. Comoy EE, Mikol J, Luccantoni-Freire S. Transmission of scrapie prions to primate after an extended silent incubation period. Sci Rep. 2015;5:11573. doi:10.1038/srep11573
137. Wisconsin Legislative Audit Bureau. An evaluation: Chronic wasting disease, Department of Natural Resources: Wisconsin Legislative Audit Bureau Report. 2006. Available from: http://legis.wisconsin.gov/lab/reports/06-13full.pdf.
138. Arnot C, Laate E, Unterschultz J, Adamowicz W. Chronic wasting disease (CWD) potential economic impact on cervid farming in Alberta. J Toxicol Environ Health A. 2009;72(17–18):1014–1017. doi:10.1080/15287390903084223
139. Bishop RC. The economic impacts of chronic wasting disease (CWD) in Wisconsin. Hum Dimens Wildl. 2004;9(3):181–192. doi:10.1080/10871200490479963
140. Fish and Wildlife Service (US) editors. 2016 National Survey of Fishing, Hunting and Wildlife-Associated Recreation. Fish & Wildlife Service; 2018.
141. Mysterud A, Edmunds DR. A review of chronic wasting disease in North America with implications for Europe. Eur J Wildl Res. 2019;65(2):26. doi:10.1007/s10344-019-1260-z
142. Mateus-Pinilla N, Weng HY, Ruiz MO, Shelton P, Novakofski J. Evaluation of a wild white-tailed deer population management program for controlling chronic wasting disease in Illinois, 2003–2008. Prev Vet Med. 2013;110(3–4):541–548. doi:10.1016/j.prevetmed.2013.03.002
143. CWD herd certification program standard [USDA-APHIS]. Chronic Wasting Disease program standards. 2019. Available from: https://www.aphis.usda.gov/animal_health/animal_diseases/cwd/downloads/cwd-program-standards.pdf.
144. Uehlinger FD, Johnston AC, Bollinger TK, Waldner CL. Systematic review of management strategies to control chronic wasting disease in wild deer populations in North America. BMC Vet Res. 2016;12(1):173. doi:10.1186/s12917-016-0813-6
145. Potapov A, Merrill E, Lewis MA. Wildlife disease elimination and density dependence. Proc Biol Sci. 2012;279(1741):3139–3145. doi:10.1098/rspb.2012.0520
146. Wasserberg G, Osnas EE, Rolley RE, Samuel MD. Host culling as an adaptive management tool for chronic wasting disease in white‐tailed deer: a modeling study. J Appl Ecol. 2009;46(2):457–466. doi:10.1111/j.1365-2664.2008.01576.x
147. Wild MA, Hobbs NT, Graham MS, Miller MW. The role of predation in disease control: a comparison of selective and nonselective removal on prion disease dynamics in deer. J Wildl Dis. 2011;47(1):78–93. doi:10.7589/0090-3558-47.1.78
148. Pilon JL, Rhyan JC, Wolfe LL, et al. Immunization with a synthetic peptide vaccine fails to protect mule deer (Odocoileus hemionus) from chronic wasting disease. J Wildl Dis. 2013;49(3):694–698. doi:10.7589/2012-07-200
149. Wolfe LL, Kocisko DA, Caughey B, Miller MW. Assessment of prospective preventive therapies for chronic wasting disease in Mule Deer. J Wildl Dis. 2012;48(2):530–533. doi:10.7589/0090-3558-48.2.530
150. Goñi F, Mathiason CK, Yim L, et al. Mucosal immunization with an attenuated Salmonella vaccine partially protects white-tailed deer from chronic wasting disease. Vaccine. 2015;33(5):726–733. doi:10.1016/j.vaccine.2014.11.035
151. Manjerovic MB, Green ML, Mateus-Pinilla N, Novakofski J. The importance of localized culling in stabilizing chronic wasting disease prevalence in white-tailed deer populations. Prev Vet Med. 2014;113(1):139–145. doi:10.1016/j.prevetmed.2013.09.011
152. Evans TS, Schuler KL, Walter WD. Surveillance and monitoring of white-tailed deer for chronic wasting disease in the northeastern United States. J Fish Wildl Manage. 2014;5(2):387–393. doi:10.3996/032014-JFWM-021
153. Mysterud A, Viljugrein H, Solberg EJ, Rolandsen CM. Legal regulation of supplementary cervid feeding facing chronic wasting disease. J Wildlife Manage. 2019:1–9. doi:10.1002/jwmg.21746
154. Sivitz LB, Erdtmann R, editors. Advancing Prion Science: Guidance for the National Prion Research Program. National Academies Press; 2004.
155. Spickler AR. Chronic wasting disease. 2016 (Last Updated). Available from: http://www.cfsph.iastate.edu/Factsheets/pdfs/chronic_wasting_disease.pdf.
156. Imran M, Mahmood S. An overview of animal prion diseases. Virol J. 2011;8(1):493. doi:10.1186/1743-422X-8-493
157. Spickler AR Feline spongiform encephalopathy; 2016 (Last Updated). Available from: http://www.cfsph.iastate.edu/Factsheets/pdfs/feline_spongiform_encephalopathy.pdf.
158. Babelhadj B, Di Bari MA, Pirisinu L, et al. Prion disease in dromedary camels, Algeria. Emerg Infect Dis. 2018;24(6):1029. doi:10.3201/eid2408.171312
159. Helms KL. PrPC and PrPSC structures. Wildlife Veterinary Epidemiology Laboratory – Illinois Natural History Survey. Available from: https://publish.illinois.edu/wildlifevetlab/projects/chronic-wasting-disease/.
160. Richards B. Distribution of Chronic Wasting Disease in North America. United States Geological Survey (USGS) National Wildlife Health Center. Available from: https://www.usgs.gov/media/images/distribution-chronic-wasting-disease-north-america-0.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.