")
Back to Journals » Journal of Pain Research » Volume 8
Chronic pain in hypermobility syndrome and Ehlers–Danlos syndrome (hypermobility type): it is a challenge
Authors Scheper M, de Vries J, Verbunt J, Engelbert R
Received 19 March 2015
Accepted for publication 11 June 2015
Published 20 August 2015 Volume 2015:8 Pages 591—601
DOI https://doi.org/10.2147/JPR.S64251
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Michael Schatman
Mark C Scheper,1,2 Janneke E de Vries,1–3 Jeanine Verbunt,3,4 Raoul HH Engelbert1,2
1School of Physiotherapy, Amsterdam University of Applied Sciences, Amsterdam, 2Department of Rehabilitation, Academic Medical Center, University of Amsterdam, Amsterdam, 3Department of Rehabilitation Medicine, CAPHRI School for Public Health and Primary Care, Maastricht University, Maastricht; 4Adelante, Center of expertise in Rehabilitation and Audiology, Hoensbroek, the Netherlands
Abstract: Generalized joint hypermobility (GJH) is highly prevalent among patients diagnosed with chronic pain. When GJH is accompanied by pain in ≥4 joints over a period ≥3 months in the absence of other conditions that cause chronic pain, the hypermobility syndrome (HMS) may be diagnosed. In addition, GJH is also a clinical sign that is frequently present in hereditary diseases of the connective tissue, such as the Marfan syndrome, osteogenesis imperfecta, and the Ehlers–Danlos syndrome. However, within the Ehlers–Danlos spectrum, a similar subcategory of patients having similar clinical features as HMS but lacking a specific genetic profile was identified: Ehlers–Danlos syndrome hypermobility type (EDS-HT). Researchers and clinicians have struggled for decades with the highly diverse clinical presentation within the HMS and EDS-HT phenotypes (Challenge 1) and the lack of understanding of the pathological mechanisms that underlie the development of pain and its persistence (Challenge 2). In addition, within the HMS/EDS-HT phenotype, there is a high prevalence of psychosocial factors, which again presents a difficult issue that needs to be addressed (Challenge 3). Despite recent scientific advances, many obstacles for clinical care and research still remain. To gain further insight into the phenotype of HMS/EDS-HT and its mechanisms, clearer descriptions of these populations should be made available. Future research and clinical care should revise and create consensus on the diagnostic criteria for HMS/EDS-HT (Solution 1), account for clinical heterogeneity by the classification of subtypes within the HMS/EDS-HT spectrum (Solution 2), and create a clinical core set (Solution 3).
Keywords: chronic musculoskeletal pain, generalized joint hypermobility, hypermobility syndrome, Ehlers–Danlos hypermobility type
Introduction
In 1967, the first scientific observation on the association between chronic musculoskeletal pain and hypermobile joints, also known as generalized joint hypermobility (GJH), was documented.1,2 The presence of GJH is traditionally scored by the Beighton score. The Beighton score is a standardized test consisting of five clinical maneuvers performed bilaterally, in which a positive score in ≥4 joints indicates the presence of GJH. When GJH is accompanied by chronic musculoskeletal pain, lasting for longer than 3 months in ≥4 joints, hypermobility syndrome (HMS) may be diagnosed. A prevalence rate of 41% has been reported initially by Kirk et al2,3 for HMS, whereas in later publications, the prevalence ranged between 2% and 89%.4 Pain is often characterized from mild to severe, in the absence of any other condition that could account for the chronic musculoskeletal pain.5 The diagnosis of HMS is derived from the Brighton criteria, a valid set of clinical criteria and morphological features in terms of concurrent validity6 and reproducibility (Table 1: left panel).7 The clinical presentation of HMS not only encompasses the combination of pain and GJH (major criteria) but also includes abnormalities in other body structures such as the skin (hyperextensibility and smooth, velvety skin), marfanoid habitus, and other signs of connective tissue laxity (minor criteria).3,5,8 HMS may be established when a patient is positive on two major criteria, one major with two minor criteria, four minor criteria, or two minor when a first-degree relative is affected by HMS (Table 1).
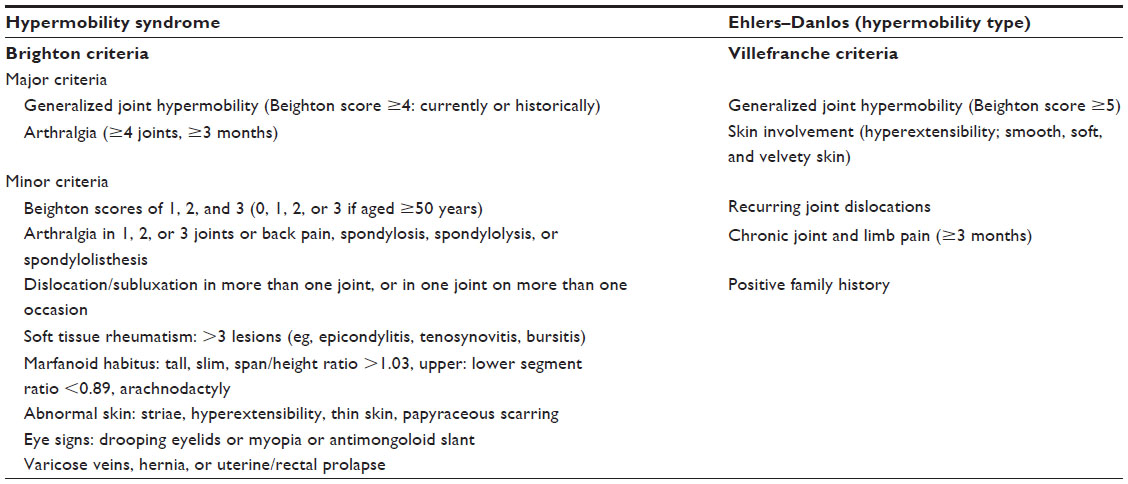 | Table 1 Brighton and Villefranche diagnostic criteria |
When considering the multisystemic nature of HMS, it shows overlap with other hereditary diseases of the connective tissue (HDCT) such as the Marfan syndrome, osteogenesis imperfecta, Loeys–Dietz syndrome, or Ehlers–Danlos syndrome (classical and vascular type).9–11 These serious and sometimes life-threatening conditions have, besides the high incidence of GJH, also highly recognizable clinical features such as aortic dilatation. Within the Ehlers–Danlos phenotype, a subgroup of patients who shared the same clinical features but also lacked specific genetic profiles, also known as Ehlers–Danlos syndrome hypermobility type (EDS-HT), was identified.12 The diagnosis of EDS-HT is based on the Villefranche criteria (Table 1: right panel). Within the Villefranche criteria, the presence of GJH (Beighton score ≥5) is also a major criterion, as is the presence of hyperextensible, smooth, and velvety skin.8 There are differences between the diagnostic criteria for HMS and EDS-HT. In the Villefranche criteria, the emphasis is focused on laxity in terms of GJH (at a cutoff level of ≥5 instead of ≥4) and skin hyperextensibility. In contrast to the Brighton criteria, less emphasis is put on chronic musculoskeletal pain, joint dislocations, and morphological features such as marfanoid habitus or varicose veins.5 Despite the differences in diagnostic criteria, the similarities between conditions have created the notion that both HMS and EDS-HT may constitute the same pathological entity. In contradiction to the before-mentioned HDCT, both HMS and EDS-HT lack specific genetic profiles by which the diagnosis can be confirmed.3,9
GJH is a prevalent clinical characteristic that is present in 2%–57% of the healthy population6 and is dependent on age, sex, and ethnicity.3,13 Not all individuals with GJH develop chronic complaints. It has been estimated that among individuals with GJH (when accounting for age, ethnicity, and current diagnostic criteria), approximately 3.3% of women and 0.6% of men will develop chronic complaints in terms of HMS/EDS-HT.2,3,14 However, these incidence rates may be underestimated due to the general unawareness of clinicians regarding GJH-related chronic pain4,15 and due to the lack of consensus on the operationalization of measures within the diagnostic criteria for HMS/EDS-HT.10 For some, the nature of complaints is temporary and for others, GJH is even a predisposition for high-level athletic performance (eg, dance, martial arts, and gymnastics).3,16,17 Similarly, in these individuals, dysfunctions are present in terms of muscle weakness, pain, fatigue, psychological distress, and sometimes even mild disability. These individuals succeed in keeping the condition manageable and may even excel in human performance.16,18,19 The reasons why individuals with GJH develop complaints remain elusive and are highly debated.
When considering the highly prevalent nature of GJH and the potential severity of disability that it is associated with HMS and EDS-HT, it remains surprising that these diagnoses are often overlooked and often not even considered. Patients tend to search for years within the medical system before a diagnosis is established.4 In children, late diagnosis might even add to the severity of complaints and disability at later ages.3,20,21 Even within society, the diagnoses of HMS and EDS-HT are often received with skepticism and disbelieved due to lack of awareness of these diseases, which further adds to the suffering of patients. In addition, the absence of objective diagnostic markers also adds to the societal unawareness for these individuals.
The first objective of this topical review is to provide an overview of the major challenges within hypermobility-related patient care and research, focusing on chronic pain. The second objective is to propose and describe potential solutions for the development of more accurate diagnostic procedures and more effective treatment.
Part 1: challenges
Challenge 1: clinical heterogeneity and subtypes
From a diagnostic perspective, only one type of HMS/EDS-HT was historically distinguished.22 When considering the highly prevalent nature of GJH, and the (estimated) incidence rate of HMS/EDS-HT, not all subjects with GJH become patients. It has been acknowledged that one of the trademarks of HMS/EDS-HT is the clinical heterogeneity;23 however, this can be problematic for clinicians during diagnosis and treatment.4 The presence of clinical heterogeneity could imply the existence of subtypes within the spectrum of HMS/EDS-HT (Figure 1).24,25
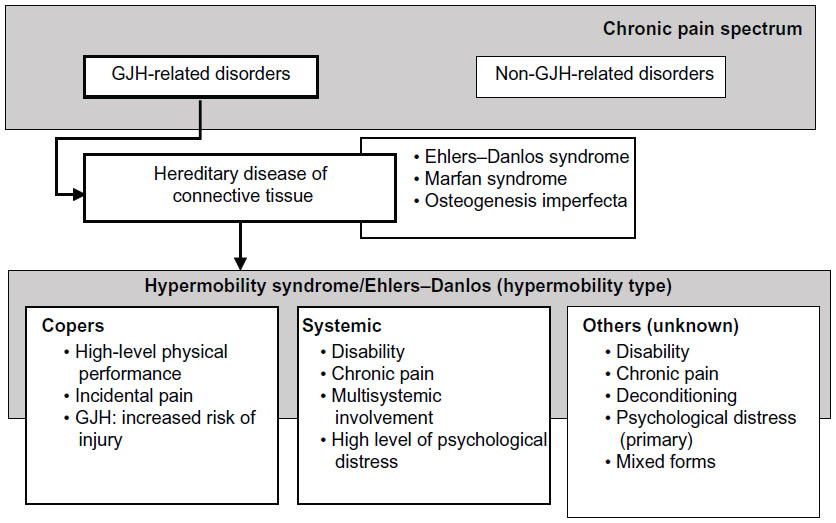 | Figure 1 Clinical heterogeneity. |
For some, GJH is not seen as a clinical sign that is associated with disease. GJH is highly prevalent among athletes, dancers, martial artists, and gymnasts, as also among other professionals in performing arts, such as musicians.26 Although these groups are more at risk of developing musculoskeletal pain, they are still able to demonstrate high levels of physical performance and motor control. This group can be designated as “copers.”16 Copers can be defined as subjects with GJH, but without chronic musculoskeletal complaints lasting longer than 3 months. However, this does not necessarily indicate that these individuals are symptom free.
High-performance athletes are more susceptible to musculoskeletal injury, in general, due to performance-related trauma or overload (strain injury).27 Although high levels of physical performance alone could account for a higher injury rate,18 among individuals with GJH, profound effects on health status,16–18 in terms of musculoskeletal pain, fatigue, deconditioning, and psychological distress, have been reported.16–18 Although these individuals spent a lot of effort on physical training, GJH was still associated with decreased muscle strength and endurance, as well as lower functional capacities.16,17 Although continuous musculoskeletal complaints seem to be present, they are less profound and more periodically present. The strategies on how “copers” manage these complaints and maintain functional status remain elusive but could be vital for the understanding on why some individuals develop chronicity.
Among patients with HMS/EDS-HT, distinct subgroups have been identified in the clinical presentation in which nonmusculoskeletal symptoms are dominant: dysautonomia, intestinal complaints, reflux, and other organ-related dysfunction.28,29 In this group, musculoskeletal pain and psychological distress were also significantly higher. This group could be designated as “systemic.”25
Although not all patients do present systemic symptoms, the remaining patients are characterized by a mixture of psychological and biomechanical factors that could be related to pain; however, evidence is still very limited.
In general, literature does seem to indicate the existence of subtypes within the HMS/EDS-HT phenotype. However, the literature does have limitations, as the information available is only limited to the existence of specific clusters of symptoms/patient characteristics and not all included subjects were categorized into one single cluster. This may imply the existence of other subtypes. In addition, when considering the clinical heterogeneity, sample sizes were small. Further research in larger populations is essential.
Challenge 2: lacking evidence on pathological mechanism
Although many theories have been proposed regarding why individuals with GJH develop chronic pain, no single pathophysiological model has been identified. The fol- lowing paragraph discusses the most dominant theories (Figure 2).
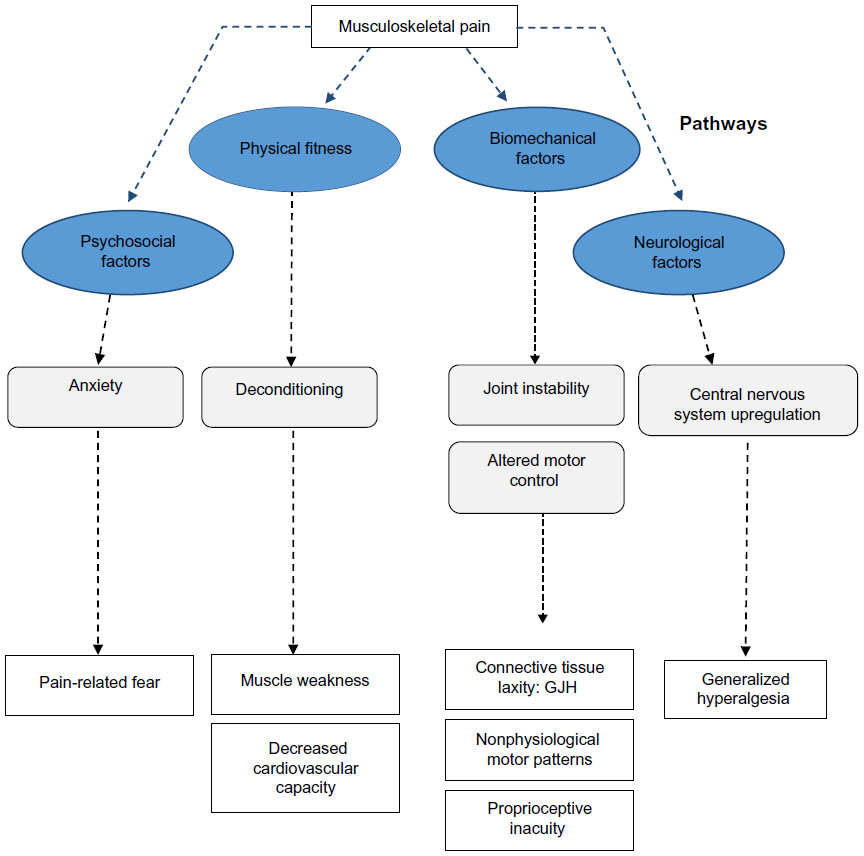 | Figure 2 Potential pathways of chronic pain in GJH-related disorders. |
Biomechanical and physical determinants of musculoskeletal pain
The primary hypothesis regarding the development of musculoskeletal complaints is localized biomechanical overload during activity, with a high risk of repetitive trauma.3 Generalized joint instability may cause the occurrence of microtraumas on joint surfaces, leading to adaptation and compensation of movement patterns, consequently causing overload in other areas of the movement apparatus.30
Recent evidence in subjects with GJH proves that wal- king patterns are different.31–34 In children and adults, measures of joint stability, for instance, of the trunk and head, have shown decreased lateral joint stability.33 However, different stabilization strategies (kinetic patterns) have also been shown.31,32,34,35 In children36 with nonsymptomatic GJH, only deviations in gait kinematics were observed in terms of lowered joint momentum of the lower extremity. Similar findings were also shown in adults.37 Moreover, altered neuromuscular activation patterns have been reported.1,38 These findings support the assumption that GJH has direct consequences for joint biomechanics; however, the clinical importance of these findings remains unknown. In HMS and EDS-HT, nonphysiological gate patterns (absolute comparisons: spatiotemporal, kinematics, and kinetics) have been demonstrated in children and adults. These patterns were not consistent and a high variability among patients was present.31,32,34,35 In addition, soft tissue laxity could also contribute to localized biomechanical overload of tendons and other soft tissue structures.39,40 Although it is assumed that soft-tissue laxity is an important factor for both GJH and the observed musculoskeletal pain, only limited evidence is available.
Another important factor within the biomechanical pathway in HMS/EDS-HT patients may be proprioceptive inacuity, which has been suggested to be important for the occurrence of gait abnormalities and musculoskeletal pain.3,41 Proprioception provides the brain with positional information, originating from joint capsules and tendons, which is of importance for automated processes such as joint stabilization.41 However, the importance of proprioceptive inacuity in relation to musculoskeletal pain, as for the occurrence of gait abnormalities, remains unknown. Associations between reduced joint forces and musculoskeletal fatigue have been established; however, most studies are of cross-sectional nature with high variability in terms of both biomechanical determinants and musculoskeletal complaints.
Although evidence is inconclusive, it can be assumed that biomechanical factors do play a role in the pathological mechanism leading to the development of complaints. The exact mechanism and its consequences are unclear and may even be population specific (GJH in elite sports versus other populations with GJH).
Deconditioning
In children and adults with HMS, preventing and reducing deconditioning is currently the rationale for physical interventions. Enhancing physical fitness, in terms of endurance and muscle strength training, are effective treatment strategies for pain reduction.3,20 Overuse injuries occur with minimal provocation and may lead to activity impairment to preserve joints. This results in “pay later” behavior for participating in certain activities, characterized by overactivity, followed by underactivity to recover. Consequently, there is a downward spiral of less activity due to fear and more pain with less provocation, leading to deconditioning.42
Muscle weakness was observed in not only HMS/EDS- HT patients30,43–46 but also among subjects with GJH without chronic musculoskeletal pain.16,17 The presence of decreased muscle strength in high-level athletes with GJH indicates that muscle weakness is not solely the result of deconditioning.16,17,46 The amount of force a muscle can generate is dependent not only on the contraction force of muscle fibers but also on the elasticity of tendons and intrinsic muscle structures.39 The elasticity of the connective tissue within muscle structures is important for the transfer of muscle force, from contracting fibers and fascia through tendons and joints. Before joint momentum can be generated, the elasticity has to be overcome, which leads to loss of some amount of the generated muscle force. Although connective tissue laxity cannot be directly influenced by physical training, it could be hypothesized that some degree of muscle weakness is a natural phenomenon.16,47 Rombaut et al39 confirmed the existence of increased tendon laxity, which is in line with the assumption that altered structural integrity of connective tissue is an independent factor that explains some of the observed muscle weakness. When comparing the differences in muscle strength between subjects with HMS/EDS-HT and those with nonsymptomatic GJH, subjects diagnosed with HMS/EDS-HT have higher levels of muscle weakness. These observations might imply that pain negatively interferes with muscle testing in patients with chronic pain. However, it could also imply that deconditioning could be explained by the difference between HMS/EDS-HT and nonsymptomatic GJH.3,16,17,47
In addition, other factors such as proprioception41 and reflex inhibition48 have also been implicated as factors causing reduced muscle strength.49 In adults with EDS-HT, muscle atrophy has been observed, which might not only affect muscle strength but may also decrease the amount of proprioceptive fibers, with a negative effect on proprioception.44
Reduced endurance has also been demonstrated in both HMS/EDS-HT and nonsymptomatic forms of GJH.16,45 However, its contribution to the development of chronic pain or functional decline is unclear. Cardiovascular function is dependent on the efficiency of the movement apparatus to convert energy into sustained momentum; still, much is unclear on its importance for this specific group of patients. Systematic disruption of homeostatic function, such as downregulated metabolism, blood pressure instability, and gastric intestinal dysfunction, have been documented. Cardiovascular dysfunction could be related to the extent of the multisystemic problems that are frequently seen in this category of patients.3,21,28,29
Neurological mechanisms
The presence of generalized hyperalgesia in adult patients with EDS-HT has been described recently.50 Subjects with EDS-HT had considerably lower pressure thresholds in symptomatic and asymptomatic areas, compared to healthy controls. Similar results have been shown in subjects diagnosed with HMS.51 It was hypothesized that central-oriented upregulating processes are present within the central nervous system. Due to centralized sensitization, subjects with HMS/EDS-HT may be more susceptible to pain and fatigue. Recently, signs of upregulation of the central nervous system were found in individuals diagnosed with HMS.15 Furthermore, in patients with HMS and EDS-HT, a reduced effectiveness of (lidocaine-based) analgesia was observed,52 which is also in line with the central nervous system upregulation. In addition, more severe pain medications such as opiates are frequently prescribed, but still, these patients can experience high levels of pain and discomfort.53 In concurrence with this discussion, other literature on chronic pain has also identified a role of central nervous system sensitization in the chronicity of pain.54
The reasons for why upregulation of brain sensitivity does occur in patients with HMS and EDS-HT are unknown. This could be a time-dependent process in which pain sensitivity thresholds become lowered as pain flare-ups occur, until the point that only minimal provocation will cause a central-oriented pain response. Pain becomes chronic from a neurological origin. Although these observations were recorded in a cross-sectional study, it was hypothesized that the process of pain-induced central nervous system upregulation could cause the onset of chronicity. If central sensitization is an adaptation of the central nervous system to counter adverse events due to joint instability (and the lack of proprioceptive feedback), this adaptation could be viewed as a compensatory mechanism and might even be protective.
To date, much is unclear on the role of the central nervous system in this group of patients in terms of the clinical relevance of lowered pain thresholds, as well as treatment with medication.
Challenge 3: psychosocial determinants of musculoskeletal pain
HMS can lead to considerable impairments in daily life functioning. However, the impact of HMS on daily life performance seems not solely explained by a person’s level of hypermobility. Higher Beighton scores alone do not automatically account for more impairments in daily life functioning. As in other chronic pain syndromes, it seems that apart from biomedical factors, psychosocial factors also contribute to a person’s eventual level of disability. In the literature on adult chronic pain, the fear-avoidance model has been introduced to explain the disabling role of pain-related fear55 and has been confirmed by numerous studies.55–57 It states that highly fearful persons who tend to think in terms of catastrophes will avoid activities they perceive as harmful or pain provoking. In the long term, this avoidance behavior can result in disability and depression, further fueling the vicious circle of disabling musculoskeletal pain. This disabling effect of pain-related fear on the ability to perform physical activities/movements has recently also been confirmed in adolescents with pain.58,59 In highly anxious adolescents, anxiety instead of pain appeared to be associated with restrictions in social and physical functioning.60
It could be that pain-related fear will also have an accumulating disabling effect in hypermobile persons with pain. In case of a new onset of musculoskeletal pain, fear of pain will trigger avoidance of painful muscle contractions, leading to submaximal muscle performance. For persons with joint hypermobility, submaximal muscle performance will, however, have the immediate negative consequence that their compensation mechanism, essential for joint stability, will fail. Functional consequences, as impaired balance and lower balance confidence, will further fuel fear of movement and catastrophizing thoughts about pain and vice versa. In fearful hypermobile patients, a painful stimulus can thus, in the very short term, lead to a high level of disability, depression, and disuse.
The high prevalence scores of both anxiety and joint hypermobility in patients with musculoskeletal pain could indicate that this hypothesized mechanism may explain disability in a substantial subgroup of patients. A finding that seems to support a common pathway for hypermobility and anxiety is an increased prevalence score for joint hypermobility in patient populations with other anxiety-related problems. Thus, 61.8% of patients with a panic disorder appeared to be hypermobile.61 In a recent cohort study, persons with joint hypermobility had an increased risk for developing a panic disorder or simple phobia.62 In addition, the severity of joint hypermobility and anxiety even appeared positively associated.16,62 Whether fear of injury is indeed associated with the occurrence of hypermobility and whether both phenomena indeed negatively interact with disabling consequences for daily life functioning are currently unknown.
Part 2: potential solutions and topics for further research
For clinicians, the diagnostic process can be viewed as challenging due to the current diagnostic criteria and due to the complex patient profiles. The lack of standardization for some of the main clinical features (pain and connective tissue laxity) and the lack of inclusion of other important clinical trademarks of HMS and EDS-HT are the most frequently mentioned problems in clinical practice. In addition to the issues regarding diagnostics, the lack of consensus on treatment or guidelines on which treatment can be tailored is also problematic. Even for experienced physicians, it remains hard to correctly identify patients and to determine which factors should be modified to get positive treatment outcomes. Increasing recognition of this specific phenotype of musculoskeletal pain would be the first, and an essential, step enabling the development of new and more effective treatment programs. This could be achieved by revising diagnostic criteria and establishing international consensus (Solution 1), accounting for clinical heterogeneity (Solution 2), and development of a clinical core set (Solution 3).
Solution 1: revising diagnostic criteria and establishing international consensus
Currently, the diagnostic criteria for GJH, HMS, and EDS-HT are highly debated, with uncertainty for researchers, clinicians, and patients. Although these diagnostic criteria have been in place for decades, there are still many controversies surrounding these criteria, which are still not addressed.23
It is assumed that GJH is an expression of generalized connective tissue laxity, in which joint capsules, ligaments, tendons, and muscle structures are hyperextensible.4,22,47 Therefore, in both HMS and EDS-HT, the presence of GJH according to the Brighton criteria is a primary clinical feature (Table 1). The Beighton score is considered the golden standard from infancy to old age and has been the most used instrument to classify GJH. Although several studies confirmed good psychometric characteristics in terms of reliability and face validity, there is considerable variation among the test procedures described. This variation concerns not only the practical instruction of how to perform the various tests but, more importantly, also the cutoff level for a positive test and the definition of GJH.63 It remains unclear which cutoff level is the most appropriate. In the current classification, a score of ≥4 is considered to be the minimum level for GJH, independent of age, sex, and ethnicity.6 Other scores of ≥5, ≥6, and ≥7 have also been suggested; nevertheless, the validity of these cutoff values is debated. Recent studies have shown that a Beighton score of ≥6 at the age of 10 years is a predictor for pain recurrence and persistence at 14 years,64–66 and a Beighton score of ≥6 at the age of 14 years is a predictor for general pain at 18 years of age.67 This would suggest that higher cutoff scores of ≥5 would be more appropriate. However, with increasing age, joint laxity decreases, which may imply that a cutoff level of ≥4 may be more appropriate.68 In addition, sex- and ethnicity-specific effects on joint laxity have been documented and should also be incorporated into the classification of GJH. Within the current diagnostic criteria, no such distinction is made. Furthermore, there is little knowledge on the natural course of GJH with increasing age, which also complicates clinical diagnostic procedures.
Similarly, other features of connective tissue laxity have been suggested for incorporation in the diagnostic process, such as the involvement of the skin (hyperelasticity, soft silk-like skin, scarring, and bruising), multisystemic dysfunction (obstipation, incontinence, gastrointestinal pain, and reflux), and blood vessel hyperelasticity (low blood pressure, orthostatic intolerance, and dysautonomia). Currently, only skin features are incorporated in the diagnostic criteria for HMS and EDS-HT. These measures of skin elasticity and morphological features are not well documented and also lack standardization, despite the availability of more objective measures and clearer descriptions of the disease-specific deviation of skin morphology in HMS and EDS-HT patients.69,70
Pain and fatigue are considered to be the most prevalent and dominant symptoms in HMS and EDS-HT. However, only arthralgia is included as a diagnostic measure. Regarding pain, only the number of pain locations and the duration have been considered, without a specified method of assessment. When considering the dominance of pain within this phenotype, the use of more standardized pain measures is warranted. Other aspects of pain, in terms of severity, may be beneficial when considering the diagnosis of HMS and EDS-HT. The addition of fatigue to the diagnostic criteria may also be beneficial, as it is considered one of the most disabling HMS-related complaints by many patients.71
There is much discussion on the overlap between the diagnostic criteria for HMS and EDS-HT. When comparing both Brighton and Villefranche criteria, there seems to be considerable overlap between both phenotypes.12 It has been suggested that both diagnoses may be synonymous; it still may be that the pathways that cause the onset of pain and functional decline are different. However, when considering the potential existence of subtypes within both HMS and EDS-HT, one might consider the usefulness of having two different classification systems for a disease that is characterized by large heterogeneity in clinical presentation. Although many authors, clinicians, and research faculties have stressed the need for a revision of the diagnostic criteria, no changes in the diagnostic criteria have been considered yet.
Solution 2: accounting for clinical heterogeneity
It has been recognized that the heterogeneity of clinical manifestation of HMS/EDS-HT and related syndromes proves to be problematic. Patients can differ in terms of morphological features and severity of complaints; complaints may occur episodically with minimal provocation or may even migrate through the human movement apparatus. The variability in the clinical presentation might also be one of the defining trademarks. Using subgroup classifications, the variability could be reduced and even may result in more possibilities for effective disease management. However, currently only limited information on the different types of subgroups is available and no formal subclassification can be made. Future research may focus on the identification of subgroups in disability needing differentiated forms of treatment. As has been done before for back pain, identification of subgroups in disability (for back pain based on the STarTBack Tool) can lead to the possibility of providing a stepped-care approach in the treatment for HMS/EDS-HT based on the level of subject disability.
Solution 3: development of clinical core set
The incidence of GJH and HMS fluctuates around the world due to the impact of ethnicity, age, and differences in diagnostic criteria and procedures. To compare patients from different environments (topological, social), with different genetic backgrounds, and influenced by various psychosocial factors, a standardized set of clinical measures with adequate psychometric properties should be available. This will not only enable clinicians and researchers to compare results around the world, it can eventually lead to potential improvement in patient care. The development of a clinical core set would enable creation of disease-specific reference values.
Currently, there is great abundance of generic clinical measures that are being used within research; therefore, the use of a common framework seems to be recommended. The International Classification of Functioning (ICF) could serve as the basic framework for the development of a clinical core set for adults, whereas the International Classification of Functioning for Child and Youth (ICF-CY) would be more suitable for children and youth. The ICF/ICF-CY is a multidimensional model of functioning, with participation as the key construct.72,73 This model provides a framework to describe limitations associated with a person’s individual functioning and identifies the influencing personal and environmental factors. It has logical coherent content, aids in determining classification and effective decision making, and is easily adopted in rehabilitation service.74
The content of measures that could be included within the core set should 1) span all dimensions of the ICF/ICF-CY (body structures and functioning, activity and participation, as well as personal and environmental issues), 2) include a combination of objective measures and subjective (self-report) measures,75 3) contain measures that not only have good psychometric properties (reproducibility, validity, and responsiveness) but also should be able to cope with cross-cultural aspects of psychometric quality (culture, language), 4) be easy to use within clinical practice, independent of the setting in which it is being used, 5) be embedded into a clinical decision model/treatment regime to monitor the progression of the disease or treatment effects, and 6) distinguish between disabled patients and high-level performance athletes, as they may have other clinical profiles and may have musculoskeletal injury of a different nature.
From this core set, not only can disease progression be monitored, but also patient profiles and trajectories may be derived. Patient profiles could reduce clinical heterogeneity and more clearly identify patients, in addition to enabling future research investigating the predictive value for functional decline and musculoskeletal injury. This would enable early detection and may even prevent the onset of chronicity.
However, before such a data set can be developed or implemented, international consensus on diagnostics with a generic core set is indicated. Support is needed from the scientific community, as well as from clinicians (from all involved disciplines) and patients.
Conclusion
When considering the accumulation of knowledge and GJH-related syndromes, much has been discovered and even more crucial questions have been identified. HMS and EDS-HT are multifactorial diseases that affect all levels of human functioning. Dysfunction can be the result of chronic pain but also due to multisystemic involvement, psychological distress, and related disability. How chronic pain and multisystemic deficits come into effect and interact with each other is currently unknown. The specific problems associated with the GJH-related syndromes, as compared to other chronic pain syndromes, are still challenging for most physicians and other health personnel due to many issues surrounding etiology, disease classification, diagnostics, and treatment. To develop new innovative ways of treating chronic pain, interdisciplinary cooperation should be stimulated and issues concerning clinical heterogeneity, disease classification, and diagnostics should be addressed. In addition, lessons learned from other fields of chronic pain management should be considered.
Acknowledgments
This study was supported by the School of Physiotherapy, University of Applied Sciences–Amsterdam, and by a grant from the National Dutch Board of Scientific Research.
Disclosure
The authors report no conflicts of interest in this work.
References
Greenwood NL, Duffell LD, Alexander CM, McGregor AH. Electromyographic activity of pelvic and lower limb muscles during postural tasks in people with benign joint hypermobility syndrome and non hypermobile people. A pilot study. Man Ther. 2011;16(6):623–628. | |
Kirk JA, Ansell BM, Bywaters EG. The hypermobility syndrome. Musculoskeletal complaints associated with generalized joint hypermobility. Ann Rheum Dis. 1967;26(5):419–425. | |
Scheper MC, Engelbert RH, Rameckers EA, Verbunt J, Remvig L, Juul-Kristensen B. Children with generalised joint hypermobility and musculoskeletal complaints: state of the art on diagnostics, clinical characteristics, and treatment. Biomed Res Int. 2013;2013:121054. | |
Grahame R. Hypermobility: an important but often neglected area within rheumatology. Nat Clin Pract Rheumatol. 2008;4(10):522–524. | |
Grahame R, Bird HA, Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol. 2000;27(7):1777–1779. | |
Remvig L, Jensen DV, Ward RC. Are diagnostic criteria for general joint hypermobility and benign joint hypermobility syndrome based on reproducible and valid tests? A review of the literature. J Rheumatol. 2007;34(4):798–803. | |
Juul-Kristensen B, Rogind H, Jensen DV, Remvig L. Inter-examiner reproducibility of tests and criteria for generalized joint hypermobility and benign joint hypermobility syndrome. Rheumatology. 2007; 46(12):1835–1841. | |
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77(1):31–37. | |
Malfait F, Hakim AJ, De Paepe A, Grahame R. The genetic basis of the joint hypermobility syndromes. Rheumatology. 2006;45(5):502–507. | |
De Paepe A, Malfait F. The Ehlers-Danlos syndrome, a disorder with many faces. Clin Genet. 2012;82(1):1–11. | |
Grahame R, Hakim AJ. Hypermobility. Curr Opin Rheumatol. 2008; 20(1):106–110. | |
Tinkle BT, Bird HA, Grahame R, Lavallee M, Levy HP, Sillence D. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (aka hypermobility syndrome). Am J Med Genet A. 2009;149A(11):2368–2370. | |
Wordsworth P, Ogilvie D, Smith R, Sykes B. Joint mobility with particular reference to racial variation and inherited connective tissue disorders. Br J Rheumatol. 1987;26(1):9–12. | |
Murray KJ. Hypermobility disorders in children and adolescents. Best Pract Res Clin Rheumatol. 2006;20(2):329–351. | |
Mallorquí-Bagué N, Bulbena A, Roé-Vellvé N, et al. Emotion processing in joint hypermobility: a potential link to the neural bases of anxiety and related somatic symptoms in collagen anomalies. Eur Psychiatry. 2015;30(4):454–458. | |
Scheper MC, de Vries JE, de Vos R, Verbunt J, Nollet F, Engelbert RH. Generalized joint hypermobility in professional dancers: a sign of talent or vulnerability? Rheumatology. 2013;52(4):651–658. | |
Scheper MC, de Vries JE, Juul-Kristensen B, Nollet F, Engelbert RH. The functional consequences of generalized joint hypermobility: a cross-sectional study. BMC Musculoskelet Disord. 2014;15:243. | |
Day H, Koutedakis Y, Wyon MA. Hypermobility and dance: a review. Int J Sports Med. 2011;32(7):485–489. | |
Foley EC, Bird HA. Hypermobility in dance: asset, not liability. Clin Rheumatol. 2013;32(4):455–461. | |
Palmer S, Bailey S, Barker L, Barney L, Elliott A. The effectiveness of therapeutic exercise for joint hypermobility syndrome: a systematic review. Physiotherapy. 2014;100(3):220–227. | |
Pacey V, Tofts L, Wesley A, Collins F, Singh-Grewal D. Joint hypermobility syndrome: a review for clinicians. J Paediatr Child Health. 2014;51(4):373–380. | |
Grahame R. Heritable disorders of connective tissue. Baillieres Best Pract Res Clin Rheumatol. 2000;14(2):345–361. | |
Remvig L, Engelbert RH, Berglund B, et al. Need for a consensus on the methods by which to measure joint mobility and the definition of norms for hypermobility that reflect age, gender and ethnic-dependent variation: is revision of criteria for joint hypermobility syndrome and Ehlers-Danlos syndrome hypermobility type indicated? Rheumatology. 2011;50(6):1169–1171. | |
Pacey V, Adams RD, Tofts L, Munns CF, Nicholson LL. Joint hypermobility syndrome subclassification in paediatrics: a factor analytic approach. Arch Dis Child. 2015;100(1):8–13. | |
De Wandele I, Rombaut L, Malfait F, De Backer T, De Paepe A, Calders P. Clinical heterogeneity in patients with the hypermobility type of Ehlers-Danlos syndrome. Res Dev Disabil. 2013;34(3):873–881. | |
Grahame R. Hypermobility – not a circus act. Int J Clin Pract. 2000; 54(5):314–315. | |
Hogan KA, Gross RH. Overuse injuries in pediatric athletes. Orthop Clin North Am. 2003;34(3):405–415. | |
De Wandele I, Rombaut L, Leybaert L, et al. Dysautonomia and its underlying mechanisms in the hypermobility type of Ehlers-Danlos syndrome. Semin Arthritis Rheum. 2014;44(1):93–100. | |
De Wandele I, Calders P, Peersman W, et al. Autonomic symptom burden in the hypermobility type of Ehlers-Danlos syndrome: a comparative study with two other EDS types, fibromyalgia, and healthy controls. Semin Arthritis Rheum. 2014;44(3):353–361. | |
Ferrell WR, Tennant N, Sturrock RD, et al. Amelioration of symptoms by enhancement of proprioception in patients with joint hypermobility syndrome. Arthritis Rheum. 2004;50(10):3323–3328. | |
Rombaut L, Malfait F, De Wandele I, et al. Balance, gait, falls, and fear of falling in women with the hypermobility type of Ehlers-Danlos syndrome. Arthritis Care Res (Hoboken). 2011;63(10):1432–1439. | |
Fatoye FA, Palmer S, van der Linden ML, Rowe PJ, Macmillan F. Gait kinematics and passive knee joint range of motion in children with hypermobility syndrome. Gait Posture. 2011;33(3):447–451. | |
Falkerslev S, Baagø C, Alkjær T, et al. Dynamic balance during gait in children and adults with generalized joint hypermobility. Clin Biomech. 2013;28(3):318–324. | |
Celletti C, Galli M, Cimolin V, Castori M, Albertini G, Camerota F. Relationship between fatigue and gait abnormality in joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type. Res Dev Disabil. 2012;33(6):1914–1918. | |
Rigoldi C, Galli M, Cimolin V, et al. Gait strategy in patients with Ehlers-Danlos syndrome hypermobility type and Down syndrome. Res Dev Disabil. 2012;33(5):1437–1442. | |
Nikolajsen H, Larsen PK, Simonsen EB, et al. Gait pattern in 9-11-year-old children with generalized joint hypermobility compared with controls; a cross-sectional study. BMC Musculoskelet Disord. 2013;14:341. | |
Simonsen EB, Tegner H, Alkjaer T, et al. Gait analysis of adults with generalised joint hypermobility. Clin Biomech. 2012;27(6):573–577. | |
Junge T, Wedderkopp N, Thorlund JB, Sogaard K, Juul-Kristensen B. Altered knee joint neuromuscular control during landing from a jump in 10-15year old children with generalised joint hypermobility. A substudy of the CHAMPS-study Denmark. J Electromyogr Kinesiol. 2015;25(3):501–507. | |
Rombaut L, Malfait F, De Wandele I, et al. Muscle-tendon tissue properties in the hypermobility type of Ehlers-Danlos syndrome. Arthritis Care Res (Hoboken). 2012;64(5):766–772. | |
Magnusson SP, Julsgaard C, Aagaard P, et al. Viscoelastic properties and flexibility of the human muscle-tendon unit in benign joint hypermobility syndrome. J Rheumatol. 2001;28(12):2720–2725. | |
Smith TO, Jerman E, Easton V, et al. Do people with benign joint hypermobility syndrome (BJHS) have reduced joint proprioception? A systematic review and meta-analysis. Rheumatol Int. 2013;33(11):2709–2716. | |
Russek LN. Hypermobility syndrome. Phys Ther. 1999;79(6):591–599. | |
Sahin N, Baskent A, Ugurlu H, Berker E. Isokinetic evaluation of knee extensor/flexor muscle strength in patients with hypermobility syndrome. Rheumatol Int. 2008;28(7):643–648. | |
Rombaut L, Malfait F, De Wandele I, et al. Muscle mass, muscle strength, functional performance, and physical impairment in women with the hypermobility type of Ehlers-Danlos syndrome. Arthritis Care Res (Hoboken). 2012;64(10):1584–1592. | |
Engelbert RH, van Bergen M, Henneken T, Helders PJ, Takken T. Exercise tolerance in children and adolescents with musculoskeletal pain in joint hypermobility and joint hypomobility syndrome. Pediatrics. 2006;118(3):e690–e696. | |
Fatoye F, Palmer S, Macmillan F, Rowe P, van der Linden M. Proprioception and muscle torque deficits in children with hypermobility syndrome. Rheumatology. 2009;48(2):152–157. | |
Scheper M, Vries J, Beelen A, Vos R, Nollet F, Engelbert R. Generalized joint hypermobility, muscle strength and physical function in healthy adolescents and young adults. Curr Rheumatol Rev. 2015; 10(2):117–125. | |
Ferrell WR, Tennant N, Baxendale RH, Kusel M, Sturrock RD. Musculoskeletal reflex function in the joint hypermobility syndrome. Arthritis Rheum. 2007;57(7):1329–1333. | |
Holla JF, van der Leeden M, Peter WF, et al. Proprioception, laxity, muscle strength and activity limitations in early symptomatic knee osteoarthritis: results from the CHECK cohort. J Rehabil Med. 2012; 44(10):862–868. | |
Rombaut L, Scheper M, De Wandele I, et al. Chronic pain in patients with the hypermobility type of Ehlers-Danlos syndrome: evidence for generalized hyperalgesia. Clin Rheumatol. 2014;34(6):1121–1129. | |
Clark CK, Khattab A, Carr E. Chronic widespread pain and neurophysiological symptoms in joint hypermobility syndrome (JHS). Int J Ther Rehabil. 2014;21(2):9. | |
Hakim AJ, Grahame R, Norris P, Hopper C. Local anaesthetic failure in joint hypermobility syndrome. J R Soc Med. 2005;98(2):84–85. | |
Rombaut L, Malfait F, De Wandele I, et al. Medication, surgery, and physiotherapy among patients with the hypermobility type of Ehlers-Danlos syndrome. Arch Phys Med Rehabil. 2011;92(7):1106–1112. | |
Nijs J, Malfliet A, Ickmans K, Baert I, Meeus M. Treatment of central sensitization in patients with ‘unexplained’ chronic pain: an update. Expert Opin Pharmacother. 2014;15(12):1671–1683. | |
Vlaeyen JW, Linton SJ. Fear-avoidance and its consequences in chronic musculoskeletal pain: a state of the art. Pain. 2000;85(3):317–332. | |
Leeuw M, Goossens ME, Linton SJ, Crombez G, Boersma K, Vlaeyen JW. The fear-avoidance model of musculoskeletal pain: current state of scientific evidence. J Behav Med. 2007;30(1):77–94. | |
Vlaeyen JW, Linton SJ. Fear-avoidance model of chronic musculoskeletal pain: 12 years on. Pain. 2012;153(6):1144–1147. | |
Simons LE, Kaczynski KJ. The fear avoidance model of chronic pain: examination for pediatric application. J Pain. 2012;13(9):827–835. | |
Eccleston C, Wastell S, Crombez G, Jordan A. Adolescent social development and chronic pain. Eur J Pain. 2008;12(6):765–774. | |
Cohen LL, Vowles KE, Eccleston C. The impact of adolescent chronic pain on functioning: disentangling the complex role of anxiety. J Pain. 2010;11(11):1039–1046. | |
Moreno-Peral P, Conejo-Cerón S, Motrico E, et al. Risk factors for the onset of panic and generalised anxiety disorders in the general adult population: a systematic review of cohort studies. J Affect Disord. 2014;168:337–348. | |
Bulbena A, Gago J, Pailhez G, Sperry L, Fullana MA, Vilarroya O. Joint hypermobility syndrome is a risk factor trait for anxiety disorders: a 15-year follow-up cohort study. Gen Hosp Psychiatry. 2011; 33(4):363–370. | |
Remvig L, Flycht L, Christensen KB, Juul-Kristensen B. Lack of consensus on tests and criteria for generalized joint hypermobility, Ehlers-Danlos syndrome: hypermobile type and joint hypermobility syndrome. Am J Med Genet A. 2014;164A(3):591–596. | |
El-Metwally A, Salminen JJ, Auvinen A, Macfarlane G, Mikkelsson M. Risk factors for development of non-specific musculoskeletal pain in preteens and early adolescents: a prospective 1-year follow-up study. BMC Musculoskelet Disord. 2007;8:46. | |
El-Metwally A, Salminen JJ, Auvinen A, Kautiainen H, Mikkelsson M. Lower limb pain in a preadolescent population: prognosis and risk factors for chronicity – a prospective 1- and 4-year follow-up study. Pediatrics. 2005;116(3):673–681. | |
El-Metwally A, Salminen JJ, Auvinen A, Kautiainen H, Mikkelsson M. Prognosis of non-specific musculoskeletal pain in preadolescents: a prospective 4-year follow-up study till adolescence. Pain. 2004;110(3):550–559. | |
Tobias JH, Deere K, Palmer S, Clark EM, Clinch J. Joint hypermobility is a risk factor for musculoskeletal pain during adolescence: findings of a prospective cohort study. Arthritis Rheum. 2013;65(4):1107–1115. | |
Medeiros HB, de Araujo DS, de Araujo CG. Age-related mobility loss is joint-specific: an analysis from 6,000 flexitest results. Age. 2013;35(6):2399–2407. | |
Remvig L, Duhn PH, Ullman S, et al. Skin extensibility and consistency in patients with Ehlers-Danlos syndrome and benign joint hypermobility syndrome. Scand J Rheumatol. 2009;38(3):227–230. | |
Remvig L, Duhn P, Ullman S, et al. Skin signs in Ehlers-Danlos syndrome: clinical tests and para-clinical methods. Scand J Rheumatol. 2010;39(6):511–517. | |
Voermans NC, Knoop H, van de Kamp N, Hamel BC, Bleijenberg G, van Engelen BG. Fatigue is a frequent and clinically relevant problem in Ehlers-Danlos syndrome. Semin Arthritis Rheum. 2010; 40(3):267–274. | |
Atkinson HL, Nixon-Cave K. A tool for clinical reasoning and reflection using the international classification of functioning, disability and health (ICF) framework and patient management model. Phys Ther. 2011;91(3):416–430. | |
Chien CW, Rodger S, Copley J, Skorka K. Comparative content review of children’s participation measures using the International Classification of Functioning, Disability and Health-Children and Youth. Arch Phys Med Rehabil. 2014;95(1):141–152. | |
Raghavendra P, Bornman J, Granlund M, Bjorck-Akesson E. The World Health Organization’s International Classification of Functioning, Disability and Health: implications for clinical and research practice in the field of augmentative and alternative communication. Augment Altern Commun. 2007;23(4):349–361. | |
Wilson GC, Quillin RC 3rd, Hanseman DJ, Lewis JD, Edwards MJ, Shaughnessy EA. Incidence and predictors of neuropathic pain following breast surgery. Ann Surg Oncol. 2013;20(10):3330–3334. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.