Back to Journals » Breast Cancer: Targets and Therapy » Volume 15
Chromosomal Instability as Enabling Feature and Central Hallmark of Breast Cancer
Authors Castellanos G ![]() , Valbuena DS, Pérez E, Villegas VE
, Valbuena DS, Pérez E, Villegas VE ![]() , Rondón-Lagos M
, Rondón-Lagos M ![]()
Received 26 July 2022
Accepted for publication 11 October 2022
Published 9 March 2023 Volume 2023:15 Pages 189—211
DOI https://doi.org/10.2147/BCTT.S383759
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Giovanny Castellanos,1,2 Duván Sebastián Valbuena,2 Erika Pérez,2 Victoria E Villegas,3 Milena Rondón-Lagos2
1Maestría en Ciencias Biológicas, Universidad Pedagógica y Tecnológica de Colombia, Tunja, Colombia; 2School of Biological Sciences, Universidad Pedagógica y Tecnológica de Colombia, Tunja, Colombia; 3Centro de Investigaciones en Microbiología y Biotecnología-UR (CIMBIUR), Facultad de Ciencias Naturales, Universidad del Rosario, Bogotá, Colombia
Correspondence: Milena Rondón-Lagos, Tunja, 150003, Colombia, Tel +57 608 7428263, Fax +57 608 7405626, Email [email protected]
Abstract: Chromosomal instability (CIN) has become a topic of great interest in recent years, not only for its implications in cancer diagnosis and prognosis but also for its role as an enabling feature and central hallmark of cancer. CIN describes cell-to-cell variation in the number or structure of chromosomes in a tumor population. Although extensive research in recent decades has identified some associations between CIN with response to therapy, specific associations with other hallmarks of cancer have not been fully evidenced. Such associations place CIN as an enabling feature of the other hallmarks of cancer and highlight the importance of deepening its knowledge to improve the outcome in cancer. In addition, studies conducted to date have shown paradoxical findings about the implications of CIN for therapeutic response, with some studies showing associations between high CIN and better therapeutic response, and others showing the opposite: associations between high CIN and therapeutic resistance. This evidences the complex relationships between CIN with the prognosis and response to treatment in cancer. Considering the above, this review focuses on recent studies on the role of CIN in cancer, the cellular mechanisms leading to CIN, its relationship with other hallmarks of cancer, and the emerging therapeutic approaches that are being developed to target such instability, with a primary focus on breast cancer. Further understanding of the complexity of CIN and its association with other hallmarks of cancer could provide a better understanding of the cellular and molecular mechanisms involved in prognosis and response to treatment in cancer and potentially lead to new drug targets.
Keywords: breast cancer, genomic instability, chromosomal instability, clonal heterogeneity, resistance to therapy, therapeutic targets
Introduction
In recent years, CIN has gained great importance due to its implications for both, the diagnosis and prognosis of cancer. Recent studies have suggested that both CIN and clonal heterogeneity, influence cancer progression, aggressiveness, prognosis, and response to therapy.1–4 The above because both, CIN and clonal heterogeneity, lead to variations in gene regulation (proto-oncogenes and/or tumor suppressor genes),5 as well as to variations in the levels of the proteins for which they encode.6 The available evidence on the effect of CIN and clonal heterogeneity levels on survival and response to therapy in cancer is paradoxical,7 since while some studies have shown associations between CIN and clonal heterogeneity with proliferative advantages and survival,8–10 other studies have shown associations with decreased proliferation11 and better outcomes in cancer patients.12,13 This evidences the complex role played by CIN and clonal heterogeneity in the prognosis and response to cancer treatment. The studies carried out to date suggest that CIN and clonal heterogeneity could be considered as therapeutic targets, aimed at improving response to treatment, reducing side effects, and overcoming resistance to cancer therapy.14 This review focuses on recent studies on the role of CIN in cancer, the cellular mechanisms leading to CIN, its relationship with other hallmarks of cancer, and the emerging therapeutic approaches that are being developed to target such instability, with a primary focus on breast cancer.
General Overview of Genomic Instability and CIN
Genomic Instability as a Distinctive Hallmark of Cancer
The hallmarks of cancer are the set of biological characteristics acquired by neoplastic cells during the different stages of tumor development,15 including sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, tumor promoting inflammation, reprogramming energy metabolism, evading immune destruction, genomic instability and mutation.16 Recently, additional emerging hallmarks and enabling features were added, including unlocking phenotypic plasticity, non-mutational epigenetic reprogramming, polymorphic microbiomes, and senescent cells.17 Together, these constitute “the fourteen hallmarks of cancer”.
Genomic instability refers to a greater tendency for alterations in the genome during the life cycle of cells. It is a major driving force for heterogeneity and tumorigenesis.18,19 Genomic instability leads an accumulation of genetic alterations, which allow tumor cells to acquire proliferative and survival advantages.20 According to the genetic level of the alteration, genomic instability is classified into three types, including: nucleotide instability (NIN), microsatellite instability (MSI), and CIN.21 NIN refers to an increased frequency of substitutions, deletions and insertions of one or a few nucleotides. The MSI is characterized by a decreasing or increasing in microsatellite length as a consequence of errors in the missing base repair system.22 Finally, CIN is the most prevalent but least understood form of genomic instability.
CIN has gained great importance in recent years, due to its implications in both the diagnosis and prognosis of cancer.10,23 This type of genomic instability is classified into numerical CIN (gain or loss of whole chromosomes - aneuploidy) and structural CIN (gain or loss of chromosome segments)14 (Figure 1). Aneuploidy is a state in which the cell has an abnormal total number of chromosomes, which may be stable or unstable. When cells exhibit unstable aneuploidy, the simultaneous growth of several genetically distinct tumor subpopulations is favored, increasing inter and intra-tumoral genomic heterogeneity.2
 |
Figure 1 Types of Chromosomal Instability (CIN). The CIN is classified into Numerical CIN and Structural CIN. Numerical CIN, corresponds to the gain or loss of whole chromosomes (aneuploidy, and polyploidy), while structural CIN refers to the gain or loss of chromosome segments. |
Cellular Mechanisms Leading to CIN in Cancer
Exposure to different endogenous factors such as reactive oxygen species (ROS), and exogenous factors such as physical (ionizing and ultraviolet radiation), chemical (intercalating and alkylating agents), and biological (viruses and bacteria), cause up to one million of DNA changes per day in somatic cells.24
The damage to the genetic material, can be corrected by the DNA repair machinery, without generating repercussions to the cells.25 However, such repair mechanisms are prone to error and can drive CIN, producing chromosomal rearrangements26 that affect key genes, or regulatory elements, for cell division and apoptosis.27,28 Thus, the cellular mechanisms leading to CIN in cancer, vary depending on whether they correspond to numerical or structural CIN. Numerical CIN is caused by the erroneous segregation of chromosomes during somatic cell division (mitotic nondisjunction), which gives rise to two daughter cells with an aneuploid karyotype.21,29 This nondisjunction is triggered by defects in chromosomal cohesion (Figure 2A), kinetochore-microtubule junction dynamics (Figure 2B), centrosome copy number (Figure 2C), cell cycle regulation (Figure 2D) and spindle assembly checkpoint (SAC)28 (Figure 2E). Of the above mechanisms, the SAC is quite important for cells, because it helps them maintain genome stability by delaying their division until ensuring that anaphase occurs only after all kinetochores are amphitelically connected to spindle microtubules,30 thus guaranteeing a correct chromosomal segregation.31
 |
Figure 2 Cellular mechanisms leading to numerical Chromosomal Instability (CIN) in cancer. Numerical CIN is caused by the erroneous segregation of chromosomes during somatic cell division (mitotic nondisjunction), which gives rise to two daughter cells with an aneuploid karyotype. The mechanisms associated with mitotic nondisjunction include: (A) Incorrect chromosomal cohesion. In metaphase cells, sister chromatids (purple) are linked together by cohesin (blue). Chromosomes lacking cohesin do not pair at metaphase, and individual sister chromatids fail to achieve stable bipolar attachment at the spindle. When anaphase begins, the chromosomes fail to segregate at opposite Poles, leading to errors in chromosome segregation. (B) Errors in the dynamics of the kinetochore-microtubule junction. For chromosome segregation to occur with high fidelity, prior to the onset of anaphase, kinetochores must bind to spindle microtubules and connect sister chromatids of each chromosome to opposite spindle Poles. If chromosomes do not attach correctly to the spindle, for example when only one kinetochore is connected to one spindle pole, sister chromatids migrate to the same spindle pole in anaphase, leading to chromosome missegregation. (C) Defects in centrosome copy number. Cells with more than two centrosomes significantly increase the rate of kinetochore junction formation. The presence of extra centrosomes elevates lagging chromosomes and induces chromosome missegregation. (D) Defects in cell cycle regulation. Cell cycle checkpoints are directly involved in supporting the fidelity of chromosome segregation during mitosis. Alterations in these checkpoints may allow cells that have poorly segregated chromosomes to continue cell cycle progression. (E) Spindle assembly checkpoint (SAC) errors. When chromosomes do not properly attach to the spindle (misjunctions), kinetochores activate the SAC network, which inhibits the initiation of anaphase and preserves sister chromatid cohesion. If the mismatches are not detected by the SAC, chromosome missegregation results. |
However, when the SAC is defective, three types of misjunctions between the mitotic spindle and chromosomes can occur, including: monotelic union (only one kinetochore of the chromatid pair is anchored to a spindle microtubule), syntelic union (the kinetochore of the two chromatids is attached to microtubules of the same pole), and merothelic union (at least one kinetochore of the pair of chromatids is anchored to the microtubules of the two poles). These mismatches, lead to early onset of anaphase, significantly increasing the probability of incorrect chromosome segregation. This suggests, that defects in the SAC may cause CIN.28 In fact, merotelic anchoring is considered to be the main mechanism that induces CIN in neoplastic cells.32 In addition, it has been suggested, that the non-correction of this type of junctions, is responsible for the CIN observed in approximately 85% of all sporadic cancers.33
Moreover, has also been indicated that the main mechanism associated with the induction of structural CIN is correlated with failures in DNA repair34 (Figure 3), especially those involved in double-strand break (DSB) repair processes, including homologous recombination repair (HR) (Figure 3A) and non-homologous end joining (NHEJ) repair35,36 (Figure 3B). Considering that these repair mechanisms are prone to error, their activation can lead to chromosomal rearrangements.37 Specifically, HR detects and repairs DSBs during the G2 and late S phases of the cell cycle.38,39 In this mechanism, the pair of homologous chromosomes are brought together and the region of the undamaged homolog or chromatid is taken as a template to reconstruct the DSB of the affected chromosome6,40 (Figure 3A). NHEJ instead, allows strand joining at their ends without requiring a homologous or complementary sequence to guide repair41 (Figure 3B). When either of these mechanisms functions improperly, it leaves the affected DNA strand ends free, which can then lead to structural chromosomal alterations through a variety of processes such as splicing, resection, alignment, invasion and/or replication,37 thus favoring the induction of CIN.
 |
Figure 3 DNA double-strand break (DSB) repair mechanism leading to structural Chromosomal Instability (CIN) in cancer. (A) Homologous recombination repair (HR). In this mechanism, the pair of homologous chromosomes are brought together and the region of the undamaged homolog or chromatid is taken as a template to reconstruct the DSB of the affected chromosome. (B) Non-homologous end joining (NHEJ) repair. NHEJ allows strand joining at their ends without requiring a homologous or complementary sequence to guide repair. When either of these mechanisms functions improperly, it leaves the affected DNA strand ends free, which can then lead to structural chromosomal alterations including: chromosomal deletions (del), chromosomal duplications (dup), derivatives chromosomes (der), isochromosomes (i), isodicentric chromosomes (idic), dicentric chromosomes (dic), acentric fragments chromosomes (ace), and chromosomal translocations (t). Some of the processes involved in the formation of the above chromosomal alterations are: splicing, resection, alignment, invasion and/or replication, all of which favor the induction of CIN. |
Associations Between CIN and Clonal Heterogeneity
Clonal heterogeneity has important clinical implication, since it is directly related to cancer progression and metastases.42 Clonal heterogeneity is often described as the presence of a large variety of cells that behave differently within a tumor, thus favoring the adaptation of tumor cells to stressful environments,14 and promoting both, tumor progression and resistance to therapy.43–45 In fact, tumorigenesis occurs under a mechanism of natural selection, in which only tumor cells that can adapt to the changing adverse conditions of their microenvironment, manage to survive and proliferate46 (Figure 4). Consequently, these tumor cells take advantage of their genetic variability to develop traits that give them biological adaptability. Under these conditions, CIN ensuring the diversification of cell clones and enabling the acquisition of other cancer hallmarks through the acquisition of new mutations16,21,47 (Figure 4).
 |
Figure 4 Chromosomal Instability (CIN) as the main promoter of Clonal Heterogeneity. Clonal heterogeneity reflects the general instability of the system and leads to the clonal evolution of tumor cells with specific mutations, thus favoring the adaptation of tumor cells to stressful environments, and promoting both, tumor progression and resistance to therapy. Under these conditions, CIN ensuring the diversification of cell clones and enabling the acquisition of other cancer hallmarks through the acquisition of new mutations. |
CIN and clonal heterogeneity, have a directly proportional association, where an increase in CIN leads to an increase in clonal heterogeneity. The above is evidenced by the increase in molecular and genetic diversity between cells belonging to the same tumor. Therefore, each cell subpopulation can present unique characteristics, increasing heterogeneity and consequently diversifying the therapeutic response. In fact, clinical longitudinal studies based on the analysis of biopsies from different regions of the same tumor, showed changes in the cellular heterogeneity, from initial stages to metastatic stages, also observing an exponential increase in clonal heterogeneity. This increase in both, cellular heterogeneity and clonal heterogeneity, has been associated with decreased survival after endocrine therapy (tamoxifen) and chemotherapy.44
For instance, recent evidence has postulated CIN as a bet-hedging mechanism in tumor evolution, upon which the selective pressures induced by pharmacological treatments can act, thus CIN could be exploited therapeutically to achieve better patient outcomes.48 In this regard, it has been indicated that the pharmacological reduction of CIN levels can stop the adaptability of cancer cells and prevent the acquisition of aggressive characteristics.48 For example, recent studies have indicated that therapeutic reduction of CIN can be achieved through inhibition of the anaphase-promoting complex, which together with the use of mitosis-targeting chemotherapeutics, can act synergistically to inhibit the proliferation of ovarian cancer cells.49 Of highlighting what recent studies have also shown that intrinsic levels of CIN correlate with poor responses to numerous therapies in human tumors. In fact, it was indicated that CIN provides phenotypic plasticity to cancer cells that can allow them to adapt to diverse stressful environments.50 Furthermore, it was demonstrated that inducing exogenous CIN can accelerate the acquisition of resistance to both targeted and cytotoxic therapies.50–52 Overall, studies to date highlight CIN as a cause of resistance to cancer therapy, and highlight the importance of identifying reliable methods to directly examine it and thus support novel strategies aimed at target CIN and improving outcomes in cancer.
CIN as an Enabling Feature of Other Hallmarks of Cancer
CIN has acquired great importance in recent years, given its involvement in the fixation of random mutations that confer proliferation, survival and metastasis advantages to tumor cells. The studies carried out to date, show the important role that CIN plays in prognosis and response to therapy in cancer, as well as evidence the ability of CIN to improve tumor fitness and enable the acquisition of other hallmarks of cancer. Below we discuss the associations between CIN with eight of the fourteen hallmarks of cancer, and its relevance to cancer prognosis and response to therapy.
Sustaining Proliferative Signaling
One of the main mechanisms that lead tumor cells to maintain permanent activation of cell proliferation, is through an increase in the number of gene copies (gene amplification). A typical example is the BRAF and PIK3CA genes, which code for serine/threonine-protein kinase B-Raf and phosphatidylinositol 3-Kinase (PI3K) transcription factors, respectively. The amplification of these genes, leads to the overexpression of their proteins, which favors the permanent activation of cell proliferation.53–55 The permanent activation of the cell proliferation could lead to chromosome missegregation during mitosis, and therefore to the gain and/or loss of whole chromosomes (numerical CIN) or chromosome fragments (structural CIN) (Figure 5). In fact, it has been suggested that while in stable diploid cell lines, one chromosome mis-segregate per hundred cell divisions, cancer cells with CIN, mis-segregate a chromosome approximately once every one to five cell divisions.56,57 The above explains why permanent cell cycle activation leads to an increase in CIN. For instance, this behaviour has been observed in breast cancer (BC) patients with human epidermal growth factor receptor 2 (HER2) gene amplification. HER2 gene is amplified and overexpressed in up to 15% of BC, and is associated with poor prognosis.58 Preclinical models have indicated a role for HER2 signaling in CIN initiation and defective cell cycle control, and evidence suggests that targeting HER2 may attenuate this process. In fact, has been indicated that HER2 positive (HER2+) breast tumors show significantly increased CIN relative to HER2 negative tumors (HER2-).59
 |
Figure 5 Chromosomal Instability (CIN) as an enabling hallmark in cancer. CIN is the main type of instability that promotes neoplastic cells. CIN favors the adaptation of tumor cells to stressful environments, thus facilitating the activation of other hallmarks of cancer through the acquisition of new mutations. In the same way, the hallmarks of cancer (sustaining proliferative signaling, resisting cell death, enabling replicative immortality, angiogenesis, invasion and metastasis, reprogramming of energy metabolism, evading immune destruction and tumor promoting inflammation), favors the induction and/or increase of CIN. Both, CIN as an enabling hallmark of cancer, and the ability of hallmarks of cancer to induce and/or increase CIN, allow cells to survive and acquire resistance to therapy. Abbreviations: AKT, AKT Serine/Threonine Kinase; A-NHEJ, Alternative non-homologous end joining pathway; Apaf1, Apoptosis protease-activating factor-1; ATM, ATM Serine/Threonine Kinase; ATR, ATR Serine/Threonine Kinase; BAX, BCL2 Associated X Apoptosis Regulator; BID, BH3 Interacting Domain Death Agonist; CDKN1A, Cyclin Dependent Kinase Inhibitor 1A; CDKN1B, Cyclin Dependent Kinase Inhibitor 1B; CENPF, Centromere Protein F; C-NHEJ, Canonical non-homologous end joining pathway; COXA, Cytochrome c oxidase subunit 1; cGAS, Cyclic GMP-AMP synthase; cGAMP, Cyclic guanosine monophosphate–adenosine monophosphate; ENO2, Enolase 2; ERK, Extracellular signal-regulated kinase; GAPDH, Glyceraldehyde-3-Phosphate Dehydrogenase; GLUT4, Glucose transporter type 4; GTP, Guanosine-triphosphate; HDR, homology-directed repair; HER2, epidermal growth factor receptor 2; HIF, hypoxia-inducible factor-1; IFN, Interferon; LDHB, lactate dehydrogenase B; MASTL, Microtubule Associated Serine/Threonine Kinase Like; MDM2, Mouse double minute 2 homolog; MDM4, Regulator of P53; MEK, Mitogen-activated protein kinase kinase; mTOR, Mechanistic Target Of Rapamycin Kinase; MYC, MYC Proto-Oncogene BHLH Transcription Factor; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; NOXA, pro-apoptotic BH3-only protein; PI3K, phosphatidylinositol 3-Kinase; PTEN, Phosphatase And Tensin Homolog; PUMA, p53 upregulated modulator of apoptosis; p53, tumor suppressor protein; RAS, Rat sarcoma virus protein; STING, Stimulator of interferon genes protein; SAC, Spindle Assembly Checkpoint; TERT, Telomerase Reverse Transcriptase; TWIST, Twist Family BHLH Transcription Factor; TBK1, TANK Binding Kinase 1; VEGF, Vascular Endothelial Growth Factor A. |
Interestingly, HER2 overexpression in tumoral cells has also been associated with centrosome abnormalities,60,61 which through chromosome missegregation during mitosis, may promote CIN62 (Figure 5). Furthermore, several lines of preclinical and clinical evidence have supported a functional role for HER2 signaling in mediating and sustaining CIN. Indeed, has been indicated that HER2 protein overexpression constitutively activates the PI3K/AKT/mTOR signal pathway, promoting chromosome segregation errors in mouse tumors63 and the survival and proliferation of aneuploid in mouse mammary epithelium.64 In accordance with the role of HER2 signaling in the induction of CIN and, consistent with the heterogeneity of CIN in individual tumors, additional studies showed higher HER2 expression in aneuploid compared to diploid components of HER2+ tumors.65 These findings suggest an important role for HER2 in the induction and maintenance of CIN, through the permanent activation of cell proliferation (Figure 5).
Other markers related to CIN induction and associated with poor prognosis in BC, include the centromere protein F (CENP-F). CENP-F is a cell cycle-regulated protein implicated in kinetochore assembly and/or the spindle checkpoint66,67 and maximally expressed at the G2/M phase of the cell cycle.68,69 Upregulation of CENP-F in primary BC samples was significantly associated with markers of CIN including cyclin E, increased telomerase activity, C-MYC amplification and aneuploidy (Figure 5), as well as with worse overall survival and reduced metastasis free survival. Of note, a significant proportion of tumors over-expressing CENP-F were aneuploid, strengthening the associations between CENP-F expression and markers of CIN.70 Taking into the account the above, CENP-F could be considered as an important, clinically significant target in the treatment of BC patients with CIN.
Resisting Cell Death
Genomic instability and apoptosis are closely related processes, with important implications for the survival of tumor cells. Such associations have been established based on reports indicating that CIN causes inactivation of proapoptotic pathways.71 Inhibition of apoptosis can lead to the survival and subsequent proliferation of cells with altered and potentially unstable genomes (due to non-activation of DNA double-strand break repair pathways) or that are in an abnormal polyploid state. Interestingly, it has been indicated that the associations between genomic integrity and regulation of cell death could be dual, that is, just as CIN can lead to mutation or altered expression levels of cell death regulators, and disabled apoptosis, can also lead to and/or favor CIN.71 For instance, the inhibition of apoptosis increases the risk of CIN, which could provide proliferative advantages to cells that are fit enough to survive, favoring the development of cancer. Since, disabling apoptosis could favor CIN, it is plausible that both mechanisms could cooperate to increase the oncogenic and metastatic potential of the transformed cells. Considering the above, a single process could be involved in the control of both apoptosis and CIN, which allows “crosstalk” between the two processes.
In BC, has been indicated that CIN may favor the deletion and/or mutation of tumor suppressor genes involved in the regulation of apoptosis, including the TP53 gene.72 TP53 gene, known as the guardian of the genome, codes for a transcription factor, p53, that initiates transcription of genes involved in cell cycle arrest, cellular senescence, apoptosis, metabolism, DNA repair and other processes following cellular stress.73 p53 can engage several pro-apoptotic pathways and promotes cell death by transactivating a wide array of apoptosis inducing genes, including BAX, BID, PUMA and NOXA, and also for repressing the anti-apoptotic protein BCL2.74 TP53 is mutated in approximately 20–40% of all BC cases.75,76 Mutations in TP53 can predispose cells to an increase in CIN in BC. In fact, associations between mutations in the TP53 and genomic instability in primary BC have been reported.77,78 For example, studies in primary BC cells, showed that cells with abnormal p53 protein expression, had a statistically significant higher numerical instability of chromosome 17 (aneuploidy), than cells without p53 protein staining, in the same BC samples. The results of this study strongly implicate TP53 mutation as a cause for CIN and a crucial step in mammary carcinogenesis79 (Figure 5). The connection between TP53 mutation and CIN has also been evidenced due to the role that p53 plays in the regulation of centrosome duplication.80 For instance, a study showed that mouse embryonic cells lacking the p53 protein, generate multiple copies of centrosomes in a single cell cycle, while controls show no such abnormalities81 (Figure 5). Centrosome hyper-amplification has also been detected in human cancers with TP53 mutation or deletion, leading to extensive CIN.81 The above results point to a complex, dual relationship between apoptosis inhibition and CIN in cancer.
Enabling Replicative Immortality
Telomeres and associated telomeric proteins play an important role in maintaining the integrity of the genome. Telomere length is stabilized by the expression of telomerase which allows for continued replication or cell immortality. Indeed, telomere crisis (telomere loss or dysfunction) results in complex types of genomic abnormalities, including loss of heterozygosity, aneuploidy and gene amplifications and deletions.82–84 In addition, has been indicated that telomerase is activated by tumor cells to ensure survival of the cancer cells with genomic instability.85 Given the above, telomere crisis has been considered one of the mechanisms underlying genomic instability and an important source of CIN in cancer86 (Figure 5), also associated with aggressiveness and poor prognosis.87,88 For instance, breast tumor cells generally have shorter telomeres than corresponding cells in normal tissue,89,90 and such telomere shortening has been considered a negative prognostic indicator in BC patients.91,92 In addition, specific associations between telomere shortening and BC tumor subtypes have been established. For example, it has been indicated that telomere length is shorter in more aggressive BC subtypes, such as luminal B, HER2+ and TNBC tumors, suggesting that tumor telomere length may have utility as a prognostic and/or risk marker for CIN in BC.93
Additional studies, aimed at investigating the association of telomere dysfunction and CIN with tumor prognostic factors, such as histopathological grades and hormone receptor status, found higher telomerase activity in grade III than in grade I, as well as, higher telomere dysfunction in estrogen receptor negative (ER-) breast tumors.94 Furthermore, telomere attrition was associated with telomere dysfunction, as revealed by the presence of significantly higher anaphase bridges in tumor cells, which was dependent on tumor grade (Figure 5). Considering that anaphasic bridges are indicative of CIN,43 the above results suggest an association between CIN, telomere attrition and higher grades (grades II and III) of breast cancers. In addition, was reported that ER- tumors displayed higher anaphase and internuclear bridges.94 These data support the idea that telomere dysfunction and CIN, might have value as a marker of tumor aggressiveness in BC patients.
Angiogenesis
Angiogenesis is important, as the proliferation and the metastatic spread of cancer cells depends on an adequate supply of oxygen and nutrients. Since angiogenesis is associated with aggressive tumor phenotypes, establishing the mechanisms that favour this process could allow the development of targeted therapies aimed at improving survival. One of the mechanisms associated with the induction of angiogenesis in BC is CIN. In this sense, it has been indicated that the amplification and overexpression of Twist Family BHLH Transcription Factor 1 gene (TWIST1), in breast tumor samples, and in the breast cancer cell line MCF-7/Twist, correlated strongly with CIN, with high-grade invasive carcinomas, and with an increase in vascular endothelial growth factor (VEGF).95 TWIST1 gene codes for a transcription factor belonging to the basic helix-loop-helix (bHLH) family of proteins,96 and is essential for normal vertebrate development.97,98 Twist is overexpressed in breast,95,99 gastric,100 and prostate cancers,101 among others. Functionally, Twist overexpression promotes BC by increasing angiogenesis and CIN.95,102 The associations between CIN and TWIST1 gene in BC, were established on the basis that the development of a highly invasive BC phenotype requires the coordination of many different molecular changes, which are a consequence of genomic alterations. Indeed, associations between increased cytogenetic alterations and Twist overexpression in breast tumors have already been reported. These results show that Twist overexpression promotes angiogenesis and correlates with CIN in BC, and could be considered as a potential therapeutic target to prevent invasion and metastasis95 (Figure 5).
Reprogramming Energy Metabolism
As has previously been indicated, CIN is related to progression, adverse prognosis, and resistance to therapy, among others. Such resistance has been associated with the ability of CIN to induce the growth of tumor cell populations with different chromosomal alterations. It should be noted that resistance to treatment generates a high level of stress in tumor cells. Such level of stress, is tolerated by tumor cells by modifying their energy use, which allows them to adapt to genetic changes and promote their survival and growth. In fact, has been indicated that tumor cells reprogram their energy metabolism because they constantly need to drive cell growth and division through alterations that positively regulate the upregulation of glucose transporters and multiple enzymes of the glycolytic pathway.103,104 Metabolism reprogramming is an important feature of the cancer phenotype with great potential for prognosis and targeted therapy. Some studies have shown that genomic alterations, such as oncogenic MYC amplification and mutations in PIK3CA or TP53 genes, could drive metabolic reprogramming in triple negative breast cancer (TNBC).105,106 The C-MYC oncogene is overexpressed in 40% of TNBC and encodes a transcription factor, c-myc, that links metabolic reprogramming to tumorigenesis.75,105 C-MYC, in addition to directly transactivating genes involved in metabolic pathways, can also cooperate with other crucial metabolic drivers to facilitate critical cellular processes for survival, including hypoxia-inducible factor-1-alpha (HIF-1α)107,108 (Figure 5). Therefore, MYC amplification is crucial for metabolic rewiring of TNBC cells to promote tumorigenesis. In addition, a recent study109 found that in TNBC, the glycolytic subtype with upregulated carbohydrate and nucleotide metabolism, had a relatively high CIN level. Specifically, in this study, it was observed that this glycolytic subtype had frequent somatic copy number alterations involving mainly the chromosomal region 12p12.1–12p13.3. In this chromosomal region are located several glycolytic genes, including ENO2, GAPDH, LDHB, and TPI1, and several cancer-related genes, including FOXM1, FOXJ2, KRAS, and TIGAR. Alterations in this chromosomal region might influence the overexpression of the above glycolytic genes and transcriptional factors, creating subtype-specific metabolic reprogramming (Figure 5). These results revealed that alterations in specific chromosomal regions, which are associated with CIN, might influence the metabolic reprogramming and heterogeneity of TNBCs.
Tumor Promoting Inflammation
Inflammation is the immune system’s response to infection and injury and has been implicated in the pathogeneses of cancer. In fact, it has been indicated that in the tumor microenvironment, latent inflammation contributes to the survival of malignant cells, angiogenesis, metastasis, subversion of adaptive immunity, reduced response to hormones and chemotherapeutic agents.110 Interestingly, has been indicated that CIN, represented among others by the formation of micronuclei (MNs), causes activation of immune responses. MNs, are small nuclei that form when a chromosome or chromosome fragment fails to incorporate into one of the daughter nuclei during cell division. It is usually a sign of genotoxic events and CIN. Specifically, DNA is normally located strictly within the nucleus and mitochondria, however, under conditions of CIN, DNA can be released into the cytoplasm, predominantly through the formation of extranuclear DNA containing MN111 (Figure 6). Due to the collapse of the MN membrane, the DNA leaks into the cytoplasm,111,112 for which MNs are increasingly recognized as a critical source of cytoplasmic DNA fragments.113,114 The release of DNA fragments into the cytoplasm, product of CIN, trigger innate inflammatory responses through cGAS/STING signaling and related pathways.115–117 Induction of CIN, triggers activation of the cGAS/STING pathway or other inflammatory signaling, also due to chromosome missegregation caused by defective function of the SAC118 (Figure 5). Chromosome missegregation leads to the induction of aneuploidies, indicating that cells with chromosomal trisomies accumulate cytoplasmic double stranded DNA (dsDNA), thus leading to the activation of the cGAS/STING pathway.118 Of note, recent studies have shown that chromosomal aneuploidy induces local inflammation by promoting a senescence-associated secretory phenotype (SASP).119 In particular, it has been suggested that the chronic inflammatory response induced by CIN can lead to tissue destruction and cancer.120 In fact, in vivo studies in cancer showed that CIN promoted chronic inflammatory signaling through cGAS/STING and caused aggressive metastatic tumor growth.121 Additionally, in cancer-related inflammation, the induction of CIN by inflammatory mediators, has been suggested as an additional mechanism involved in this process.110,122 The induction of CIN leads to, or favors, the accumulation of random genetic alterations in cancer cells. For instance, overexpression of cyclooxygenase-2 (COX2) in BC cells, has been associated with a significant increase in numerical (tetraploidy) and structural (fusions, breaks) chromosomal alterations123 (Figure 5). COX2 is expressed in immune cells, and is a key player in initiating the inflammatory response.124 The up-regulation and over-expression of COX2 is mainly associated with inflammation, loss of apoptosis, uncontrolled cell proliferation, growth, metastasis, neovascularization, and angiogenesis.124 Upregulation of COX2 has been detected in approximately 40% of human breast carcinoma cases, as well as in preinvasive ductal carcinoma in situ (DCIS) lesions. In addition, COX2 overexpression in BC has been correlated with high-grade, highly proliferative tumors, negative hormone receptor status, HER2 overexpression and disease aggressiveness.125–127 The above results suggested direct associations between COX2 overexpression with an increase in CIN, and strongly supported the validity of COX2 signaling as an anticancer target. In fact, epidemiological analyses have suggested a protective effect of COX-inhibiting drugs in BC.128 Taken together, the above results suggest a reciprocal interaction between CIN and inflammatory signaling in determining genomic integrity and tumor progression.
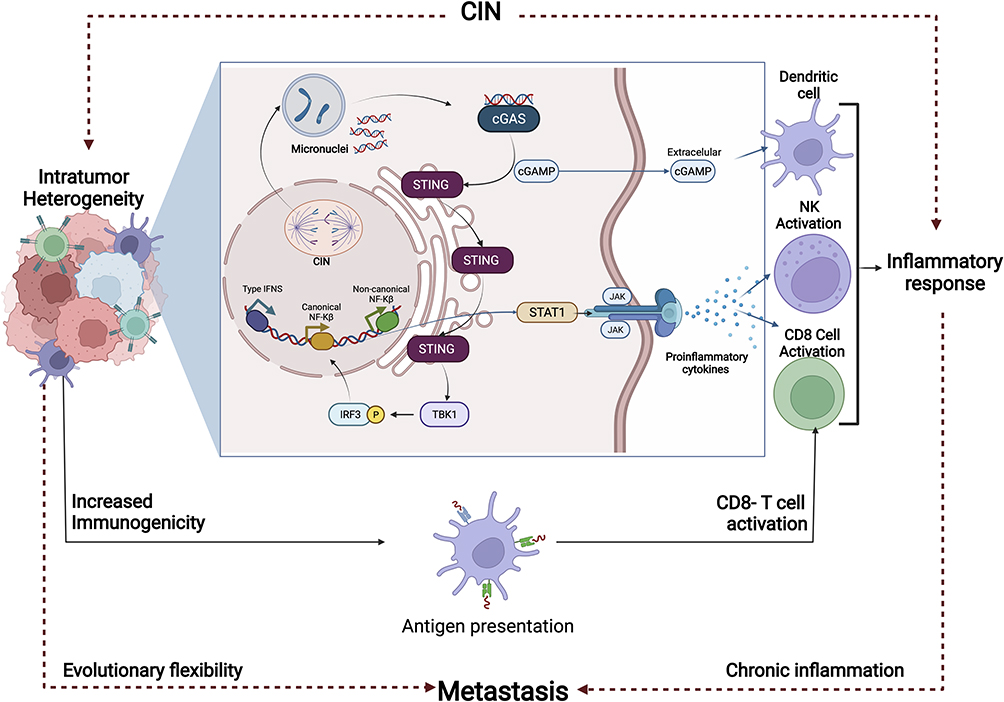 |
Figure 6 Activation of the immune system’s response and induction of metastasis as response to the Chromosomal Instability (CIN). Inflammation is the immune system’s response to infection and injury, and has been implicated in the pathogeneses of Breast Cancer (BC). CIN causes activation of immune responses, due among other, to the formation of micro nuclei (MN). MNs are usually a sign of genotoxic events and CIN. Due to the collapse of the MN membrane, the DNA leaks into the cytoplasm. The release of DNA fragments into the cytoplasm, trigger innate inflammatory responses through cGAS/STING signaling and non-canonical NF-kB. In the tumor microenvironment, activation of the latent inflammatory response contributes to malignant cell survival, angiogenesis, metastasis, and resistance to chemotherapeutic agents, among others. Abbreviations: CD8, cluster of differentiation 8 glycoprotein; cGAS, Cyclic GMP-AMP synthase; cGAMP, Cyclic guanosine monophosphate–adenosine monophosphate; IFNS, Interferons; IRF3, Interferon Regulatory Factor 3; JAK, Janus Kinase; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; NK, Natural killer; STING, Stimulator of interferon genes protein; STAT1, Signal Transducer And Activator Of Transcription 1; TBK1, TANK Binding Kinase 1. |
Evading Immune Destruction
Antitumor immune responses prevent tumor formation, and cancers have developed many mechanisms of immune evasion. The above has been evidenced by studies that have shown that human tumors with high CIN are significantly less immunogenic (tumors characterized by reduced lymphocyte infiltration), compared to those with more stable genomes.129 The above results are paradoxical and interesting, since GI is expected to elicit an innate immune response. In fact, genomic datasets from cancerous tissues and cell lines, combined with analysis of an experimental model of tumor aneuploidy, support a CIN model of tumor evolution, where initial induction of CIN activates cGAS/STING and proinflammatory signaling pathways, leading to a proimmunogenic state in the tumor (Figures 5 and 6). However, as tumors evolve under immune pressure, they suppress the signaling machinery of the innate immune response and antigen presentation, and acquire a cold immune state. Consistent with this hypothesis, human and mouse breast tumors with CIN, that do not experience significant immune pressure during their evolution, retain their proinflammatory character and show high STING expression and, an innate response compared with more genomically stable tumors. Taken together, these results support an important mechanism of immune evasion in chromosomally unstable tumors.
In addition, has been indicated that genomic instability favors the selection of cell clones resistant to immune system, activity due to acquired mutations that facilitate evasion of immune destruction.130–132 Specific examples are the results of in silico studies that demonstrated that overexpression of ABCG2 in BC samples, has a greater potential for CIN and a greater ability to evade immune surveillance. The above genetic characteristics have been associated with highly aggressive and resistant tumors.133 ABCG2 is an efflux transporter implicated in broad-spectrum chemoresistance.134,135
Invasion and Metastasis
Invasion and metastasis are the most defining feature of cancer malignancy and the leading cause of patient mortality. Comparative reports of metastatic and primary tumors suggest that CIN may contribute to the development of cancer metastases via diverse mechanisms.136 One such mechanism includes chromosome segregation errors that lead to the formation of MN (Figure 6). Specifically, the defective and delayed DNA replication that occurs within MN, with impaired checkpoint arrest, leads to the pulverization of incompletely replicated MN DNA in subsequent mitosis.113 Cytosolic DNA in MN leads to activation of non-canonical NF-kB and cGAS-STING signaling pathways in CIN rich cancer cells, thereby stimulating metastasis.121 In BC, the MASTL gene has been associated with metastasis. MASTL gene encodes a microtubule-associated serine/threonine kinase essential for proper progression through mitosis. MASTL gene amplification and protein overexpression have been correlated with increased CIN in BC (Figure 5) and poor patient survival, while knockdown of MASTL has been shown to suppress BC metastasis in vivo.137
CIN and Clonal Heterogeneity in BC
BC is the most common neoplasm around the world and the most prevalent type of cancer in women. This type of cancer had an estimated of 2.3 million new cases diagnosed in 2020, accounting for 11.7% of new cases of all cancer’s types.138 Breast tumor karyotypes are characterized by a high degree of complexity, with multiple chromosomes showing both, numerical and structural alterations,139,140 indicative of CIN. In addition, cytogenetic and molecular studies show that breast tumors are characterized by being multiclonal, suggesting the existence of a high degree of intratumoral heterogeneity, mainly supported by CIN.
Although CIN has been identified in BC, the associations established between CIN with the BC prognosis is variable.7 Thus, while some studies suggest that high levels of CIN are related to better outcome, other studies associate high levels of CIN with resistance to therapy.141 The above because many segregation errors could impact the number of cellular disorders producing a proinflammatory response and leading to cell apoptosis.23 In fact, a recent study reported that the various responses triggered by CIN and clonal heterogeneity depended on each tumor subtype.142 Specifically, this study demonstrated the impact of CIN and clonal heterogeneity on the response to various treatments (endocrine therapy, chemotherapy, combined therapies). The results of this study suggest that intermediate CIN in BC cells has prognostic value and can predict response to chemotherapy, hormonal therapy or combination therapy. In the same way, this study suggests the existence of CIN threshold that, when exceeded, can generate resistance or sensitivity to treatments, and this CIN threshold depends on the status of the ER and HER2.142 In this study the importance of evaluating CIN and clonal heterogeneity in establishing of new therapeutic treatments in BC is shown. Although the studies developed to date have identified some associations between CIN levels with BC subtypes, CIN is highly variable among BC cases, which could explain the characteristic heterogeneous nature of this neoplasm.
CIN and Clonal Heterogeneity in BC Subtypes and Their Associations with Prognosis
BC comprises a group of diseases that are morphologically, molecularly, genomically, and clinically distinct. Clinical classification of BCs is based on expression of ER, progesterone receptor (PR), and HER2. Tumors that lack these receptors are called triple negative BC (TNBC). BCs are also molecularly classified according to their gene expression profiles in Luminal A, Luminal B, HER2 enriched and TNBC. Most TNBC are of the basal subtype143,144 (Table 1).
 |
Table 1 Immunohistochemical, Clinicopathological and Genomic Characteristics of the Intrinsic Tumor Subtypes of Breast Cancer |
CIN in Hereditary BC
Approximately 10% of all BC cases are related to genetic predisposition or family history, with variation by geographic region and ethnicity.145 Familial cancer predisposition generally correlates with mutations in genes involved in DNA double-strand break repair mechanisms, including the BRCA1 and BRCA2 genes. Germline mutations in these two genes have been correlated with a high rate of somatic gene mutations and with a risk factor for breast and ovarian cancer.146 Of note, while BRCA1 mutations have been associated with the more aggressive receptor-negative phenotype, BRCA2 mutations have been associated with significantly lower survival rates than BRCA-/ER+ BC patients.147 Regarding the existing associations between CIN and hereditary BC, interesting results have been reported, such as the associations between CIN and the CHEK2 gene. Germline mutations in the CHEK2 gene represent the second most common cause of this neoplasm after BRCA1/2 lesions. Recently, a study reported somatic loss of the wild-type CHEK2 allele in approximately half of CHEK2-driven BC patients. Interestingly, the results of this study showed that, tumors without CHEK2 LOH were chromosomally stable, whereas BC patients with LOH showed some signs of CIN.148 These results demonstrate the important implications of CIN in hereditary BC and suggest CIN as a possible therapeutic target for the treatment of patients with this neoplasm.
CIN in ER+ and ER- BC
Regarding ER status, it has been indicated that ER-negative (ER-) breast tumors have greater genomic instability than that observed in ER+ breast tumors.13,144 In fact, recent studies have indicated that in patients with ER- BC, CIN is a common event that plays an important role in the development of the disease, due to the close relationships between CIN with the integrity of the genome.144,149 In addition, some studies have showed that extreme CIN was associated with improved long-term survival in ER- BC.13 For instance, a recent study evaluating CIN in 1173 tumors from the TACT breast cancer trial (CRUK01/001) reported an association between extreme CIN and improved outcome in ER- and ER- /HER2- BC.12
In ER+ tumors, CIN has been associated with disease progression,144 due to the instability in the copy number of genes involved in cancer progression and in the evolutionary adaptations for the cell survival.149 Of note, some studies have shown that the Luminal B molecular subtype presents higher levels of CIN when compared to the Luminal A subtype.144
BC patients classified according to their CIN and clonal heterogeneity levels, could provide a better understanding of alterations at the molecular level and potentially lead to new drug targets, as well as distinguish groups with good prognosis from those with poor prognosis.
CIN in HER2+ BC
In HER2+ tumors, it has been indicated that the presence of heterogeneous CIN (intra and intertumoral) could have important therapeutic implications, which include the use of chemotherapeutic agents directed at karyotypically different tumor subpopulations within this type of heterogeneous disease.150 In fact, has been indicated that HER2+ BC tumors show significantly increased CIN relative to HER2- tumors.59
CIN in TNBC
In TNBC (ER-/PR-/HER2-) has been indicated that the CIN is higher, suggesting a possible association between CIN with the negative status of such receptors.144 This is not surprising, considering that the TNBC subtype is characterized by being aggressive and generally poor prognosis, suggesting associations between CIN with poor prognosis in triple-negative tumors. Various gene expression studies have shown that representative cell lines of the triple-negative (TNBC) and HER2+ subtypes present the majority of genetic clusters with a tendency to instability and with the most unstable karyotypes represented by the accumulation of structural and numerical abnormalities compared to luminal subtypes.151,152
In fact, a recent study in TNBC, showed that most of the mutations present in patients resistant to therapy were already present during the pre-treatment stages, although with lower frequencies after treatment. Additionally, they found that resistant patients had higher levels of aneuploidy (high CIN) before treatment than patients who responded to chemotherapy.153 The above results suggest an important role for CIN in response to therapy in TNBC.
CIN and Clonal Heterogeneity in the Treatment of BC
Tumor resistance and recurrence remains to date, one of the main causes of death related to BC. In fact, it has been indicated that intratumoral heterogeneity provides the tumor with the necessary characteristics to survive in stressful environments, thus favoring the generation of clones resistant to therapy.44 Thus, after treatment, the most favorable cellular response corresponds to the decrease in tumor size due to the cytotoxic effects of treatment on the entire tumor population. However, when a tumor presents variable subpopulations, represented by high levels of CIN and clonal heterogeneity, the cells continue to grow and activate signaling pathways involved among others, in angiogenesis and metastasis.154,155 Therefore, although there are various cellular responses to treatment, it has been suggested that the response depends on the presence of specific molecular markers and the levels of CIN and clonal heterogeneity contained in each tumor.156 In fact, the inter- and intratumoral heterogeneity observed in BC patients is the subject of research to be implemented in the histological classification of the tumor and their tumor characterization.14,47 Indeed, important clinical features such as tumor size, grade, lymph node metastasis, and ER status have been correlated with DNA index, an indicator of karyotypic heterogeneity.157
Considering the above, one of the main challenges in the study of BC, is the application of effective target-specific therapies aimed to overcoming resistance and reducing side effects. For this reason, the identification of potential therapeutic targets becomes a priority in BC research and in cancer in general. One of these potential targets is CIN, given the evidence showing its association with response or resistance to therapy in various types of cancer. The identification of pharmacological factors that induce intolerable levels of CIN for tumor cells could lead to highly effective therapies for BC. However, it is important to highlight that although CIN and its associated intratumoral heterogeneity, contribute to cancer cell survival and therapeutic recurrence in BC,158,159 CIN may also be a therapeutic vulnerability that can be exploited to induce cell death and tumor regression or suppression.160–162
Although the role of CIN and clonal heterogeneity in the response to treatment in BC has been demonstrated in cell lines, studies establishing these relationships in patient cohorts are scarce. One of these studies corresponds to a recent publication by Scribano et al162 who studied BC patients undergoing paclitaxel treatment as part of a clinical trial to elucidate the drug’s mechanism of action, showing that paclitaxel treatment increased cell division with chromosomal missegregation to induce cytotoxicity. Additionally, they found that increased CIN in tumor cells before treatment, predicted response to taxane therapy. Similar results were reported by us in BC cell lines,142 where intermediate CIN was linked to drug sensitivity according to among other, the pre-existing CIN level in cancer cells, and the CIN induced by the treatments. For instance, a recent study demonstrated, by using a CIN model of Kras-driven BC, that the presence of CIN during primary tumor development increases the chance to develop therapy resistance,163 which supports the hypothesis of the selection of recurrent aneuploidies for the survival of tumor cells. In addition, it has been indicated that karyotypic heterogeneity might be responsible for drug resistance compared to karyotypically stable tumors164 also associated with worse prognosis.10 The above results suggest that treatment resistance can occur in tumors showing high levels of CIN. In fact, it has been indicated that in an environment with strong selective pressure such as therapy, unstable tumors develop resistance by selecting for recurrent aneuploidies.165
In summary, research to date suggests that CIN acts as a source of genetic variability that under strong selective pressure, such as during targeted therapy, confers an evolutionary advantage to tumor cells.166 Likewise, it is important to highlight that according to these studies, the few initially chromosomally stable cancers that manage to persist during treatment, do so concomitantly with the acquisition of CIN, which increases the risk of developing secondary resistance. Although these findings require further validation, it is clear that CIN could be considered a predictive biomarker of response to therapy and therefore a potential therapeutic target in BC patients. In fact, attempts have been made to therapeutically counteract genomic instability and CIN in early stages as well as in the different tumor subtypes of the disease.
For instance, in the treatment of BC patients with mutations in the BRCA1 and BRCA2 genes, inhibitors of Poly ADP-ribose polymerase (PARP) are used to induce, among others, high levels of CIN to achieve synthetic lethality167 (Figure 7). Another example is the application of monopolar spindle 1 (MPS1) inhibitors, used with the aim of allowing tumor cells to evade mitotic controls and generate non-viable karyotypes. However, this therapeutic approach may be limited by the rapid acquisition of resistance.168 Additional therapeutic strategies include: forcing cells with additional centrosomes to enter multipolar divisions, preventing clustering and thus chromosome segregation by kinesin-like proteins (KIFC1),169 reduce aneuploidy in tumor cells using compounds that inhibit proteotoxic stress such as heat shock protein 90 (HSP90) and metabolic stress such as AMPK associated with the generation of aneuploidy.170,171
 |
Figure 7 Chromosomal Instability (CIN) as a therapeutic target in molecular subtypes of Breast Cancer (BC). Some of the current treatments in BC seek through the application of specific drugs, to induce high levels of CIN to achieve the lethality of tumor cells. The identification of pharmacological factors that induce intolerable levels of CIN for tumor cells, could lead to highly effective therapies for BC. Abbreviations: ER, Estrogen Receptor; TAM, Tamoxifen; PICH, Plk1-interacting checkpoint helicase; PARP, Poly ADP-ribose polymerase. |
In the treatment of HER2+ BC tumors, it has been suggested that both, anthracyclines and platinum agents, have preferential activity in tumors with defined patterns of karyotypic complexity (structural CIN).150 Additional therapies suggested for the treatment of this tumor subtype (HER2+), include the use of taxane therapy (paclitaxel, docetaxel, etc.) based on CIN status (Figure 7). Indeed, associations between CIN and taxane resistance in vitro172 and, between CIN and the prediction of intrinsic resistance to taxanes in vivo173 have been indicated. Additionally, and taking into account the high heterogeneity (intra and inter heterogeneity) observed in HER2+ BC patients, has been raised the possibility of using combined therapies (for example taxanes and HER2 directed therapies) in order to target distinct tumor populations with variations in the CIN levels. In fact, clinical trials in advanced BC patients, have consistently shown that taxane therapy in combination with trastuzumab or lapatinib is associated with additional clinical benefit in HER2+ BC patients,174–178 compared to expected response rates when are used individual therapies (monotherapy) with trastuzumab or lapatinib179,180 or taxane alone.174–178,181 The results of these studies show the benefit of the combined use of trastuzumab and taxanes in the treatment of HER2+ BC patients, since these treatments target karyotypically different subpopulations within the same tumor.182,183 Taking into account the results obtained to date, it is possible to consider that anthracyclines and platinum agents target karyotypically more complex tumors (CIN) in HER2+ tumors, in contrast to taxanes, where previous studies suggest greater activity in tumors with chromosomal stability.150
Regarding to the TNBC treatment, and highlighting the utility of targeting CIN to induce excessive genomic alteration, this molecular BC subtype is often treated with taxanes and radiation therapy, both of which induce DNA and chromosomal damage.184,185 Radiotherapy induces a high frequency of structural chromosomal alterations (structural CIN) and causes chaotic mitosis that can lead to the loss and/or gain of complete chromosomes (numerical CIN),186,187 while taxanes stabilize microtubules by affecting the chromosome segregation thus leading to numerical CIN.184,185 The extreme CIN induction due to these therapies cause cytotoxicity and therefore apoptosis.162,173,188 However, although these therapies are initially very effective, resistance and recurrence are common, suggesting that the level of CIN is not sufficient to uniformly induce cell death. In addition, recent studies have shown that the induction of CIN through the depletion of Plk1-interacting checkpoint helicase (PICH), promotes cell death in TNBC189 (Figure 7). PICH is a DNA-dependent ATPase associated with cell cycle and proliferation, highly expressed in TNBC compared to other BC subtypes. High expression of PICH, in TNBC cells, has been correlated with an increased risk of distal metastasis and worse clinical outcomes. In TNBC, cells depend on PICH for proper chromosome segregation, so loss of PICH induces CIN (as evidenced by chromatin bridging, chromosomes lagging in anaphase, chromosomal abnormalities, micronuclei, or binucleation).190,191 The above results evidence the clinical potential of PICH and suggest it as a promising therapeutic target for the treatment of patients with TNBC.189
An additional drug target associated with CIN in TNBC is the centrosome itself. Evolving therapies postulate to centrosomes as a promising prognostic tool and therapeutic target in the treatment of BC. In fact, it has been indicated that centrosome defects such as amplification, overelongated centrioles, structural defects, and loss of primary cilium nucleation are common in BC.192,193 Considering that centrosome aberrations cause chromosome missegregation which is associated with CIN and tumorigenesis, this cellular structure constitutes a promising prognostic tool and a therapeutic target in the treatment of BC. The emerging centrosome-targeting therapies currently being evaluated in BC clinical trials include: centrosome kinase inhibitors such as those that target polo-like kinases (PLK),194 cell cycle related kinases, and Aurora kinases.195,196 However, although many therapies are in various stages of development to target aberrant centrosomes in BC, a major looming limitation of anti-centrosome therapies is the development of drug resistance.
A greater understanding of the role that CIN plays in the response to therapy in BC could provide a rational basis to understand the role of individual cytotoxic therapies in the management of this neoplasm.
Conclusion
Studies conducted to date, show the important role that CIN play in the prognosis and treatment of cancer. Although CIN is being approached from different perspectives as a therapeutic target, the development of robust biomarkers that take advantage of the great predictive power and evaluation of CIN is crucial for the treatment and management of resistance of this disease. Tumors classified according to their CIN level, could provide a better understanding of alterations at the molecular level and potentially lead to new drug targets. Further understanding of the complexity of CIN and identification of methods to directly examine it could support novel strategies aimed at target CIN and improving outcomes in cancer.
Acknowledgments
We thank Ministerio de Ciencia, Tecnología e Innovación, Universidad Pedagógica y Tecnológica de Colombia and Universidad del Rosario for their support in the development of this research.
Funding
This research was funded by the Ministerio de Ciencia, Tecnología e Innovación 874 call and by the Universidad Pedagógica y Tecnológica de Colombia.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chandrakasan S, Ye CJ, Chitlur M, et al. Malignant fibrous histiocytoma two years after autologous stem cell transplant for Hodgkin lymphoma: evidence for genomic instability. Pediatr Blood Cancer. 2011;56(7):1143–1145. doi:10.1002/pbc.22929
2. Gagos S, Irminger-Finger I. Chromosome instability in neoplasia: chaotic roots to continuous growth. Int J Biochem Cell Biol. 2005;37(5):1014–1033. doi:10.1016/j.biocel.2005.01.003
3. Heng HH, Bremer SW, Stevens JB, et al. Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013;32(3–4):325–340. doi:10.1007/s10555-013-9427-7
4. Thomson AB, Wallace WHB. Treatment of paediatric Hodgkin’s disease. Eur J Cancer. 2002;38(4):468–477. doi:10.1016/S0959-8049(01)00335-5
5. Weaver BAA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy Acts Both Oncogenically and as a Tumor Suppressor. Cancer Cell. 2007;11(1):25–36. doi:10.1016/j.ccr.2006.12.003
6. Dayal JHS, Albergante L, Newman TJ, South AP. Quantitation of multiclonality in control and drug-treated tumour populations using high-throughput analysis of karyotypic heterogeneity. Converg Sci Phys Oncol. 2015;1(2):025001. doi:10.1088/2057-1739/1/2/025001
7. Birkbak NJ, Eklund AC, Li Q, et al. Paradoxical Relationship between Chromosomal Instability and Survival Outcome in Cancer. Cancer Res. 2011;71(10):3447–3452. doi:10.1158/0008-5472.CAN-10-3667
8. Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38(9):1043–1048. doi:10.1038/ng1861
9. Nakamura H, Saji H, Idiris A, et al. Chromosomal instability detected by fluorescence in situ hybridization in surgical specimens of non-small cell lung cancer is associated with poor survival. Clin Cancer Res. 2003;9(6):2294–2299.
10. Walther A, Houlston R, Tomlinson I. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut. 2008;57(7):941–950. doi:10.1136/gut.2007.135004
11. Williams BR, Prabhu VR, Hunter KE, et al. Aneuploidy Affects Proliferation and Spontaneous Immortalization in Mammalian Cells. Science. 2008;322(5902):703–709. doi:10.1126/science.1160058
12. Jamal-Hanjani M, A’Hern R, Birkbak NJ, et al. Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: a prospective validation cohort study from the TACT trial. Ann Oncol. 2015;26(7):1340–1346. doi:10.1093/annonc/mdv178
13. Roylance R, Endesfelder D, Gorman P, et al. Relationship of Extreme Chromosomal Instability with Long-term Survival in a Retrospective Analysis of Primary Breast Cancer. Cancer Epidemiol Biomarkers Prevention. 2011;20(10):2183–2194. doi:10.1158/1055-9965.EPI-11-0343
14. Tanaka K, Hirota T. Chromosomal instability: a common feature and a therapeutic target of cancer. Biochimica et Biophysica Acta. 2016;1866(1):64–75. doi:10.1016/j.bbcan.2016.06.002
15. Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100(1):57–70. doi:10.1016/S0092-8674(00)81683-9
16. Hanahan D, Weinberg RA. Hallmarks of Cancer: the Next Generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
17. Hanahan D. Hallmarks of Cancer: new Dimensions. Cancer Discov. 2022;12(1):31–46. doi:10.1158/2159-8290.CD-21-1059
18. Shen Z. Genomic instability and cancer: an introduction. J Mol Cell Biol. 2011;3(1):1–3. doi:10.1093/jmcb/mjq057
19. Duijf PHG, Nanayakkara D, Nones K, Srihari S, Kalimutho M, Khanna KK. Mechanisms of Genomic Instability in Breast Cancer. Trends Mol Med. 2019;25(7):595–611. doi:10.1016/j.molmed.2019.04.004
20. Pikor L, Thu K, Vucic E, Lam W. The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 2013;32(3–4):341–352. doi:10.1007/s10555-013-9429-5
21. Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396(6712):643–649. doi:10.1038/25292
22. Gadgil R, Barthelemy J, Lewis T, Leffak M. Replication stalling and DNA microsatellite instability. Biophys Chem. 2017;225:38–48. doi:10.1016/j.bpc.2016.11.007
23. Xu Z, Verma A, Naveed U, Bakhoum SF, Khosravi P, Elemento O. Deep learning predicts chromosomal instability from histopathology images. iScience. 2021;24(5):102394. doi:10.1016/j.isci.2021.102394
24. Clancy S. DNA Damage & Repair: mechanisms for Maintaining DNA Integrity. Nature Educ. 2015;1(1):103.
25. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58(5):235–263. doi:10.1002/em.22087
26. Bailey SM, Cornforth MN, Ullrich RL, Goodwin EH. Dysfunctional mammalian telomeres join with DNA double-strand breaks. DNA Repair (Amst). 2004;3(4):349–357. doi:10.1016/j.dnarep.2003.11.007
27. Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349(6255):1483–1489. doi:10.1126/science.aab4082
28. Thompson SL, Bakhoum SF, Compton DA. Mechanisms of Chromosomal Instability. Curr Biol. 2010;20(6):R285–R295. doi:10.1016/j.cub.2010.01.034
29. Torres EM, Williams BR, Amon A. Aneuploidy: cells Losing Their Balance. Genetics. 2008;179(2):737–746. doi:10.1534/genetics.108.090878
30. Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. Chromosome Missegregation and Apoptosis in Mice Lacking the Mitotic Checkpoint Protein Mad2. Cell. 2000;101(6):635–645. doi:10.1016/S0092-8674(00)80875-2
31. Lara-Gonzalez P, Westhorpe FG, Taylor SS. The Spindle Assembly Checkpoint. Curr Biol. 2012;22(22):R966–R980. doi:10.1016/j.cub.2012.10.006
32. Gregan J, Polakova S, Zhang L, Tolić-Nørrelykke IM, Cimini D. Merotelic kinetochore attachment: causes and effects. Trends Cell Biol. 2011;21(6):374–381. doi:10.1016/j.tcb.2011.01.003
33. Martínez-A C, van Wely KHM. Are aneuploidy and chromosome breakage caused by a CINgle mechanism? Cell Cycle. 2010;9(12):2275–2280. doi:10.4161/cc.9.12.11865
34. Janssen A, van der Burg M, Szuhai K, Kops GJPL, Medema RH. Chromosome Segregation Errors as a Cause of DNA Damage and Structural Chromosome Aberrations. Science. 2011;333(6051):1895–1898. doi:10.1126/science.1210214
35. Obe G, Durante M. DNA Double Strand Breaks and Chromosomal Aberrations. Cytogenet Genome Res. 2010;128(1–3):8–16. doi:10.1159/000303328
36. Pastink A, Eeken JCJ, Lohman PHM. Genomic integrity and the repair of double-strand DNA breaks. Mutation Res. 2001;480-481:37–50. doi:10.1016/S0027-5107(01)00167-1
37. Kasparek TR, Humphrey TC. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin Cell Dev Biol. 2011;22(8):886–897. doi:10.1016/j.semcdb.2011.10.007
38. San Filippo J, Sung P, Klein H. Mechanism of Eukaryotic Homologous Recombination. Annu Rev Biochem. 2008;77(1):229–257. doi:10.1146/annurev.biochem.77.061306.125255
39. Thompson LH, Schild D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutation Res. 2001;477(1–2):131–153. doi:10.1016/S0027-5107(01)00115-4
40. Daley JM, Kwon Y, Niu H, Sung P. Investigations of homologous recombination pathways and their regulation. Yale J Biol Med. 2013;86(4):453–461.
41. Cahill D. Mechanisms of eukaryotic DNA double strand break repair. Front Bioscience. 2006;11(1):1958. doi:10.2741/1938
42. Chu E. Tumor heterogeneity and implications for clinical practice. Oncology. 2014;28(9):726.
43. McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep. 2012;13(6):528–538. doi:10.1038/embor.2012.61
44. McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: past, Present, and the Future. Cell. 2017;168(4):613–628. doi:10.1016/j.cell.2017.01.018
45. Zasadil LM, Britigan EMC, Weaver BA. 2n or not 2n: aneuploidy, polyploidy and chromosomal instability in primary and tumor cells. Semin Cell Dev Biol. 2013;24(4):370–379. doi:10.1016/j.semcdb.2013.02.001
46. Nowell PC. The Clonal Evolution of Tumor Cell Populations. Science. 1976;194(4260):23–28. doi:10.1126/science.959840
47. Bakhoum SF, Landau DA. Chromosomal Instability as a Driver of Tumor Heterogeneity and Evolution. Cold Spring Harb Perspect Med. 2017;7(6):a029611. doi:10.1101/cshperspect.a029611
48. Lukow DA, Sheltzer JM. Chromosomal instability and aneuploidy as causes of cancer drug resistance. Trends Cancer. 2022;8(1):43–53. doi:10.1016/j.trecan.2021.09.002
49. Raab M, Sanhaji M, Zhou S, et al. Blocking Mitotic Exit of Ovarian Cancer Cells by Pharmaceutical Inhibition of the Anaphase-Promoting Complex Reduces Chromosomal Instability. Neoplasia. 2019;21(4):363–375. doi:10.1016/j.neo.2019.01.007
50. Lukow DA, Sausville EL, Suri P, et al. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev Cell. 2021;56(17):2427–2439.e4. doi:10.1016/j.devcel.2021.07.009
51. Ippolito MR, Martis V, Hong C, et al. Aneuploidy-driven genome instability triggers resistance to chemotherapy. bioRxiv. 2020;9:313924. doi:10.1101/2020.09.25.313924
52. Ippolito MR, Martis V, Martin S, et al. Gene copy-number changes and chromosomal instability induced by aneuploidy confer resistance to chemotherapy. Dev Cell. 2021;56(17):2440–2454.e6. doi:10.1016/j.devcel.2021.07.006
53. Davies MA, Samuels Y. Analysis of the genome to personalize therapy for melanoma. Oncogene. 2010;29(41):5545–5555. doi:10.1038/onc.2010.323
54. Jiang B, Chapter LL. 2 PI3K/PTEN Signaling in Angiogenesis and Tumorigenesis. In:. 2009;19–65. doi:10.1016/S0065-230X(09)02002-8
55. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–5510. doi:10.1038/onc.2008.245
56. Cimini D, Tanzarella C, Degrassi F. Differences in malsegregation rates obtained by scoring ana-telophases or binucleate cells. Mutagenesis. 1999;14(6):563–568. doi:10.1093/mutage/14.6.563
57. Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol. 2008;180(4):665–672. doi:10.1083/jcb.200712029
58. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human Breast Cancer: correlation of Relapse and Survival with Amplification of the HER-2/ neu Oncogene. Science. 1987;235(4785):177–182. doi:10.1126/science.3798106
59. Ellsworth RE, Ellsworth DL, Patney HL, et al. Amplification of HER2 is a marker for global genomic instability. BMC Cancer. 2008;8(1):297. doi:10.1186/1471-2407-8-297
60. Montagna C, Andrechek ER, Padilla-Nash H, Muller WJ, Ried T. Centrosome abnormalities, recurring deletions of chromosome 4, and genomic amplification of HER2/neu define mouse mammary gland adenocarcinomas induced by mutant HER2/neu. Oncogene. 2002;21(6):890–898. doi:10.1038/sj.onc.1205146
61. Schneeweiss A, Sinn HP, Ehemann V, et al. Centrosomal aberrations in primary invasive breast cancer are associated with nodal status and hormone receptor expression. Int J Cancer. 2003;107(3):346–352. doi:10.1002/ijc.11408
62. Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460(7252):278–282. doi:10.1038/nature08136
63. Aoki K, Tamai Y, Horiike S, Oshima M, Taketo MM. Colonic polyposis caused by mTOR-mediated chromosomal instability in Apc+/Δ716 Cdx2+/− compound mutant mice. Nat Genet. 2003;35(4):323–330. doi:10.1038/ng1265
64. Wang X, Zhou YX, Qiao W, et al. Overexpression of Aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene. 2006;25(54):7148–7158. doi:10.1038/sj.onc.1209707
65. Smith CA, Pollice AA, Gu LP, et al. Correlations among p53, Her-2/neu, and ras overexpression and aneuploidy by multiparameter flow cytometry in human breast cancer: evidence for a common phenotypic evolutionary pattern in infiltrating ductal carcinomas. Clin Cancer Res. 2000;6(1):112–126.
66. Chan GKT, Schaar BT, Yen TJ. Characterization of the Kinetochore Binding Domain of CENP-E Reveals Interactions with the Kinetochore Proteins CENP-F and hBUBR1. J Cell Biol. 1998;143(1):49–63. doi:10.1083/jcb.143.1.49
67. Jablonski SA, Chan GKT, Cooke CA, Yen TJ, Earnshaw WC. The hBUB1 and hBUBR1 kinases sequentially assemble onto kinetochores during prophase with hBUBR1 concentrating at the kinetochore plates in mitosis. Chromosoma. 1998;107(6–7):386–396. doi:10.1007/s004120050322
68. Liao H, Winkfein RJ, Mack G, Rattner JB, Yen TJ. CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J Cell Biol. 1995;130(3):507–518. doi:10.1083/jcb.130.3.507
69. Zhu X, Mancini MA, Chang KH, et al. Characterization of a novel 350-kilodalton nuclear phosphoprotein that is specifically involved in mitotic-phase progression. Mol Cell Biol. 1995;15(9):5017–5029. doi:10.1128/MCB.15.9.5017
70. O’Brien SL, Fagan A, Fox EJP, et al. CENP-F expression is associated with poor prognosis and chromosomal instability in patients with primary breast cancer. Int J Cancer. 2007;120(7):1434–1443. doi:10.1002/ijc.22413
71. Zhivotovsky B, Kroemer G. Apoptosis and genomic instability. Nat Rev Mol Cell Biol. 2004;5(9):752–762. doi:10.1038/nrm1443
72. Hanel W, Moll UM. Links between mutant p53 and genomic instability. J Cell Biochem. 2012;113(2):433–439. doi:10.1002/jcb.23400
73. Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170(6):1062–1078. doi:10.1016/j.cell.2017.08.028
74. Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2(8):594–604. doi:10.1038/nrc864
75. The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi:10.1038/nature11412
76. Børresen-Dale AL. TP53 and breast cancer. Hum Mutat. 2003;21(3):292–300. doi:10.1002/humu.10174
77. Eyfjord JE, Thorlacius S, Steinarsdottir M, Valgardsdottir R, Ogmundsdottir HM, Anamthawat-Jonsson K. p53 Abnormalities and Genomic Instability in Primary Human Breast Carcinomas. Cancer Res. 1995;1:458.
78. Gretarsdottir S, Thorlacius S, Valgardsdottir R, et al. BRCA2 and p53 mutations in primary breast cancer in relation to genetic instability. Cancer Res. 1998;58(5):859–862.
79. Sigurdsson S, Bödvarsdottir SK, Anamthawat-Jonsson K, et al. p53 Abnormality and Chromosomal Instability in the Same Breast Tumor Cells. Cancer Genet Cytogenet. 2000;121(2):150–155. doi:10.1016/S0165-4608(00)00260-0
80. Fukasawa K, Choi T, Kuriyama R, Rulong S. Abnormal Centrosome Amplification in the Absence of p53. Science. 1996;271(5256):1744–1747. doi:10.1126/science.271.5256.1744
81. Carroll PE, Okuda M, Horn HF, et al. Centrosome hyperamplification in human cancer: chromosome instability induced by p53 mutation and/or Mdm2 overexpression. Oncogene. 1999;18(11):1935–1944. doi:10.1038/sj.onc.1202515
82. Murnane JP, Sabatier L. Chromosome rearrangements resulting from telomere dysfunction and their role in cancer. BioEssays. 2004;26(11):1164–1174. doi:10.1002/bies.20125
83. Sabatier L, Ricoul M, Pottier G, Murnane JP. The Loss of a Single Telomere Can Result in Instability of Multiple Chromosomes in a Human Tumor Cell Line. Mol Cancer Res. 2005;3(3):139–150. doi:10.1158/1541-7786.MCR-04-0194
84. Lo AWI, Sprung CN, Fouladi B, et al. Chromosome Instability as a Result of Double-Strand Breaks near Telomeres in Mouse Embryonic Stem Cells. Mol Cell Biol. 2002;22(13):4836–4850. doi:10.1128/MCB.22.13.4836-4850.2002
85. Campisi J. Cellular senescence, cancer and aging: the telomere connection. Exp Gerontol. 2001;36(10):1619–1637. doi:10.1016/S0531-5565(01)00160-7
86. Raptis S, Bapat B. Genetic instability in human tumors. In: Cancer: Cell Structures, Carcinogens and Genomic Instability. Birkhäuser-Verlag; 2006:303–320. doi:10.1007/3-7643-7378-4_13
87. Kronenwett U, Ploner A, Zetterberg A, et al. Genomic Instability and Prognosis in Breast Carcinomas. Cancer Epidemiol Biomarkers Prevention. 2006;15(9):1630–1635. doi:10.1158/1055-9965.EPI-06-0080
88. Griffith JD, Comeau L, Rosenfield S, et al. Mammalian Telomeres End in a Large Duplex Loop. Cell. 1999;97(4):503–514. doi:10.1016/S0092-8674(00)80760-6
89. Kurabayashi R, Takubo K, Aida J, et al. Luminal and cancer cells in the breast show more rapid telomere shortening than myoepithelial cells and fibroblasts. Hum Pathol. 2008;39(11):1647–1655. doi:10.1016/j.humpath.2008.04.005
90. Radpour R, Barekati Z, Haghighi MM, et al. Correlation of telomere length shortening with promoter methylation profile of p16/Rb and p53/p21 pathways in breast cancer. Modern Pathol. 2010;23(5):763–772. doi:10.1038/modpathol.2009.195
91. Bisoffi M, Heaphy CM, Griffith JK. Telomeres: prognostic markers for solid tumors. Int J Cancer. 2006;119(10):2255–2260. doi:10.1002/ijc.22120
92. Heaphy CM, Baumgartner KB, Bisoffi M, Baumgartner RN, Griffith JK. Telomere DNA Content Predicts Breast Cancer–Free Survival Interval. Clin Cancer Res. 2007;13(23):7037–7043. doi:10.1158/1078-0432.CCR-07-0432
93. Heaphy CM, Subhawong AP, Gross AL, et al. Shorter telomeres in luminal B, HER-2 and triple-negative breast cancer subtypes. Modern Pathol. 2011;24(2):194–200. doi:10.1038/modpathol.2010.198
94. Poonepalli A, Banerjee B, Ramnarayanan K, Palanisamy N, Putti TC, Hande MP. Telomere-mediated genomic instability and the clinico-pathological parameters in breast cancer. Genes Chromosomes Cancer. 2008;47(12):1098–1109. doi:10.1002/gcc.20608
95. Mironchik Y, Winnard PT, Vesuna F, et al. Twist Overexpression Induces In vivo Angiogenesis and Correlates with Chromosomal Instability in Breast Cancer. Cancer Res. 2005;65(23):10801–10809. doi:10.1158/0008-5472.CAN-05-0712
96. Qin Q, Xu Y, He T, Qin C, Xu J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012;22(1):90–106. doi:10.1038/cr.2011.144
97. Howard TD, Paznekas WA, Green ED, et al. Mutations in TWIST, a basic helix–loop–helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15(1):36–41. doi:10.1038/ng0197-36
98. Gripp KW, Stolle CA, Celle L, McDonald-McGinn DM, Whitaker LA, Zackai EH. TWIST gene mutation in a patient with radial aplasia and craniosynostosis: further evidence for heterogeneity of Baller-Gerold syndrome. Am J Med Genet. 1999;82(2):170–176. doi:10.1002/(SICI)1096-8628(19990115)82:2<170:
99. Yang J, Mani SA, Donaher JL, et al. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell. 2004;117(7):927–939. doi:10.1016/j.cell.2004.06.006
100. Rosivatz E, Becker I, Specht K, et al. Differential Expression of the Epithelial-Mesenchymal Transition Regulators Snail, SIP1, and Twist in Gastric Cancer. Am J Pathol. 2002;161(5):1881–1891. doi:10.1016/S0002-9440(10)64464-1
101. Kwok WK, Ling M-T, Lee T-W, et al. Up-Regulation of TWIST in Prostate Cancer and Its Implication as a Therapeutic Target. Cancer Res. 2005;65(12):5153–5162. doi:10.1158/0008-5472.CAN-04-3785
102. Vesuna F, Winnard P, Raman V, Glackin C. Twist overexpression promotes chromosomal instability in the breast cancer cell line MCF-7. Cancer Genet Cytogenet. 2006;167(2):189–191. doi:10.1016/j.cancergencyto.2006.01.014
103. Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23(5):537–548. doi:10.1101/gad.1756509
104. Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20(1):51–56. doi:10.1016/j.gde.2009.10.009
105. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer Discov. 2015;5(10):1024–1039. doi:10.1158/2159-8290.CD-15-0507
106. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020;122(1):4–22. doi:10.1038/s41416-019-0650-z
107. Dang CV, Le A, Gao P. MYC-Induced Cancer Cell Energy Metabolism and Therapeutic Opportunities. Clin Cancer Res. 2009;15(21):6479–6483. doi:10.1158/1078-0432.CCR-09-0889
108. Dejure FR, Eilers M. MYC and tumor metabolism: chicken and egg. EMBO J. 2017;36(23):3409–3420. doi:10.15252/embj.201796438
109. Gong Y, Ji P, Yang YS, et al. Metabolic-Pathway-Based Subtyping of Triple-Negative Breast Cancer Reveals Potential Therapeutic Targets. Cell Metab. 2021;33(1):51–64.e9. doi:10.1016/j.cmet.2020.10.012
110. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–1081. doi:10.1093/carcin/bgp127
111. Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell. 2013;154(1):47–60. doi:10.1016/j.cell.2013.06.007
112. Lewis CW, Golsteyn RM. Cancer cells that survive checkpoint adaptation contain micronuclei that harbor damaged DNA. Cell Cycle. 2016;15(22):3131–3145. doi:10.1080/15384101.2016.1231287
113. Crasta K, Ganem NJ, Dagher R, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482(7383):53–58. doi:10.1038/nature10802
114. Zhang CZ, Spektor A, Cornils H, et al. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522(7555):179–184. doi:10.1038/nature14493
115. Mackenzie KJ, Carroll P, Martin CA, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548(7668):461–465. doi:10.1038/nature23449
116. Heijink AM, Talens F, Jae LT, et al. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat Commun. 2019;10(1):100. doi:10.1038/s41467-018-07927-y
117. Reisländer T, Lombardi EP, Groelly FJ, et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat Commun. 2019;10(1):3143. doi:10.1038/s41467-019-11048-5
118. Bakhoum SF, Cantley LC. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell. 2018;174(6):1347–1360. doi:10.1016/j.cell.2018.08.027
119. Andriani GA, Almeida VP, Faggioli F, et al. Whole Chromosome Instability induces senescence and promotes SASP. Sci Rep. 2016;6(1):35218. doi:10.1038/srep35218
120. Dou Z, Ghosh K, Vizioli MG, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550(7676):402–406. doi:10.1038/nature24050
121. Bakhoum SF, Ngo B, Laughney AM, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553(7689):467–472. doi:10.1038/nature25432
122. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi:10.1038/nature07205
123. Singh B, Vincent L, Berry JA, Multani AS, Lucci A. Cyclooxygenase-2 Expression Induces Genomic Instability in MCF10A Breast Epithelial Cells. J Surgical Res. 2007;140(2):220–226. doi:10.1016/j.jss.2007.01.039
124. Gandhi J, Khera L, Gaur N, Paul C, Kaul R. Role of Modulator of Inflammation Cyclooxygenase-2 in Gammaherpesvirus Mediated Tumorigenesis. Front Microbiol. 2017;8. doi:10.3389/fmicb.2017.00538
125. Boland GP, Butt IS, Prasad R, Knox WF, Bundred NJ. COX-2 expression is associated with an aggressive phenotype in ductal carcinoma in situ. Br J Cancer. 2004;90(2):423–429. doi:10.1038/sj.bjc.6601534
126. Ristimäki A, Sivula A, Lundin J, et al. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002;62(3):632–635.
127. Subbaramaiah K, Norton L, Gerald W, Dannenberg AJ. Cyclooxygenase-2 Is Overexpressed in HER-2/neu-positive Breast Cancer. J Biol Chem. 2002;277(21):18649–18657. doi:10.1074/jbc.M111415200
128. Howe LR. Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Res. 2007;9(4):210. doi:10.1186/bcr1678
129. Tripathi R, Modur V, Senovilla L, Kroemer G, Komurov K. Suppression of tumor antigen presentation during aneuploid tumor evolution contributes to immune evasion. Oncoimmunology. 2019;8(11):1657374. doi:10.1080/2162402X.2019.1657374
130. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34. doi:10.1186/s13073-017-0424-2
131. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3(11):999–1005. doi:10.1038/ni1102-999
132. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural Innate and Adaptive Immunity to Cancer. Annu Rev Immunol. 2011;29(1):235–271. doi:10.1146/annurev-immunol-031210-101324
133. Sicchieri RD, da Silveira WA, Mandarano LRM, et al. ABCG2 is a potential marker of tumor-initiating cells in breast cancer. Tumor Biol. 2015;36(12):9233–9243. doi:10.1007/s13277-015-3647-0
134. Benderra Z, Faussat AM, Sayada L, et al. Breast Cancer Resistance Protein and P-Glycoprotein in 149 Adult Acute Myeloid Leukemias. Clin Cancer Res. 2004;10(23):7896–7902. doi:10.1158/1078-0432.CCR-04-0795
135. Diestra JE, Scheffer GL, Català I, et al. Frequent expression of the multi-drug resistance-associated protein BCRP/MXR/ABCP/ABCG2 in human tumours detected by the BXP-21 monoclonal antibody in paraffin-embedded material. J Pathol. 2002;198(2):213–219. doi:10.1002/path.1203
136. Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352(6282):169–175. doi:10.1126/science.aaf2784
137. Rogers S, McCloy RA, Parker BL, et al. MASTL overexpression promotes chromosome instability and metastasis in breast cancer. Oncogene. 2018;37(33):4518–4533. doi:10.1038/s41388-018-0295-z
138. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
139. Isola JJ, Kallioniemi OP, Chu LW, et al. Genetic aberrations detected by comparative genomic hybridization predict outcome in node-negative breast cancer. Am J Pathol. 1995;147(4):905–911.
140. Persson K, Pandis N, Mertens F, et al. Chromosomal aberrations in breast cancer: a comparison between cytogenetics and comparative genomic hybridization. Genes Chromosomes Cancer. 1999;25(2):115–122.
141. Ji X, Lu Y, Tian H, Meng X, Wei M, Cho WC. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed Pharmacother. 2019;114:108800. doi:10.1016/j.biopha.2019.108800
142. Vargas-Rondón N, Pérez-Mora E, Villegas E. Role of chromosomal instability and clonal heterogeneity in the therapy response of breast cancer cell lines. Cancer Biol Med. 2020;17(4):970–985. doi:10.20892/j.issn.2095-3941.2020.0028
143. López S, Lim EL, Horswell S, et al. Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat Genet. 2020;52(3):283–293. doi:10.1038/s41588-020-0584-7
144. Smid M, Hoes M, Sieuwerts AM, et al. Patterns and incidence of chromosomal instability and their prognostic relevance in breast cancer subtypes. Breast Cancer Res Treat. 2011;128(1):23–30. doi:10.1007/s10549-010-1026-5
145. Kuchenbaecker KB, Hopper JL, Barnes DR, et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA. 2017;317(23):2402. doi:10.1001/jama.2017.7112
146. Varol U, Kucukzeybek Y, Alacacioglu A, et al. BRCA genes: BRCA 1 and BRCA 2. J BUON. 2018;23(4):862–866.
147. Kotsopoulos J. BRCA Mutations and Breast Cancer Prevention. Cancers. 2018;10(12):524. doi:10.3390/cancers10120524
148. Iyevleva AG, Aleksakhina SN, Sokolenko AP, et al. Somatic loss of the remaining allele occurs approximately in half of CHEK2-driven breast cancers and is accompanied by a border-line increase of chromosomal instability. Breast Cancer Res Treat. 2022. doi:10.1007/s10549-022-06517-3
149. Endesfelder D, Burrell RA, Kanu N, et al. Chromosomal Instability Selects Gene Copy-Number Variants Encoding Core Regulators of Proliferation in ER + Breast Cancer. Cancer Res. 2014;74(17):4853–4863. doi:10.1158/0008-5472.CAN-13-2664
150. Burrell RA, Juul N, Johnston SR, Reis-Filho JS, Szallasi Z, Swanton C. Targeting chromosomal instability and tumour heterogeneity in HER2-positive breast cancer. J Cell Biochem. 2010;111(4):782–790. doi:10.1002/jcb.22781
151. Tang Y, Wang Y, Kiani MF, Wang B. Classification, Treatment Strategy, and Associated Drug Resistance in Breast Cancer. Clin Breast Cancer. 2016;16(5):335–343. doi:10.1016/j.clbc.2016.05.012
152. Toft DJ, Cryns VL. Minireview: basal-Like Breast Cancer: from Molecular Profiles to Targeted Therapies. Molecular Endocrinol. 2011;25(2):199–211. doi:10.1210/me.2010-0164
153. Kim C, Gao R, Sei E, et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell. 2018;173(4):879–893.e13. doi:10.1016/j.cell.2018.03.041
154. Luque-Bolivar A, Pérez-Mora E, Villegas VE, Rondón-Lagos M. Resistance and Overcoming Resistance in Breast Cancer. Breast Cancer. 2020;12:211–229. doi:10.2147/BCTT.S270799
155. Sansregret L, Vanhaesebroeck B, Swanton C. Determinants and clinical implications of chromosomal instability in cancer. Nat Rev Clin Oncol. 2018;15(3):139–150. doi:10.1038/nrclinonc.2017.198
156. McGranahan N, Swanton C. Biological and Therapeutic Impact of Intratumor Heterogeneity in Cancer Evolution. Cancer Cell. 2015;27(1):15–26. doi:10.1016/j.ccell.2014.12.001
157. Dayal JHS, Sales MJ, Corver WE, et al. Multiparameter DNA content analysis identifies distinct groups in primary breast cancer. Br J Cancer. 2013;108(4):873–880. doi:10.1038/bjc.2013.42
158. Kwei KA, Kung Y, Salari K, Holcomb IN, Pollack JR. Genomic instability in breast cancer: pathogenesis and clinical implications. Mol Oncol. 2010;4(3):255–266. doi:10.1016/j.molonc.2010.04.001
159. Bakhoum SF, Compton DA. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J Clin Invest. 2012;122(4):1138–1143. doi:10.1172/JCI59954
160. Giam M, Rancati G. Aneuploidy and chromosomal instability in cancer: a jackpot to chaos. Cell Div. 2015;10(1):3. doi:10.1186/s13008-015-0009-7
161. Funk LC, Zasadil LM, Weaver BA. Living in CIN: mitotic Infidelity and Its Consequences for Tumor Promotion and Suppression. Dev Cell. 2016;39(6):638–652. doi:10.1016/j.devcel.2016.10.023
162. Scribano CM, Wan J, Esbona K, et al. Chromosomal instability sensitizes patient breast tumors to multipolar divisions induced by paclitaxel. Sci Transl Med. 2021;13:610. doi:10.1126/scitranslmed.abd4811
163. Rowald K, Mantovan M, Passos J, et al. Negative Selection and Chromosome Instability Induced by Mad2 Overexpression Delay Breast Cancer but Facilitate Oncogene-Independent Outgrowth. Cell Rep. 2016;15(12):2679–2691. doi:10.1016/j.celrep.2016.05.048
164. Lee AJX, Endesfelder D, Rowan AJ, et al. Chromosomal Instability Confers Intrinsic Multidrug Resistance. Cancer Res. 2011;71(5):1858–1870. doi:10.1158/0008-5472.CAN-10-3604
165. Thompson L, Jeusset L, Lepage C, McManus K. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers. 2017;9(12):151. doi:10.3390/cancers9110151
166. Salgueiro L, Buccitelli C, Rowald K, et al. Acquisition of chromosome instability is a mechanism to evade oncogene addiction. EMBO Mol Med. 2020;12:3. doi:10.15252/emmm.201910941
167. Rieckhoff J, Meyer F, Classen S, et al. Exploiting Chromosomal Instability of PTEN-Deficient Triple-Negative Breast Cancer Cell Lines for the Sensitization Against PARP1 Inhibition in a Replication-Dependent Manner. Cancers. 2020;12(10):2809. doi:10.3390/cancers12102809
168. Daniel J, Coulter J, Woo JH, Wilsbach K, Gabrielson E. High levels of the Mps1 checkpoint protein are protective of aneuploidy in breast cancer cells. Proce National Acad Sci. 2011;108(13):5384–5389. doi:10.1073/pnas.1007645108
169. Ogden A, Garlapati C, Li X. Multi-institutional study of nuclear KIFC1 as a biomarker of poor prognosis in African American women with triple-negative breast cancer. Sci Rep. 2017;7(1):42289. doi:10.1038/srep42289
170. Biebl MM, Buchner J. Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harb Perspect Biol. 2019;11(9):a034017. doi:10.1101/cshperspect.a034017
171. Watkins TBK, Lim EL, Petkovic M, et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature. 2020;587(7832):126–132. doi:10.1038/s41586-020-2698-6
172. Bouchet BP, Bertholon J, Falette N, et al. Paclitaxel resistance in untransformed human mammary epithelial cells is associated with an aneuploidy-prone phenotype. Br J Cancer. 2007;97(9):1218–1224. doi:10.1038/sj.bjc.6603936
173. Swanton C, Nicke B, Schuett M, et al. Chromosomal instability determines taxane response. Proce National Acad Sci. 2009;106(21):8671–8676. doi:10.1073/pnas.0811835106
174. Slamon DJ, Leyland-Jones B, Shak S, et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N Eng J Med. 2001;344(11):783–792. doi:10.1056/NEJM200103153441101
175. Marty M, Cognetti F, Maraninchi D, et al. Randomized Phase II Trial of the Efficacy and Safety of Trastuzumab Combined With Docetaxel in Patients With Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer Administered As First-Line Treatment: the M77001 Study Group. J Clin Oncol. 2005;23(19):4265–4274. doi:10.1200/JCO.2005.04.173
176. Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus Adjuvant Chemotherapy for Operable HER2-Positive Breast Cancer. N Eng J Med. 2005;353(16):1673–1684. doi:10.1056/NEJMoa052122
177. Gasparini G, Gion M, Mariani L, et al. Randomized Phase II Trial of weekly paclitaxel alone versus trastuzumab plus weekly paclitaxel as first-line therapy of patients with Her-2 positive advanced breast cancer. Breast Cancer Res Treat. 2007;101(3):355–365. doi:10.1007/s10549-006-9306-9
178. de Leo A, Gomez HL, Aziz Z, et al. Phase III, Double-Blind, Randomized Study Comparing Lapatinib Plus Paclitaxel With Placebo Plus Paclitaxel As First-Line Treatment for Metastatic Breast Cancer. J Clin Oncol. 2008;26(34):5544–5552. doi:10.1200/JCO.2008.16.2578
179. Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and Safety of Trastuzumab as a Single Agent in First-Line Treatment of HER2 -Overexpressing Metastatic Breast Cancer. J Clin Oncol. 2002;20(3):719–726. doi:10.1200/JCO.2002.20.3.719
180. Baselga J, Carbonell X, Castañeda-Soto NJ, et al. Phase II Study of Efficacy, Safety, and Pharmacokinetics of Trastuzumab Monotherapy Administered on a 3-Weekly Schedule. J Clin Oncol. 2005;23(10):2162–2171. doi:10.1200/JCO.2005.01.014
181. Gomez HL, Doval DC, Chavez MA, et al. Efficacy and Safety of Lapatinib As First-Line Therapy for ErbB2-Amplified Locally Advanced or Metastatic Breast Cancer. J Clin Oncol. 2008;26(18):2999–3005. doi:10.1200/JCO.2007.14.0590
182. Pack SD, Alper ÖM, Stromberg K, et al. Simultaneous Suppression of Epidermal Growth Factor Receptor and c-erbB-2 Reverses Aneuploidy and Malignant Phenotype of a Human Ovarian Carcinoma Cell Line. Cancer Res. 2004;64(3):789–794. doi:10.1158/0008-5472.CAN-03-1982
183. Le XF, Lammayot A, Gold D, et al. Genes Affecting the Cell Cycle, Growth, Maintenance, and Drug Sensitivity Are Preferentially Regulated by Anti-HER2 Antibody through Phosphatidylinositol 3-Kinase-AKT Signaling. J Biol Chem. 2005;280(3):2092–2104. doi:10.1074/jbc.M403080200
184. de Ruijter TC, Veeck J, de Hoon JPJ, van Engeland M, Tjan-Heijnen VC. Characteristics of triple-negative breast cancer. J Cancer Res Clin Oncol. 2011;137(2):183–192. doi:10.1007/s00432-010-0957-x
185. Mustacchi G, de Laurentiis M. The role of taxanes in triple-negative breast cancer: literature review. Drug Des Devel Ther. 2015;4303. doi:10.2147/DDDT.S86105
186. Bakhoum SF, Kabeche L, Wood MD, et al. Numerical chromosomal instability mediates susceptibility to radiation treatment. Nat Commun. 2015;6(1):5990. doi:10.1038/ncomms6990
187. Huang RX, Zhou PK. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct Target Ther. 2020;5(1):60. doi:10.1038/s41392-020-0150-x
188. Lee HS, Lee NCO, Kouprina N, et al. Effects of Anticancer Drugs on Chromosome Instability and New Clinical Implications for Tumor-Suppressing Therapies. Cancer Res. 2016;76(4):902–911. doi:10.1158/0008-5472.CAN-15-1617
189. Huang Y, Li W, Yan W, et al. Loss of PICH promotes chromosome instability and cell death in triple-negative breast cancer. Cell Death Dis. 2019;10(6):428. doi:10.1038/s41419-019-1662-6
190. Ke Y, Huh JW, Warrington R, et al. PICH and BLM limit histone association with anaphase centromeric DNA threads and promote their resolution. EMBO J. 2011;30(16):3309–3321. doi:10.1038/emboj.2011.226
191. Nielsen CF, Huttner D, Bizard AH, et al. PICH promotes sister chromatid disjunction and co-operates with topoisomerase II in mitosis. Nat Commun. 2015;6(1):8962. doi:10.1038/ncomms9962
192. Marteil G, Guerrero A, Vieira AF, et al. Over-elongation of centrioles in cancer promotes centriole amplification and chromosome missegregation. Nat Commun. 2018;9(1):1258. doi:10.1038/s41467-018-03641-x
193. Pannu V, Mittal K, Cantuaria G, et al. Rampant centrosome amplification underlies more aggressive disease course of triple negative breast cancers. Oncotarget. 2015;6(12):10487–10497. doi:10.18632/oncotarget.3402
194. Yeow ZY, Lambrus BG, Marlow R, et al. Targeting TRIM37-driven centrosome dysfunction in 17q23-amplified breast cancer. Nature. 2020;585(7825):447–452. doi:10.1038/s41586-020-2690-1
195. Schatten H, Sun QY. Functions and dysfunctions of the mammalian centrosome in health, disorders, disease, and aging. Histochem Cell Biol. 2018;150(4):303–325. doi:10.1007/s00418-018-1698-1
196. Schöffski P. Polo-Like Kinase (PLK) Inhibitors in Preclinical and Early Clinical Development in Oncology. Oncologist. 2009;14(6):559–570. doi:10.1634/theoncologist.2009-0010
197. Focke CM, van Diest PJ, Decker T. St Gallen 2015 subtyping of luminal breast cancers: impact of different Ki67-based proliferation assessment methods. Breast Cancer Res Treat. 2016;159(2):257–263. doi:10.1007/s10549-016-3950-5
198. Harbeck N, Penault-Llorca F, Cortes J, et al. Breast cancer. Nat Rev Dis Primers. 2019;5(1):66. doi:10.1038/s41572-019-0111-2
199. Prat A, Cheang MCU, Martín M, et al. Prognostic Significance of Progesterone Receptor–Positive Tumor Cells Within Immunohistochemically Defined Luminal A Breast Cancer. J Clin Oncol. 2013;31(2):203–209. doi:10.1200/JCO.2012.43.4134
200. Rouzier R, Perou CM, Symmans WF, et al. Breast Cancer Molecular Subtypes Respond Differently to Preoperative Chemotherapy. Clin Cancer Res. 2005;11(16):5678–5685. doi:10.1158/1078-0432.CCR-04-2421
201. Goldhirsch A, Winer EP, Coates AS, et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol. 2013;24(9):2206–2223. doi:10.1093/annonc/mdt303
202. Prat A, Bianchini G, Thomas M, et al. Research-Based PAM50 Subtype Predictor Identifies Higher Responses and Improved Survival Outcomes in HER2-Positive Breast Cancer in the NOAH Study. Clin Cancer Res. 2014;20(2):511–521. doi:10.1158/1078-0432.CCR-13-0239
203. Hayes DF, Thor AD, Dressler LG, et al. HER2 and Response to Paclitaxel in Node-Positive Breast Cancer. N Eng J Med. 2007;357(15):1496–1506. doi:10.1056/NEJMoa071167
204. Sørlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proce National Acad Sci. 2003;100(14):8418–8423. doi:10.1073/pnas.0932692100
205. Harrell JC, Prat A, Parker JS, et al. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat. 2012;132(2):523–535. doi:10.1007/s10549-011-1619-7
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Development of a Prognostic Stratification Model and Identification of BDH1 as an Oncoprotein in Breast Cancer Based on Subcluster-Specific Markers of B-Cell Subsets
Kang S, Li Z, Lv C, Zhang F
Breast Cancer: Targets and Therapy 2026, 18:580906
Published Date: 10 March 2026