Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Cholinergic mechanisms in an organic dust model simulating an acute exacerbation in patients with COPD
Authors Palmberg L, Sundblad B, Ji J, Karén J, Larsson K ![]()
Received 18 April 2018
Accepted for publication 23 July 2018
Published 1 November 2018 Volume 2018:13 Pages 3611—3624
DOI https://doi.org/10.2147/COPD.S171495
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Lena Palmberg,* Britt-Marie Sundblad,* Jie Ji, Jakob Karén, Kjell Larsson
Work Environment Toxicology, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden
*These authors contributed equally to this work
Background: Exposure in a pig barn induces airway inflammation that has similarities with the response observed in acute exacerbations in COPD.
Methods: A total of 15 smokers with COPD and 15 healthy non-smokers were exposed for 2 hours in a pig barn (in vivo exposure). Symptoms were assessed, lung function measured, and blood and sputum samples taken before and after exposure. Blood neutrophils were isolated and stimulated ex vivo with dust from a pig barn and acetylcholine, and inflammatory markers were analyzed.
Results: In vivo exposure caused more symptoms and greater lung function fall in COPD patients than in controls. Baseline concentrations of MMP9, TIMP1, IL6, CXCL8, in sputum and neutrophil blood count were higher in COPD patients than in controls. In vivo exposure increased MMP9, TIMP1, IL6, CXCL8, TNFα, and LTB4 in sputum and MMP9 and IL6 in blood, with no difference between the groups, and serum CRP increased more in COPD subjects. Expression of choline acetyltransferase and acetylcholinesterase on sputum and blood cells was similar in the groups and uninfluenced by in vivo exposure. Dust exposure ex vivo increased choline acetyltransferase expression in neutrophils, but the dust and acetylcholine response did not differ between the groups before and after in vivo exposure.
Conclusion: COPD patients exposed in a pig barn experience symptoms similar to those in acute exacerbations and lung function deterioration that is unrelated to bronchial responsiveness. Cholinergic mechanisms are involved in the inflammatory response to dust, with no difference between COPD and non-smokers.
Keywords: organic dust, airway inflammation, chronic obstructive pulmonary disease, COPD, pig barn
Introduction
In healthy subjects, a few hours’ exposure in a pig barn induces a substantial increase in bronchial responsiveness to methacholine,1 an intense airway inflammation with a cellular dominance of neutrophils, and a systemic reaction reflected by increased body temperature and increased levels of circulating IL6 and acute-phase proteins.2,3 In a number of studies, dust collected in pig barns has been shown to have strong pro-inflammatory properties when bronchial epithelial cells, lymphocytes, alveolar macrophages, monocytes, and neutrophils have been stimulated ex vivo.4–7 Bioaerosols in agricultural settings, including pig farming, have been recognized as risk factors for development of airway diseases, such as chronic bronchitis and COPD.8,9
Exacerbations in COPD are trigged by airway bacterial and viral infections,10 but other factors, such as air pollution, may also cause exacerbations.11 Living close to livestock farms increases respiratory symptoms in subjects with COPD, suggesting an increased risk of exacerbations,8 as exacerbations are associated with increased symptoms, more intense airway inflammation, and systemic engagement. Characteristic exacerbation features are more cough, phlegm, more severe dyspnea, and increased numbers of neutrophils and pro-inflammatory cytokines, eg, IL6, IL1β, and TNFα, and chemokines eg, CXCL8 (IL8) in the airways and systemic circulation.12,13
Ex vivo whole-blood stimulation with dust from a pig barn and its components demonstrate an increased responsiveness with augmentation of cytokine/chemokine release in COPD subjects compared with healthy volunteers.14 These data may suggest that circulating blood cells in COPD subjects may be primed to respond more to microbial/inflammatory insult. It is known that anticholinergic drugs prevent exacerbations induced by pro-inflammatory stimuli, such as microorganisms, in COPD, supporting the assumption that cholinergic mechanisms are of importance for the inflammatory response.15 Pro-inflammatory stimuli may elicit cholinergic response by direct interaction with or regulation of muscarinic receptors, by increased expression of choline acetyltransferase (ChAT) and/or by decreased expression of acetylcholinesterase (AChE).16
The inflammatory response to acute exposure in a pig barn in healthy subjects has similarities with inflammatory features of acute exacerbations in COPD. Our hypothesis was that clinical, inflammatory, and physiological responses to acute exposure to organic material in a pig barn are stronger in COPD than in healthy non-smokers. Furthermore, we hypothesized that cholinergic responses may be augmented in COPD compared with healthy non-smokers. Therefore, symptoms, airway and systemic inflammation and lung function were assessed before and after exposure in a pig barn and a bronchial methacholine challenge in subjects with COPD and healthy non-smokers. In addition, blood neutrophils were collected before and after exposure and stimulated with dust (collected in the barn) and acetylcholine, and responses to different drugs interacting with cholinergic mechanisms were studied ex vivo.
Methods
Subjects
Fifteen smokers (recruited from the local primary care and by advertisements) with mild or moderate COPD (FEV1 >50% of predicted normal value, ie, stage 1 and 2 according to GOLD)17 and 15 healthy non-smokers matched by age and sex (controls) were included in the study. None of the subjects in the COPD or control group had a history of atopy, asthma, or allergy (confirmed with negative skin-prick tests to a panel of 12 common allergens) or any other chronic diseases. None had suffered from any respiratory tract infection during the last 2 weeks prior to the study. Seven subjects in the COPD group were on pharmacological COPD treatment, five a combination of inhaled steroids and a long-acting β2-agonist, three inhaled tiotropium, one indacaterol, and one salbutamol on demand. No medication was allowed for 48 hours prior to exposure. All subjects gave written informed consent, and the study was approved by the Ethics Committee, Stockholm, Sweden (protocol 2010/2:8).
Study design
All participants visited the laboratory on three occasions ≥1 week apart. At each visit, lung function was measured, induced sputum performed, and blood samples taken. At the first visit, baseline measurements were performed. At the other two visits, measurements were conducted after a bronchial methacholine challenge or after exposure in a pig barn. Medication with short-acting β2-agonists was not allowed for 8 hours and long-acting β2-agonists and anticholinergics for 48 hours prior to each visit.
Subjects from both groups were exposed for 2 hours in the pig barn assisting the farmer who was weighing pigs. Measurements of exposure levels were carried out on each occasion. Spirometry was performed at the first visit, at the visit in connection with the methacholine bronchial challenge, and 7 hours after commencing exposure in the pig barn. Peak expiratory flow (PEF), symptoms, and body temperature were recorded before and 2, 3, and 5 hours after exposure. For study design, see Figure 1.
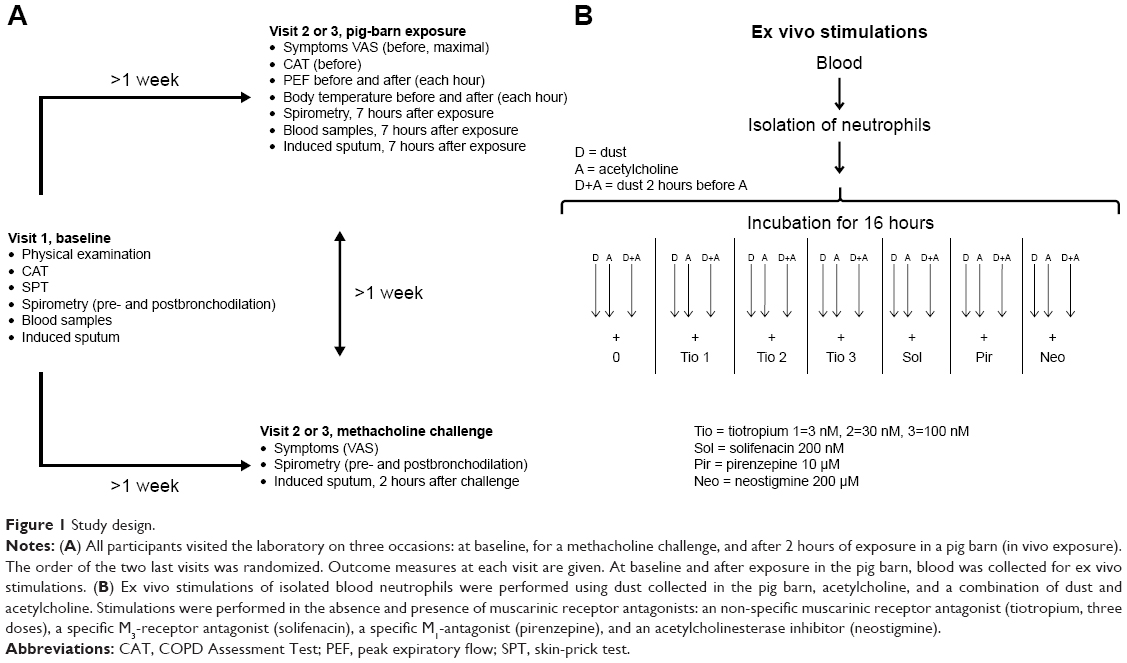 | Figure 1 Study design. |
Throughout the manuscript, “in vivo exposure” refers to 2 hours’ exposure in the pig barn. “Ex vivo exposure” refers to stimulation (dust, acetylcholine, and dust + acetylcholine) and drug intervention of isolated blood neutrophils collected from the participants before and after in vivo exposure.
Exposure measurements
IOM filter cassettes (25 mm; SKC Ltd, Blandford Forum, UK) and plastic cyclones (25 mm; Casella Solutions, London, UK) were used to monitor inhalable and respirable dust levels, respectively. The samplers were placed in the breathing zone on two subjects at each exposure. The cassettes were equipped with Teflon filters (1 μm; Millipore, Sundbyberg, Sweden).
Symptoms
The COPD Assessment Test was completed at visit 1 and before in vivo exposure. Symptoms (headache, chills, fatigue, muscle pain, nausea, sneezing, nasal congestion, nasal secretion, cough, chest tightness, shortness of breath, and wheezing) were assessed on a VAS (0–100 mm) before and immediately after conclusion of the methacholine challenge and immediately after and at the time of maximal experienced symptoms after in vivo exposure. The subjects were requested to put a cross on a scale where 0 indicated no symptoms and 100 indicated unbearable symptoms. All participants were contacted by phone 24 hours after exposure and asked about symptoms experienced.
Lung function and bronchial provocation
VC, FVC, and FEV1 were measured using a wedge spirometer (Vitalograph, Buckingham, UK) according to American Thoracic Society criteria.18 PEF was measured with a mini-Wright peak-flow meter (Clement Clarke International Ltd, Harlow, UK). Local lung function reference values were used.19,20 Bronchial provocation to methacholine was performed as previously described.21 Inhalation of the diluent was followed by inhalation of doubling concentrations of methacholine up to 32 mg/mL, starting at 0.5 mg/mL. The result was expressed as the cumulative dose causing a 20% decrease in FEV1 (PD20 FEV1).
Sputum induction and processing
Sputum induction and processing were performed as previously described,22 with minor modifications. After inhalation of salbutamol (0.4 mg) sputum was induced by inhalation of saline in increasing concentrations (0.9%, 3%, 4%, and 5%) using an ultrasonic nebulizer (UltraNeb 2000; Drive DeVilbiss Healthcare, Mannheim, Germany) with an output of 3 mg/mL. Each concentration was inhaled for 6–7 minutes, followed by FEV1 measurement. Subjects were asked to blow their nose and rinse their mouth with water before the induction and before expectorating sputum by deep coughing. The sample was considered adequate macroscopically if it appeared to be free from saliva and had a weight ≥2,000 mg.
Sputum color and weight were determined and an equal volume of 0.1% dithiothreitol added to the whole sputum sample and rocked for 15–25 minutes in a 37°C water bath and thereafter passed through a filter. The sample was centrifuged (10 minutes at 280 g) and the supernatant stored in aliquots at −70°C until analysis. Cell pellets were resuspended in 2 mL PBS, and total cell count and viability test with Trypan blue was performed.
Serum/blood
Peripheral blood was collected in supplement-free tubes and EDTA Vacutainer® tubes (BD Biosciences, San Jose, CA, USA). Samples in supplement-free tubes were left 60 minutes to clot and then centrifuged twice at 3,000 rpm for 10 minutes. The serum obtained was then aliquoted and stored in −70°C until analyses. Samples in EDTA Vacutainer tubes were used for flow-cytometry analysis.
Ex vivo stimulation of blood cells
Isolation of blood neutrophils
Whole blood was mixed with equal volumes of PBS (Thermo Fisher Scientific, Waltham, MA, USA)–dextran 2% (Sigma-Aldrich Co., St Louis, MO, USA) and left for sedimentation for 30–40 minutes. Leukocyte-containing dextran blood was put gently on top of an equal volume of Lymphoprep (STEMCELL™ Technologies, Vancouver, BC, Canada) and then centrifuged at 600 g for 25 minutes without using brake. The cell pellet was then resuspended in PBS, washed and lysed in deionized water, washed, and finally resuspended in RPMI (Sigma-Aldrich Co.) supplemented with 1% L-glutamine, 10% heat-inactivated FBS, and penicillin–streptomycin antibiotics (BioWhittaker™; Lonza, Basel, Switzerland). Cell concentration was calculated in a Bürker chamber.
Stimulation of peripheral blood neutrophils
Cells (500,000 per well) were seeded in 24-well plates (Nunc A/S, Roskilde, Denmark). Cells were stimulated with organic dust collected from pig-barn shelves and window ledges about 1.2 m above the floor (1 μg/mL), acetylcholine (100 μM), or a combination of dust and acetylcholine, which were added to the wells. To these, stimuli of tiotropium (3, 30, 100 nM; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), solifenacin (200 nM), (Santa Cruz Biotechnology Inc.), neostigmine (200 μM; Santa Cruz Biotechnology), or pirenzepine (10 μM; Sigma-Aldrich Co.) was added. After incubation at 37°C in 5% CO2 for 16 hours, cells and cell media were centrifuged at 400 g for 10 minutes and cells and supernatants collected for further analyses.
Blood-cell distribution
To determine absolute cell numbers in peripheral blood, Trucount tubes containing a specific number of beads were used. Whole blood and a four-color antibody mixture (CD3FITC/CD8PE/CD45PerCp/CD4APC; BD Biosciences, San Jose, CA, USA) were added to a Trucount tube (BD Biosciences, San Jose, CA, USA) and incubated in the dark at 20°C–22°C for 15 minutes. To lyse red blood cells, 450 μL Pharm Lyse™ (BD Biosciences) was added and an additional 10-minute incubation in the dark at 20°C–22°C performed. All samples were then analyzed on a FACSCalibur™ (BD Biosciences) using a set made in Attractor (BD Biosciences) to perform a five-part white-blood-cell differential. The results are presented as number of cells per specified volume of blood.
Cell analysis in sputum, blood, and isolated neutrophils by flow cytometry
Blood (100 μL) was incubated with CXCR1–fluorescein isothiocyanate (FITC; BD Biosciences), CXCR2–phycoerythrin (PE; BD Biosciences), CXCR3-FITC (BD Biosciences), CD25–allophycocyanin (APC; BD Biosciences), or CD69-FITC (BD Biosciences) antibodies and incubated in the dark at room temperature (RT) for 30 minutes. Then, 450 μL lysis solution (BD Biosciences) was added to each tube and incubated for 10 minutes, before samples were analyzed with the FACSCalibur.
Stimulated blood neutrophils were collected after 16 hours of stimulation by centrifugation at 300 g for 10 minutes. The cell pellet was divided into three tubes. CXCR1-FITC and CXCR2-PE antibodies were added to one tube and incubated in the dark at RT for 20 minutes, before samples were analyzed on the FACSCalibur. The other two tubes were analyzed for ChAT and AChE.
Stimulated neutrophils, 200 μL blood, and 500 μL sputum were permeabilized by addition of Cytoperm/Cytofix (BD) and incubated for 20 minutes at RT in the dark. Cells were centrifuged at 400 g for 5 minutes, before 500 μL of Cytoperm/Cytofix wash was added and samples incubated for 10 minutes. Cells were again centrifuged at 400 g for 5 minutes, before primary antibody ChAT (Abcam) or AChE (Abcam) was added and tubes incubated for 30 minutes in the dark at RT. Cells were then washed with 500 μL Cytoperm/Cytofix wash and centrifuged at 400 g for 5 minutes, before the secondary antibody FITC–antimouse (Abcam) was added. Samples were incubated an additional 30 minutes in the dark at RT before analysis with the FACSCalibur.
ELISA
MMP9, TIMP1, TGFβ, and LTB4 were measured using a DuoSet ELISA MMP9 kit, DuoSet ELISA TIMP1 kit, DuoSet ELISA TGFβ kit (R&D Systems Inc., Minneapolis, MN, USA), and LTB4 EIA kit (Cayman Chemical, Ann Arbor, MI, USA). The lower detection limit for MMP9, TIMP1, and TGFβ was 31.125 pg/mL and for LTB4 3.91 pg/mL. Serum IL6, TNFα, and CRP were measured using an HS Quantikine ELISA IL6 kit, HS Quantikine ELISA TNFα kit (R&D Systems Inc.), and high-sensitivity CRP test with detection limits of 0.2 pg/mL, 0.5 pg/mL, and 0.2 ng/mL, respectively. All analyses were performed in accordance with the manufacturer’s instructions. Measurement of IL6 and CXCL8 in sputum was performed using an in-house ELISA with detection limits of 3 pg/mL and 50 pg/mL, respectively.23
Luminex
CXCL10, CXCL8, and TNFα in the supernatant of ex vivo stimulated neutrophils were determined based on Luminex technology. A Bio-Plex Pro™ reagent kit III (Bio-Rad Laboratories Inc., Hercules, CA, USA) was used in accordance with the manufacturer’s instructions. Data were analyzed with Bio-Plex Manager™ software (Bio-Rad Laboratories Inc.). For all duplicated samples, intra-assay variation <15% was accepted. Detection limits of CXCL10 (IP10), CXCL8, and TNFα were 0.52, 0.47, and 1.29 pg/mL, respectively.
Statistical analyses
Results are presented as median (25th–75th percentiles) and mean (95% CI) for lung function, mean (SEM), or mean (range). Results from in vivo exposure (pig barn and methacholine) were compared by mean of Wilcoxon signed-rank tests within groups, Mann–Whitney U tests between groups (blood and sputum analyses), or paired and unpaired t-tests (lung function, symptoms).
Ex vivo results were compared by mean of ANOVA for repeated measurements, followed by dependent or independent t-tests post hoc for three or more comparisons. For pairwise comparisons, Wilcoxon signed-rank test was used within groups and Mann–Whitney U test between groups. P<0.05 was considered statistically significant for the in vivo part of the study. In the ex vivo part of the study, P<0.01 was considered statistically significant as a correction for multiple comparisons. Statistical analyses were performed using Statistica (Dell, Austin, TX, USA).
Results
In vivo effects of exposure in the pig barn
Exposure, symptoms, and lung function
The mean (range) level of inhalable dust was 6.3 (2.1–17.0) mg/m3 and respirable dust 0.5 (0.32–1.3) mg/m3. At baseline, COPD Assessment Test score mean (range) was 3.3 (0–7) in the controls and 17.7 (7–34) in the COPD group and before in vivo exposure 3.8 (1–11) in the controls and 17.3 (4–34) in the COPD group. Exposure in vivo induced sneezing (P=0.027) and cough (P=0.003) in the controls and headache (P=0.006), fatigue (P=0.016), cough (P=0.039), chest tightness (P=0.002), and breathlessness (P=0.003) in the COPD group. Post-exposure headache (P=0.02), fatigue (P=0.04), chest tightness (P=0.001), and breathlessness (P<0.0001) were more severe in the COPD group than in controls. Body temperature increased by 0.4°C (P=0.001) following exposure in both groups.
Baseline FEV1 and FVC were lower in subjects with COPD than in controls, and exposure in vivo induced a greater fall in FEV1 (P<0.0001) and FVC (P<0.0001) in the COPD group than in controls (Figure 2). In the controls, PEF fell from 531 (95% CI 457–604) L/min to 489 (411–566) L/min (7.9%; P=0.001) and from 325 (295–356) L/min to 240 (201–240) L/min (26.2%; P=0.0007) in the COPD group (P=0.0004 between groups).
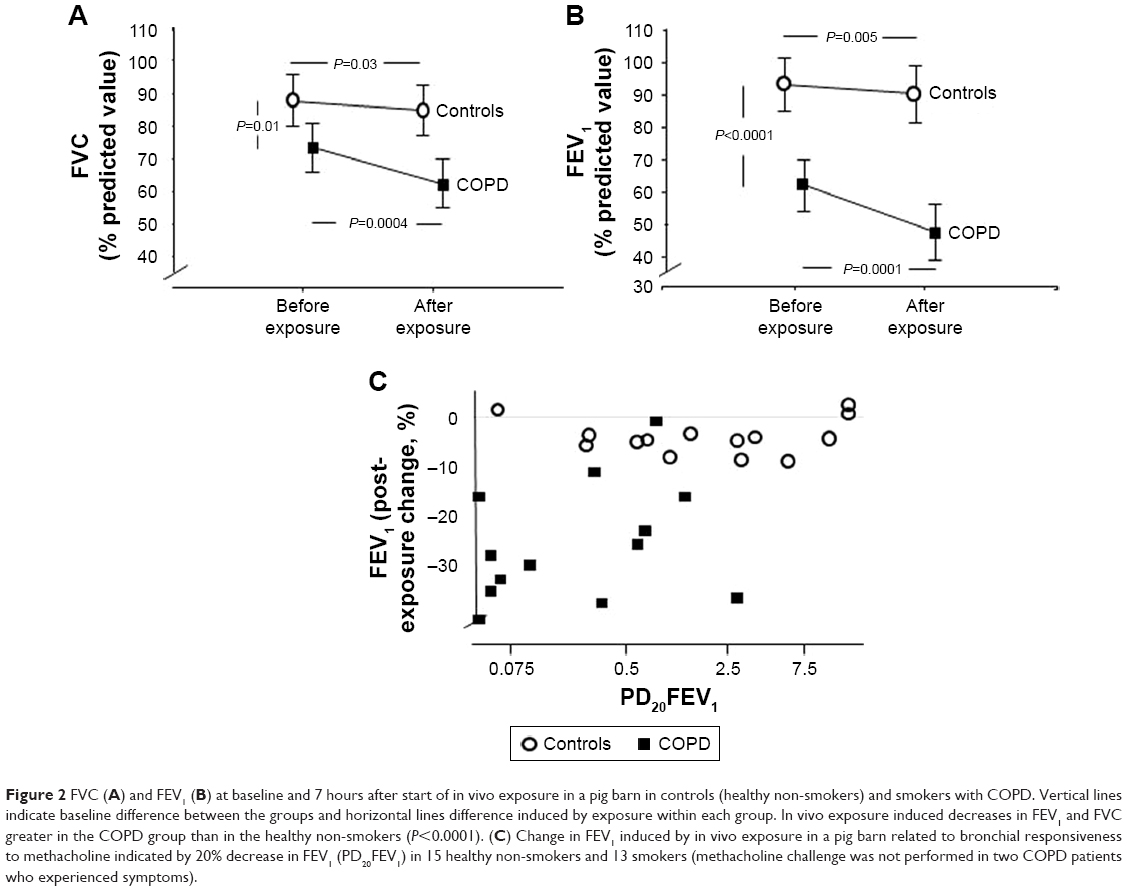 | Figure 2 FVC (A) and FEV1 (B) at baseline and 7 hours after start of in vivo exposure in a pig barn in controls (healthy non-smokers) and smokers with COPD. Vertical lines indicate baseline difference between the groups and horizontal lines difference induced by exposure within each group. In vivo exposure induced decreases in FEV1 and FVC greater in the COPD group than in the healthy non-smokers (P<0.0001). (C) Change in FEV1 induced by in vivo exposure in a pig barn related to bronchial responsiveness to methacholine indicated by 20% decrease in FEV1 (PD20FEV1) in 15 healthy non-smokers and 13 smokers (methacholine challenge was not performed in two COPD patients who experienced symptoms). |
The methacholine challenge induced chest tightness (P=0.0015) in controls and cough (P=0.049) in the COPD group, with no difference between the groups. All symptoms disappeared within 24 hours after the challenge. Bronchial responsiveness to methacholine was enhanced in COPD subjects compared with controls (P=0.004, Table 1). There was no correlation between FEV1 after in vivo exposure and bronchial responsiveness to methacholine (Figure 2).
 | Table 1 Baseline characteristics of healthy non-smokers (controls) and smokers with COPD |
Cells and mediators following in vivo exposure
Sputum
Sputum-cell concentrations did not differ between the groups at baseline (P=0.15). Exposure in vivo induced increased numbers of sputum cells in COPD subjects (P=0.002) and controls (P<0.001), with no difference between the groups (P=0.88; Figure 3).
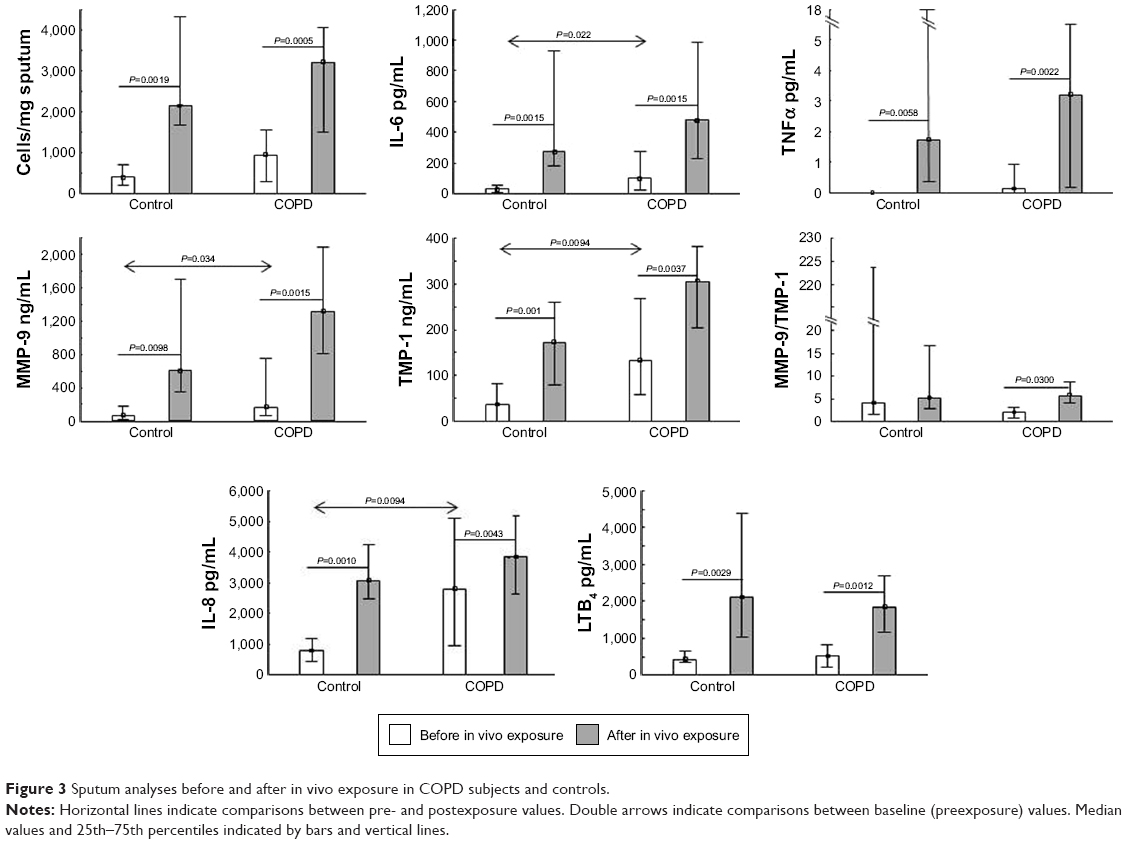 | Figure 3 Sputum analyses before and after in vivo exposure in COPD subjects and controls. |
Baseline sputum levels of MMP9, TIMP1, IL6, and CXCL8 were higher in COPD subjects than in controls. Concentrations of MMP9, TIMP1, CXCL8, IL6, TNFα, and LTB4 increased significantly and similarly in both groups (Figure 3). ChAT and AChE expression in sputum neutrophils, monocytes, and lymphocytes did not change after in vivo exposure in either group, and no differences were observed between the groups at baseline or after in vivo exposure (Table 2).
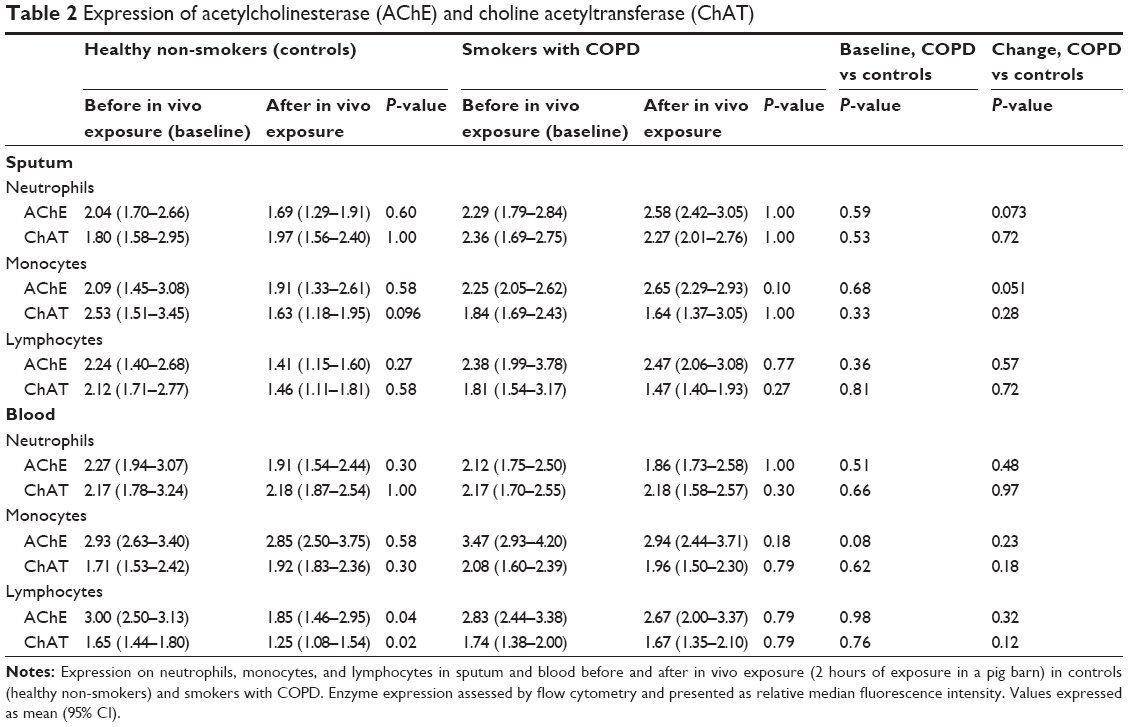 | Table 2 Expression of acetylcholinesterase (AChE) and choline acetyltransferase (ChAT) |
After methacholine provocation, total cell numbers in sputum decreased in the COPD group (P=0.03) and were unaltered in the control group (P=0.61), with no difference between the groups (P=0.19).
Blood analyses
At baseline, the COPD group had higher numbers of blood neutrophils (P=0.046) and monocytes (P=0.0014) than the controls (Figure 4). Following in vivo exposure, there was a significant increase in blood neutrophils and monocytes and a significant reduction in lymphocytes in controls (P<0.01 for all). In COPD subjects, blood neutrophils increased after exposure (P=0.01), while monocytes and lymphocytes were unaltered (P≥0.12). Blood-cell reaction to in vivo exposure did not differ significantly between COPD subjects and controls. Blood eosinophils did not differ significantly between controls and COPD subjects at baseline (35 [18–64] × 106 cells/L and 21.5 [9–71] × 106 cells/L, respectively) or after exposure (29 [17–58] × 106 cells/L and 15 [5–26] × 106 cells/L, respectively).
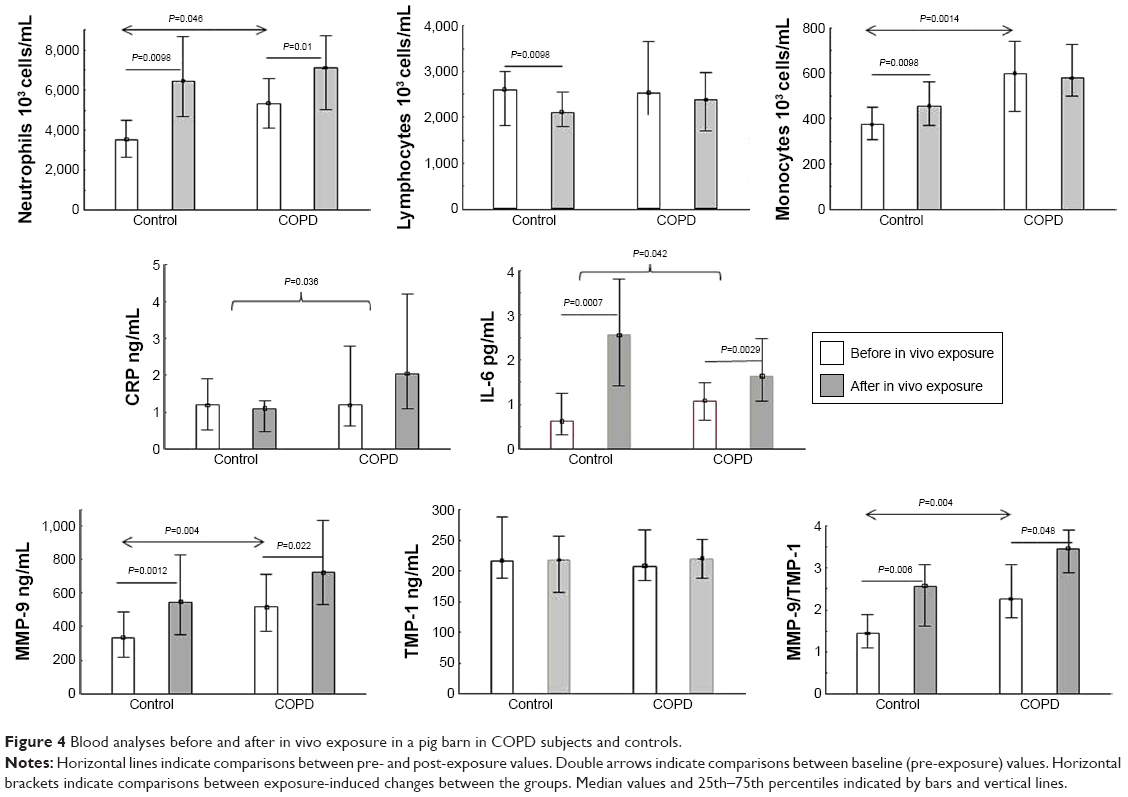 | Figure 4 Blood analyses before and after in vivo exposure in a pig barn in COPD subjects and controls. |
Baseline levels of blood MMP9 and MMP9:TIMP1 ratios were higher in COPD subjects than in controls (Figure 4). Exposure in vivo increased blood MMP9 levels and the MMP9:TIMP1 ratios in both groups, with no significant difference between the groups (P≥0.88; Figure 4). Baseline blood levels of IL6 and CRP did not differ between the groups and blood IL6 and CRP increased significantly in both groups following exposure in vivo. IL6 increase was greater in controls than in COPD subjects (P=0.042; Figure 4) and the CRP increase greater in the COPD group than in controls (P=0.036; Figure 4).
In the controls, baseline expression of CXCR1 was lower in COPD subjects (45.4 [35.0–57.0] relative median fluorescence intensity) than in controls (72.7 [61.7–83.8] relative median fluorescence intensity; P=0.014) and in vivo exposure induced relative falls in CXCR1 expression in blood neutrophils compared with COPD subjects (P=0.04). CXCR2 expression in blood neutrophils was similar at baseline in both groups and did not change after in vivo exposure in either group (data not shown). Baseline expression of ChAT and AChE in blood cells did not differ between the groups (Table 2). In vivo exposure decreased ChAT (P=0.02) and AChE (P=0.04) in lymphocytes in controls, but did not alter their expression in other cell types in either group (Table 2).
Ex vivo stimulation of blood neutrophils
With a few exceptions, no significant differences were found between before and after in vivo exposure or between COPD and controls with regard to the effects of neutrophil stimulation with dust, acetylcholine, and the combination thereof or the effect of the drugs (tiotropium, pirenzepine, solifenacin, neostigmine). Therefore, unless otherwise stated, results of measured outcomes are given for before in vivo exposure.
Dust significantly decreased neutrophil expression of CXCR1 in controls (P=0.0003). In COPD subjects, a similar pattern was observed, but did not reach statistical significance (P=0.015). A similar pattern was observed after in vivo exposure. Dust increased the expression of CXCR2 in both groups (Figure 5A and B). Dust-induced alteration of CXCR1 and CXCR2 expression was not altered by tiotropium (Figure 5A and B), pirenzepine, solifenacin, or neostigmine (data not shown).
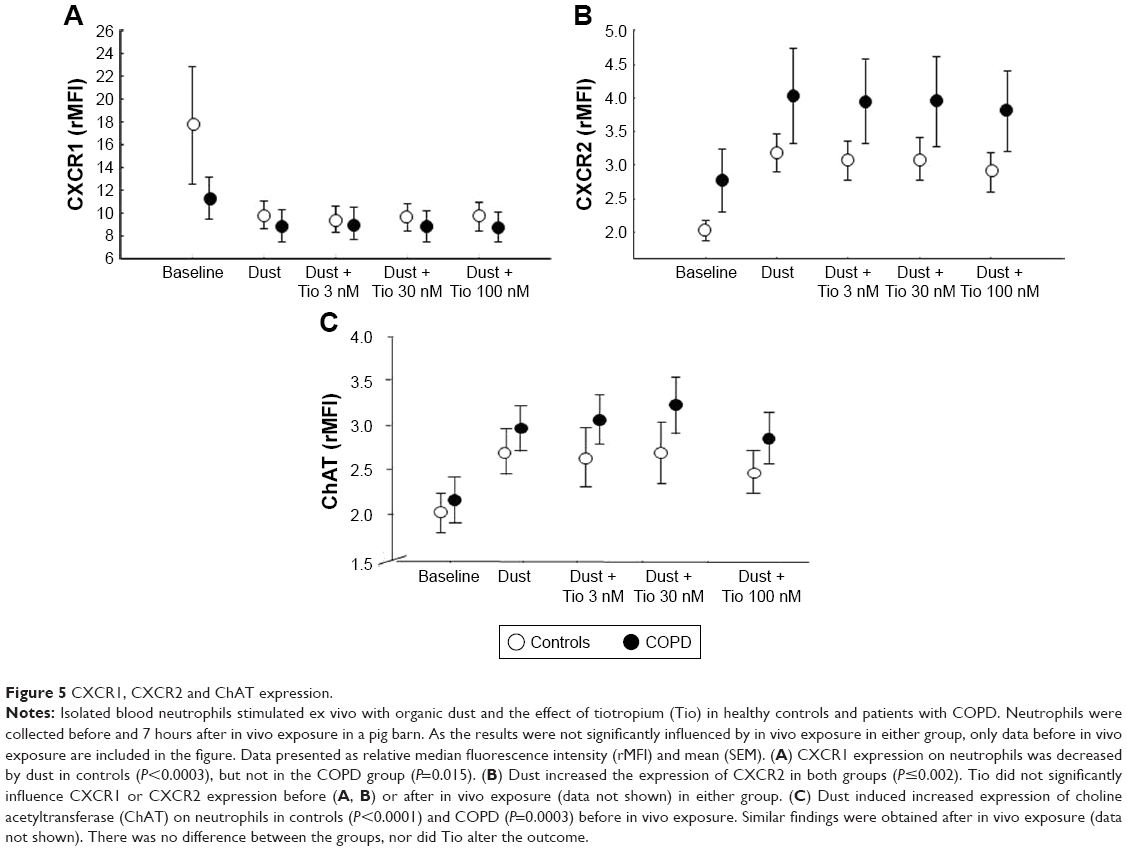 | Figure 5 CXCR1, CXCR2 and ChAT expression. |
Dust induced increased levels of ChAT in neutrophils from controls (P<0.0001) and COPD (P=0.0003; Figure 5C) before in vivo exposure and in COPD subjects after in vivo exposure (P<0.001). Before and after in vivo exposure, dust induced significantly increased secretion of TNFα in controls and COPD subjects, with no difference between the groups (P≥0.2). Dust-induced TNFα release was not altered by tiotropium (Figure 6A) or any other drug (data not shown).
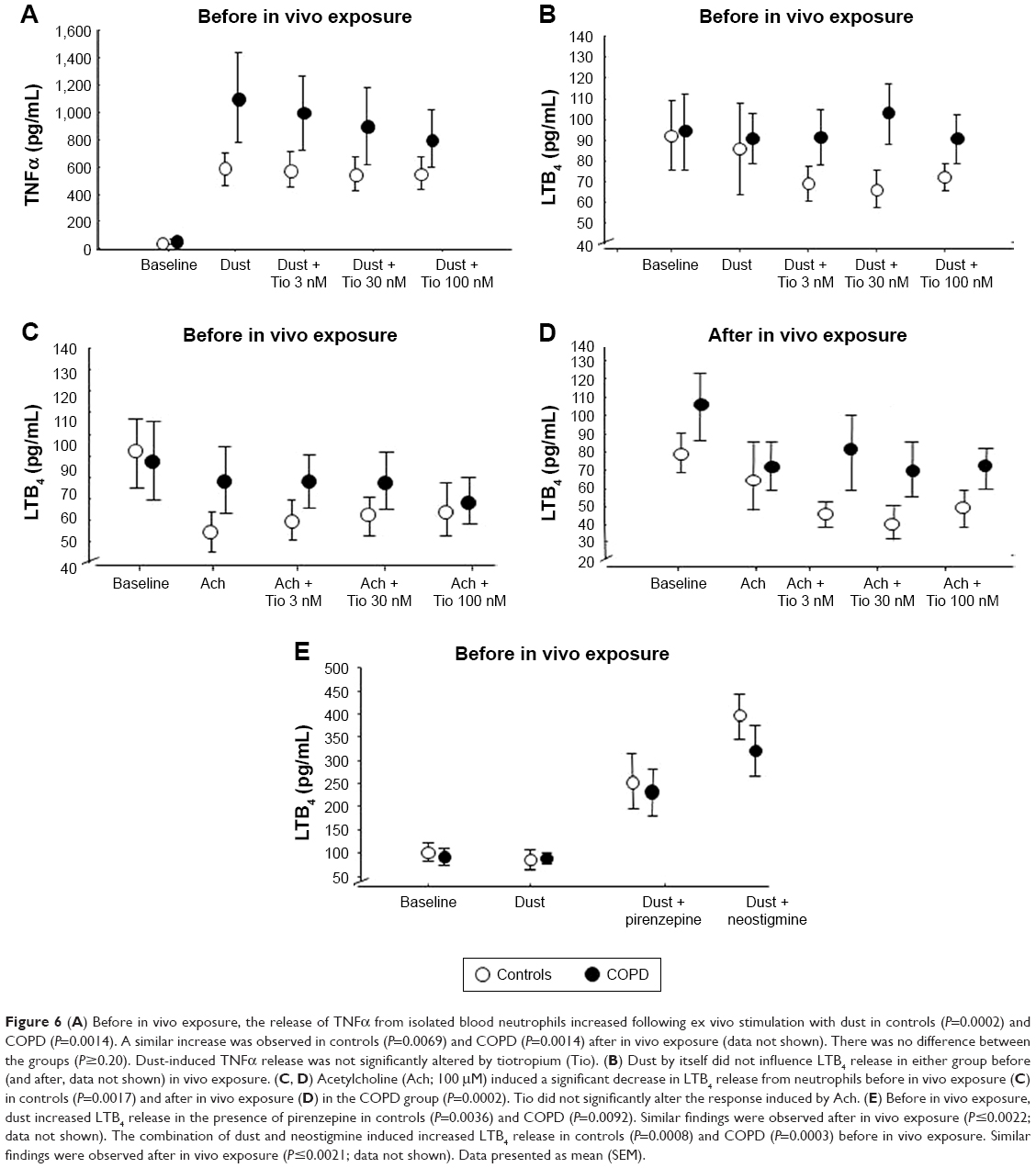 | Figure 6 (A) Before in vivo exposure, the release of TNFα from isolated blood neutrophils increased following ex vivo stimulation with dust in controls (P=0.0002) and COPD (P=0.0014). A similar increase was observed in controls (P=0.0069) and COPD (P=0.0014) after in vivo exposure (data not shown). There was no difference between the groups (P≥0.20). Dust-induced TNFα release was not significantly altered by tiotropium (Tio). (B) Dust by itself did not influence LTB4 release in either group before (and after, data not shown) in vivo exposure. (C, D) Acetylcholine (Ach; 100 μM) induced a significant decrease in LTB4 release from neutrophils before in vivo exposure (C) in controls (P=0.0017) and after in vivo exposure (D) in the COPD group (P=0.0002). Tio did not significantly alter the response induced by Ach. (E) Before in vivo exposure, dust increased LTB4 release in the presence of pirenzepine in controls (P=0.0036) and COPD (P=0.0092). Similar findings were observed after in vivo exposure (P≤0.0022; data not shown). The combination of dust and neostigmine induced increased LTB4 release in controls (P=0.0008) and COPD (P=0.0003) before in vivo exposure. Similar findings were observed after in vivo exposure (P≤0.0021; data not shown). Data presented as mean (SEM). |
Release of LTB4 from blood neutrophils was not influenced by dust stimulation (Figure 6B), but decreased after stimulation with acetylcholine in controls before and in COPD subjects after in vivo exposure (Figure 6C and D). Release of LTB4 was not influenced by tiotropium (Figure 6B–D) or solifenacin (data not shown). Both pirenzepine (P≤0.0092) and neostigmine (P≤0.0021) in combination with dust induced increased LTB4 release from neutrophils before and after in vivo exposure in both groups (Figure 6E).
Whereas dust induced increased CXCL8 secretion from neutrophils in the COPD group only after in vivo exposure (P=0.0013), the combination of dust and acetylcholine induced increased CXCL8 release from neutrophils in controls before (P=0.0039) and in COPD subjects before (P=0.0073) and after (P=0.0006) in vivo exposure. Dust did not alter MMP9 release, whereas dust in combination with acetylcholine induced increased MMP9 release in the COPD group after in vivo exposure (P=0.0069). Ex vivo stimulation did not alter the release of TGFβ or CXCL10.
Discussion
Exposure to organic dust in a pig barn (in vivo exposure) induced more serious symptoms in subjects with COPD than in healthy non-smokers. The increase in symptoms was similar to what COPD patients experience during acute exacerbations and was associated with substantial lung function impairment (assessed by FEV1, FVC, and PEF) in the COPD group. It has previously been shown that exposure in a pig barn does not alter bronchial responsiveness to an indirect stimulus, such as eucapnic hyperventilation,24 but induces a powerful increase in bronchial responsiveness to a cholinergic stimulus, such as methacholine, which is independent of baseline lung function assessed as FEV1.25 The present data showed that patients with COPD were more sensitive to the bronchoconstrictive effect of in vivo exposure than the controls, which was likely a result of lower baseline lung function. Intriguingly, this deterioration in lung function induced by in vivo exposure did not correlate with responsiveness to methacholine assessed as PD20FEV1, implying that lung-function alteration following exposure in a pig barn was not related to bronchial responsiveness.
Exposure in a pig barn induces an intense airway and systemic inflammation in healthy subjects,2,3,26,27 and in the present study it was demonstrated that the inflammatory response to exposure in a pig barn was similar in healthy non-smokers and smokers with COPD. At baseline, inflammatory markers in sputum and blood were elevated in subjects with COPD compared with the controls. Following exposure, however, similar alterations in sputum inflammatory cells, pro-inflammatory cytokines (IL6, TNFα), mediators involved in cell migration (CXCL8, LTB4), and mediators involved in tissue degradation (MMP9, TIMP1) were observed in both groups. The inflammatory characteristics induced by exposure in a pig barn with increased sputum levels of IL6, TNFα, CXCL8, and LTB4 thus very much resembled what is observed in acute exacerbations in COPD.12,13,28–34
Both MMP9 and TIMP1 in sputum were significantly higher before in vivo exposure in the COPD group, indicating ongoing degradation of the extracellular matrix, which is a key feature during airway remodeling. However, exposure in vivo induced an increase in sputum levels of both MMP9 and its specific inhibitor TIMP1 in both groups and an increase in the MMP9:TIMP1 ratio in COPD patients. This is, in part, in contrast to the finding in acute exacerbations of COPD in which sputum levels of MMP9 increase,32,33 whereas the concentration of TIMP1 decreases, yielding an increased MMP9:TIMP1 ratio.33 Previous findings are, however, not unequivocal, as other results have indicated an association between low MMP9:TIMP1 ratio and impaired lung function (FEV1) in patients with COPD and/or chronic bronchitis.35 Blood level of MMP9 increased as a result of in vivo exposure, whereas blood concentration of TIMP1 remained unaltered, leading to an increased MMP9:TIMP1 ratio in blood in both controls and COPD subjects. Also in blood, both MMP9 and TIMP1 were significantly increased at baseline in the COPD group, as previously demonstrated.36
As anticholinergic drugs constitute first-line therapy in COPD, it cannot be excluded that these drugs may also have an influence on nonneuronal cholinergic mechanisms operating in inflammatory cells. One of our aims was to explore whether the non-neuronal cholinergic system is activated by exposure to organic dust as a simulation of an exacerbation and whether this activation is altered in COPD patients compared with healthy non-smokers. With the exception of a slight downregulation of ChAT and AChE in circulating lymphocytes in controls, exposure in the pig barn did not alter cellular expression of enzymes (ChAT and AChE) in sputum or blood. The expression of AChE in neutrophils and monocytes in sputum did not change significantly following in vivo exposure, but AChE expression was regulated in opposite directions in COPD subjects and controls, which resulted in a tendency toward a difference between the groups (P=0.05 and 0.07). In addition, ex vivo data strongly support the involvement of cholinergic mechanisms in the response to dust, as ChAT expression on neutrophils clearly increased following dust exposure in both groups. The significance of this finding is difficult to interpret, as knowledge about the importance of acetylcholine turnover in inflammatory cells is sparse.16 Our results suggest that non-neuronal cholinergic responses – assessed as the capacity to catalyze the formation and breakdown of acetylcholine in inflammatory cells – are activated following exposure to dust in vivo and ex vivo. These responses do not, however, seem to be altered in COPD. We conclude that exposure to a strong pro-inflammatory stimulus (dust) that elicits an inflammatory response similar to what is observed during acute exacerbations in COPD may be associated with an alteration in cholinergic activity in inflammatory cells in the airway and in the circulation.
The number of neutrophils in blood was significantly increased in COPD patients, indicating a systemic inflammation. Exposure induced a greater CRP increase in blood but a lower increase in circulating IL6 in the COPD group compared with the healthy non-smokers. Therefore, even if patients with COPD had signs of systemic inflammation with somewhat higher levels of circulating neutrophils, the response to an acute pro-inflammatory stimulus seemed to be downregulated. It has previously been shown that concentrations of acute-phase proteins, eg, CRP, are increased at 24 hours but not 6 hours after exposure in a swine-confinement building,2 which is in agreement with the present results in the healthy non-smokers. As IL6 constitutes an endogenous stimulus for hepatic CRP production37 it would be reasonable to assume that the more pronounced increase in circulating IL6 in the controls would be associated with a greater CRP increase in this group than in the COPD group. The finding of the opposite, ie, augmented CRP increase in the COPD group, indicates that the time kinetics for acute-phase protein production and the interaction between IL6 and acute-phase protein synthesis may be altered in COPD subjects compared with healthy non-smokers.
We focused our ex vivo studies on neutrophils, as these cells are of importance in COPD and dominate the response to pig-barn exposure in vivo. Therefore, in order to find out whether or not 2 hours of exposure in a pig barn alters cellular responsiveness to dust and cholinergic stimuli, we conducted ex vivo stimulation of neutrophils isolated from peripheral blood before and 7 hours after beginning the exposure in the pig barn. At baseline, the expression of CXCR1 (but not CXCR2) on blood neutrophils was decreased in the COPD group compared with controls. In vivo exposure did not alter CXCR1 expression in blood neutrophils in either group, but slightly increased expression was found in the COPD group, which differed significantly from controls. These findings were corroborated by the ex vivo findings, with more marked downregulation of CXCR1 expression in neutrophils from controls than from patients with COPD. We thus conclude that regulation of CXCR1, but not CXCR2, on neutrophils differs between healthy non-smokers and smokers with COPD.
Both CXCR1 and CXCR2 enhance neutrophilic chemotactic activity.38 CXCR1 binds to CXCL6 and CXCL8, and is – apart from being important for neutrophil chemotaxis – also of importance for neutrophil activation.39 Stimulation of CXCR2 leads to neutrophil chemotaxis through binding to a number of chemokines, such as CXCL1–CXCL3, CXCL5, CXCL6, and CXCL8.40 While lipopolysaccharides exhibit neutrophil chemotaxis by activating both CXCR1 and CXCR2,38 it is intriguing that ex vivo activation of isolated neutrophils in the present study reduced CXCR1 and enhanced CXCR2 expression. The finding of increased sputum levels of CXCL8 induced by in vivo exposure is in agreement with previous research.26 The source of CXCL8 following exposure to dust from a pig barn has previously been shown to be macrophages and airway epithelial cells,5 and the present results show that neutrophils do not contribute to CXCL8 release induced by exposure to the dust. The explanation of this dualistic effect on CXCR1 and CXCR2 is not clear, but it is reasonable to assume that one of the mechanisms behind the powerful neutrophil chemotaxis induced by exposure in a pig barn is mediated through interaction between CXCL chemokines and CXCR2.
Also LTB4 is likely to be an important neutrophil chemoattractant, and is released upon stimulation with dust from a pig barn.41 Dust had no clear effect on LTB4 release from neutrophils, whereas acetylcholine reduced LTB4 release in both groups and no further effect of tiotropium was observed. This is in contrast to a previous study, in which acetylcholine did not alter LTB4 release from sputum cells.42 In addition, in that study an unselective M-receptor blocker (oxitropium bromide) prevented LTB4 release. The most likely explanation for these discrepancies is that isolated neutrophils were used in the present study and “sputum cells” in the study by Profita et al,42 leading to the conclusion that cells other than neutrophils were probably responsible for the outcome in that study.
Stimulation of isolated neutrophils with dust in the presence of neostigmine, an AChE inhibitor, induced a two- to fourfold increase in LTB4 release. It was also shown that dust induced increased expression of ChAT in neutrophils, which likely may augment the effect of neostigmine. These findings indicate that endogenous but not exogenous acetylcholine may activate LTB4 release. In addition, blocking the M1 receptor with pirenzepine entailed a dust-induced doubling of LTB4 release from neutrophils. The explanation for this is not clear. There are findings from animal studies indicating that activation of the M1 receptor may exhibit anti-inflammatory effects, eg, by suppressing TNFα release, an effect that seemed to be mediated through neuronal cholinergic mechanisms.43 There are data from animal experiments supporting vagal stimulation inhibiting TNFα release and leukocyte accumulation in endotoxemia, an effect that is attenuated by nicotinic receptor (α7nAChR) antagonism.44,45 We do not know of any studies on M1-receptor function in neutrophils. From the present data, it could however be hypothesized that blocking the M1 receptor on neutrophils has pro-inflammatory effects by constituting a stimulus for LTB4 release, thereby being a further chemoattractant for neutrophils.
Occupational exposure in pig farms includes gases and organic dust containing particles. Apart from particles from hay, grasses, pollen, pig epithelial cells, and feedstuffs, the dust contains high amounts of Gram-positive and Gram-negative bacteria and fungi, as well as high levels of airborne endotoxins and peptidoglycans. It is not known what components are responsible for the pro-inflammatory potency of dust in a pig farm, but it is most likely that the high microbial content plays a major role. Neutrophils possess M1, M2, and M3 receptors, of which M1 receptors are the most abundant.42 There are indications that cholinergic regulation of airway inflammation is mediated mainly through activation of M3 receptors and that M1 receptors may be less involved in this respect.46 It has previously been demonstrated that anticholinergic agents inhibit neutrophil chemotaxis through influencing chemokine release from structural cells and macrophages.46 The role of the M receptors located on neutrophils is not fully understood. In the present study, we were not able to show that acetylcholine had any influence on most mediator release from neutrophils. As such, release of MMP9, a profibrotic factor (TGFβ), chemokines regulating leukocyte migration (CXCL8, CXCL10), CXCL8 receptors (CXCR1, CXCR2), or enzymes regulating acetylcholine (ChAT, AChE) of neutrophils from healthy non-smokers and patients with COPD were not influenced by acetylcholine per se, and acetylcholine did not alter the response to dust stimulation. Possibly apart from LTB4 release, it seems likely that neutrophils do not function as important target cells for cholinergic stimuli as far as the measured mediators in the present study are concerned.
Conclusion
Dust from pig barns constitutes a very potent pro-inflammatory stimulus, both in vivo and ex vivo. Certain inflammatory markers in sputum and blood were enhanced at baseline in COPD subjects, but airway and systemic inflammatory responses to in vivo exposure did not differ substantially between the groups, despite the finding that in vivo exposure induced more severe symptoms and more severe lung-function deterioration in smokers with COPD than healthy non-smokers. Ex vivo stimulation of blood neutrophils did not reveal differences in neutrophil responses to dust before or after in vivo exposure or between controls and COPD subjects. Ex vivo exposure to dust induced cholinergic activation, assessed as increased expression of ChAT in neutrophils, but in conclusion cholinergic stimulation seems to constitute a weak pro-inflammatory stimulus in neutrophils.
Acknowledgment
The authors wish to thank Marianne Olsson for excellent technical assistance and for delivering subjects to the pig barns on the exposure days and the Swedish Heart-Lung Foundation.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Malmberg P, Larsson K. Acute exposure to swine dust causes bronchial hyperresponsiveness in healthy subjects. Eur Respir J. 1993;6(3):400–404. | ||
Larsson KA, Eklund AG, Hansson LO, Isaksson BM, Malmberg PO. Swine dust causes intense airways inflammation in healthy subjects. Am J Respir Crit Care Med. 1994;150(4):973–977. | ||
Wang Z, Larsson K, Palmberg L, Malmberg P, Larsson P, Larsson L. Inhalation of swine dust induces cytokine release in the upper and lower airways. Eur Respir J. 1997;10(2):381–387. | ||
Ek A, Larsson K, Siljerud S, Palmberg L. Fluticasone and budesonide inhibit cytokine release in human lung epithelial cells and alveolar macrophages. Allergy. 1999;54(7):691–699. | ||
Palmberg L, Larsson BM, Malmberg P, Larsson K. Induction of IL-8 production in human alveolar macrophages and human bronchial epithelial cells in vitro by swine dust. Thorax. 1998;53(4):260–264. | ||
Strandberg K, Palmberg L, Larsson K. Effect of formoterol and salmeterol on IL-6 and IL-8 release in airway epithelial cells. Respir Med. 2007;101(6):1132–1139. | ||
Wang Z, Malmberg P, Ek A, Larsson K, Palmberg L. Swine dust induces cytokine secretion from human epithelial cells and alveolar macrophages. Clin Exp Immunol. 1999;115(1):6–12. | ||
Borlée F, Yzermans CJ, van Dijk CE, Heederik D, Smit LA. Increased respiratory symptoms in COPD patients living in the vicinity of livestock farms. Eur Respir J. 2015;46(6):1605–1614. | ||
Eduard W, Pearce N, Douwes J. Chronic bronchitis, COPD, and lung function in farmers: the role of biological agents. Chest. 2009;136(3):716–725. | ||
Seemungal T, Harper-Owen R, Bhowmik A, et al. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164(9):1618–1623. | ||
Peacock JL, Anderson HR, Bremner SA, et al. Outdoor air pollution and respiratory health in patients with COPD. Thorax. 2011;66(7):591–596. | ||
Crooks SW, Bayley DL, Hill SL, Stockley RA. Bronchial inflammation in acute bacterial exacerbations of chronic bronchitis: the role of leukotriene B4. Eur Respir J. 2000;15(2):274–280. | ||
Gompertz S, O’Brien C, Bayley DL, Hill SL, Stockley RA. Changes in bronchial inflammation during acute exacerbations of chronic bronchitis. Eur Respir J. 2001;17(6):1112–1119. | ||
Harting JR, Gleason A, Romberger DJ, et al. Chronic obstructive pulmonary disease patients have greater systemic responsiveness to ex vivo stimulation with swine dust extract and its components versus healthy volunteers. J Toxicol Environ Health A. 2012;75(24):1456–1470. | ||
Wollin L, Pieper MP. Tiotropium bromide exerts anti-inflammatory activity in a cigarette smoke mouse model of COPD. Pulm Pharmacol Ther. 2010;23(4):345–354. | ||
Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir Res. 2006;7:73. | ||
Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. 2016. Available from: https://goldcopd.org/. Accessed September 26, 2018. | ||
ATS. Standardization of Spirometry, 1994 Update. American Thoracic Society. Am J Respir Crit Care Med. 1995;152(3):1107–1136. | ||
Hedenström H, Malmberg P, Agarwal K. Reference values for lung function tests in females. Regression equations with smoking variables. Bull Eur Physiopathol Respir. 1985;21(6):551–557. | ||
Hedenström H, Malmberg P, Fridriksson HV. Reference values for lung function tests in men: regression equations with smoking variables. Ups J Med Sci. 1986;91(3):299–310. | ||
Malmberg P, Larsson K, Thunberg S. Increased lung deposition and biological effect of methacholine by use of a drying device for bronchial provocation tests. Eur Respir J. 1991;4(7):890–898. | ||
In ’t Veen JC, de Gouw HW, Smits HH, et al. Repeatability of cellular and soluble markers of inflammation in induced sputum from patients with asthma. Eur Respir J. 1996;9(12):2441–2447. | ||
Larsson K, Tornling G, Gavhed D, Müller-Suur C, Palmberg L. Inhalation of cold air increases the number of inflammatory cells in the lungs in healthy subjects. Eur Respir J. 1998;12(4):825–830. | ||
Sundblad BM, Palmberg L, Larsson K. Bronchial responsiveness to eucapnic hyperventilation and methacholine following exposure to organic dust. Chest. 2002;122(1):363–368. | ||
Strandberg K, Ek A, Palmberg L, Larsson K. Fluticasone and ibuprofen do not add to the effect of salmeterol on organic dust-induced airway inflammation and bronchial hyper-responsiveness. J Intern Med. 2008;264(1):83–94. | ||
Larsson BM, Palmberg L, Malmberg PO, Larsson K. Effect of exposure to swine dust on levels of IL-8 in airway lavage fluid. Thorax. 1997;52(7):638–642. | ||
Sundblad BM, Larsson BM, Palmberg L, Larsson K. Exhaled nitric oxide and bronchial responsiveness in healthy subjects exposed to organic dust. Eur Respir J. 2002;20(2):426–431. | ||
Aaron SD, Angel JB, Lunau M, et al. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163(2):349–355. | ||
Bathoorn E, Liesker JJ, Postma DS, et al. Change in inflammation in out-patient COPD patients from stable phase to a subsequent exacerbation. Int J Chron Obstruct Pulmon Dis. 2009;4:101–109. | ||
Clark TW, Medina MJ, Batham S, Curran MD, Parmar S, Nicholson KG. C-reactive protein level and microbial aetiology in patients hospitalised with acute exacerbation of COPD. Eur Respir J. 2015;45(1):76–86. | ||
Fujimoto K, Yasuo M, Urushibata K, Hanaoka M, Koizumi T, Kubo K. Airway inflammation during stable and acutely exacerbated chronic obstructive pulmonary disease. Eur Respir J. 2005;25(4):640–646. | ||
Gao P, Zhang J, He X, Hao Y, Wang K, Gibson PG. Sputum inflammatory cell-based classification of patients with acute exacerbation of chronic obstructive pulmonary disease. PLoS One. 2013;8(5):e57678. | ||
Mercer PF, Shute JK, Bhowmik A, Donaldson GC, Wedzicha JA, Warner JA. MMP-9, TIMP-1 and inflammatory cells in sputum from COPD patients during exacerbation. Respir Res. 2005;6:151. | ||
Tufvesson E, Ekberg M, Bjermer L. Inflammatory biomarkers in sputum predict COPD exacerbations. Lung. 2013;191(4):413–416. | ||
Vignola AM, Riccobono L, Mirabella A, et al. Sputum metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio correlates with airflow obstruction in asthma and chronic bronchitis. Am J Respir Crit Care Med. 1998;158(6):1945–1950. | ||
Ji J, von Schéele I, Bergström J, et al. Compartment differences of inflammatory activity in chronic obstructive pulmonary disease. Respir Res. 2014;15:104. | ||
Castell JV, Andus T, Kunz D, Heinrich PC. Interleukin-6. The major regulator of acute-phase protein synthesis in man and rat. Ann N Y Acad Sci. 1989;557:87–99. | ||
Aul R, Patel S, Summerhill S, Kilty I, Plumb J, Singh D. LPS challenge in healthy subjects: an investigation of neutrophil chemotaxis mechanisms involving CXCR1 and CXCR2. Int Immunopharmacol. 2012;13(3):225–231. | ||
Jones SA, Wolf M, Qin S, Mackay CR, Baggiolini M. Different functions for the interleukin 8 receptors (IL-8R) of human neutrophil leukocytes: NADPH oxidase and phospholipase D are activated through IL-8R1 but not IL-8R2. Proc Natl Acad Sci U S A. 1996;93(13):6682–6686. | ||
Moser B, Wolf M, Walz A, Loetscher P. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. 2004;25(2):75–84. | ||
O’Sullivan S, Dahlen SE, Larsson K, et al. Exposure of healthy volunteers to swine house dust increases formation of leukotrienes, prostaglandin D2, and bronchial responsiveness to methacholine. Thorax. 1998;53(12):1041–1046. | ||
Profita M, Giorgi RD, Sala A, et al. Muscarinic receptors, leukotriene B4 production and neutrophilic inflammation in COPD patients. Allergy. 2005;60(11):1361–1369. | ||
Zivkovic AR, Sedlaczek O, von Haken R, et al. Muscarinic M1 receptors modulate endotoxemia-induced loss of synaptic plasticity. Acta Neuropathol Commun. 2015;3:67. | ||
Altavilla D, Guarini S, Bitto A, et al. Activation of the cholinergic anti-inflammatory pathway reduces NF-kappab activation, blunts TNF-alpha production, and protects againts splanchic artery occlusion shock. Shock. 2006;25(5):500–506. | ||
Borovikova LV, Ivanova S, Zhang M, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405(6785):458–462. | ||
Kistemaker LE, Gosens R. Acetylcholine beyond bronchoconstriction: roles in inflammation and remodeling. Trends Pharmacol Sci. 2015;36(3):164–171. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.