Back to Journals » Drug Design, Development and Therapy » Volume 10
Chloroquine improves left ventricle diastolic function in streptozotocin-induced diabetic mice
Authors YUAN X, XIAO Y, ZHANG G, HOU N, WU X, CHEN W, LUO J, Zhang G ![]()
Received 23 April 2016
Accepted for publication 30 June 2016
Published 30 August 2016 Volume 2016:10 Pages 2729—2737
DOI https://doi.org/10.2147/DDDT.S111253
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Wei Duan
Xun Yuan, Yi-Chuan Xiao, Gui-Ping Zhang, Ning Hou, Xiao-Qian Wu, Wen-Liang Chen, Jian-Dong Luo, Gen-Shui Zhang
Department of Pharmacology, Guangzhou Medical University, Guangzhou, People’s Republic of China
Abstract: Diabetes is a potent risk factor for heart failure with preserved ejection fraction (HFpEF). Autophagy can be activated under pathological conditions, including diabetic cardiomyopathy. The therapeutic effects of chloroquine (CQ), an autophagy inhibitor, on left ventricle function in streptozotocin (STZ)-induced diabetic mice were investigated. The cardiac function, light chain 3 (LC3)-II/LC3-I ratio, p62, beclin 1, reactive oxygen species, apoptosis, and fibrosis were measured 14 days after CQ (ip 60 mg/kg/d) administration. In STZ-induced mice, cardiac diastolic function was decreased significantly with normal ejection fraction. CQ significantly ameliorated cardiac diastolic function in diabetic mice with HFpEF. In addition, CQ decreased the autophagolysosomes, cardiomyocyte apoptosis, and cardiac fibrosis but increased LC3-II and p62 expressions. These results suggested that CQ improved the cardiac diastolic function by inhibiting autophagy in STZ-induced HFpEF mice. Autophagic inhibitor CQ might be a potential therapeutic agent for HFpEF.
Keywords: chloroquine, diastolic function, HFpEF, autophagy, diabetic cardiomyopathy, type 1 diabetes mellitus
Introduction
Diabetes mellitus (DM) is a potent risk factor for heart failure (HF). DM contributes to the pathophysiologic process of HF with reduced or preserved ejection fraction (EF).1 The prevalence of heart failure with preserved ejection fraction (HFpEF) has increased in the past 2 decades, and DM is frequently associated with HFpEF.2 Population-based epidemiological studies reported that almost half of the patients with congestive HF have preserved EF and 30%–40% of these HFpEF patients have DM.3 HFpEF associated with DM has a long period of irreversible cardiac damage; therefore, early recognition and treatment of the preclinical cardiac abnormalities are important.4 Recently, several studies have established the key role of autophagy in the development of diabetic cardiomyopathy. In hearts from streptozotocin (STZ)-induced type 1 diabetic mice, diastolic function was impaired, though autophagic activity was significantly increased, as evidenced by increases in microtubule-associated protein 1 light chain 3 (LC3) and LC3-II/-I ratios and SQSTM1/p62 (sequestosome-1) and by the abundance of autophagic vacuoles and lysosomes detected electron microscopically.5 Mellor et al6 reported that increased myocardial autophagic flux in fructose diet-induced type 2 diabetic mice resulted in pathological remodeling of the heart. In cultured neonatal rat cardiomyocytes, the suppression of autophagy by 3-methyladenine and/or siRNA specific for beclin 1 or Atg7 attenuated high glucose-induced cardiomyocyte injury.7 Xu et al8 demonstrated that inhibition of autophagy was cardioprotective in type 1 diabetes, which was linked to activatation of noncanonical autophagy and improvement in mitophagy.
We hypothesized that inhibition of autophagy would ameliorate cardiac diastolic function in diabetic mice. Chloroquine (CQ), an autophagy inhibitor, has been used for the treatment of various diseases for a long time. CQ inhibits lysosomal acidification and therefore prevents autophagy by blocking autophagosome fusion and degradation. It has been reported that the administration of CQ reversed cardiac dysfunction and atrophy via the suppression of autophagic activity.9 However, other results demonstrated that CQ induces mitochondrial dysfunction in cardiomyocytes and significantly impairs mitochondrial antioxidant in pressure overload hypertrophy.10
In the present study, we investigated the effect of the autophagic inhibitor CQ on cardiac function in STZ-induced diabetic mice with HFpEF.
Materials and methods
Experimental animals and protocol
All animal procedures used in this study were performed in accordance with the guidelines of the National Institutes of Health and approved by the Animal Care and Use Committee of Guangzhou Medical University (No 2010-225). Specific pathogen-free male C57BL mice (age 6 weeks, weighing 16–18 g, Guangdong Experimental Animal Center) were randomly divided into three groups (n=8): 1) control; 2) STZ; and 3) CQ. Hyperglycemia of the mice in the STZ and CQ groups was induced with STZ (ip 60 mg/kg/d prepared daily in citrate buffer pH 4.5, for maximal stability) for 6 days. Six days after STZ injection, hyperglycemia was confirmed by fasting blood glucose measurement using tail-vein blood with the FreeStyle Flash Blood Glucose Monitoring System (Ultra; LifeScan, New Brunswick, NJ, USA). Mice with blood glucose concentrations ≥16.7 mmol/L were used for the study.11 At the end of 9 weeks, the mice in the CQ group were treated with CQ (ip 60 mg/kg/d; Sigma-Aldrich Co., St Louis, MO, USA) for 14 days. Two weeks after treatment, the animals underwent echocardiography followed by heart tissue experiments.
Echocardiography
Functional and dimensional parameters were measured at the end of 11 weeks after STZ-induced diabetes using a Vevo 2100 echocardiography system (VisualSonics, Toronto, Canada). Anesthesia was induced by 1.5% isoflurane in 100% oxygen. Following anesthetic induction, the mouse was placed on a heated platform for echocardiography. The body temperature was maintained at 37.0°C, and anesthesia was maintained with 0.5%–1.0% isoflurane in 100% oxygen. The isoflurane gas volume was regulated according to the rate in order to ensure an adequate depth of anesthesia. Standard imaging planes, M-mode, color-mode, Doppler, and functional calculations were acquired according to the guidelines of the American Society of Echocardiography. The parasternal short axis view of the left ventricle (LV) was used to guide calculations of percentage EF, percentage fractional shortening (FS), and ventricular dimensions and volumes. Moreover, passive LV filling peak velocity (E, mm/s) and atrial contraction flow peak velocity (A, mm/s) were acquired from the images of mitral valve Doppler flow from apical four-chamber view. Considering that heart rate (HR) positively correlates with systolic function, the HRs of animals during echocardiographic study were maintained in the range of 500–550 beats/min for M-mode, 400–450 beats/min for Doppler studies, and 450–500 beats/min for B-mode.
Preparation for heart tissue samples
At the end of 11 weeks, the mice were weighed and anesthetized with isoflurane and sacrificed by cervical dislocation. The hearts were rapidly excised and placed into a dish containing physiological saline. All four chambers were removed and weighed separately. The LV was snap frozen in liquid nitrogen for subsequent analysis.
Western blot analysis
Proteins (30 μg) extracted from the heart tissue samples were separated on 10% polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Hoffman-La Roche Ltd., Basel, Switzerland). The membranes were then incubated using antibodies against Rabbit LC3 (16–18 kDa; 1:1,000, L8918; Sigma-Aldrich Co.), Rabbit SQSTM1 (62 kDa; 1:1,000, SC-25575; Santa Cruz Biotechnology Inc., Dallas, TX, USA), and beclin 1 (60 kDa; 1:1,000, #3738; Cell Signaling Technology, Inc., Danvers, MA, USA), after which the blots were visualized using enhanced chemiluminescence (RPN 2232; Amersham/GE Healthcare, Uppsala, Sweden). β-actin (43 kDa; 1:5,000, SC-5546; Santa Cruz Biotechnology Inc.) served as the loading control. The molecular band intensity was determined as a ratio relative to that of loading control.
Electron microscopy
Fractions (1 mm3) of the LV were prefixed in a solution of 2.5% glutaraldehyde and 1% osmium tetroxide, post fixed in 1% OsO4, dehydrated in an ascending series of alcohols, and embedded in epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate. The samples were viewed under a transmission electron microscope (H-600; Hitachi Ltd., Tokyo, Japan) and analyzed using the IBAS 2.0 Image Analysis System (Kontron, Deggendorf, Germany).
Reactive oxygen species detection in the myocardium
Heart tissue sections were harvested and directly embedded in optimal cutting temperature compound. Superoxide production in the heart was detected using dihydroethidium (DHE) staining (Sigma-Aldrich Co.). Frozen heart sections (10 μm) were incubated with 10 μM DHE for 45 minutes at 37°C in a humidified chamber protected from the light. The average fluorescence intensity of the nuclei was then analyzed using Image-Pro Plus software (Media Cybernetics, Inc., Silver Spring, MD, USA).
Assessment of apoptosis
Apoptosis was assessed using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) according to the manufacturer’s instruction. Briefly, 5 μm thick paraffin-embedded sections were prepared from the hearts of each group and deparaffinized sequentially. The sections were stained with TUNEL reagents, and the nuclei were stained by 4′,6-diamidino-2-phenylindole. The samples were analyzed using a fluorescence microscopy (TE2000; Nikon Instruments, Melville, NY, USA). The number of TUNEL-positive cardiomyocyte nuclei and the total cardiomyocyte nuclei in each sight was counted. The ratio of apoptotic cardiomyocytes was calculated by dividing the number of TUNEL-positive cardiomyocyte nuclei by the number of total cardiomyocyte nuclei.
Masson’s trichrome staining for fibrosis
The hearts were removed, perfusion fixed with 10% neutral buffered formalin overnight, and embedded in paraffin. The sections were cut with 5 μm thickness and mounted onto slides. Masson’s trichrome staining was performed for fibrosis analysis. The tissue sections were examined under a light microscope (EVOS fl; AMG, Mill Creek, WA). The relative fibrosis area was determined by Image-Pro Plus software (Version 6.0), and the mean values of relative fibrosis area were obtained by one investigator blinded to the groups.
Statistical analyses
Statistical analyses were performed in GraphPad Prism (Version 6.0; GraphPad Software, Inc., La Jolla, CA, USA). All data were expressed as mean ± standard error of the mean. A one-way analysis of variance with a Tukey post hoc analysis that calculates P-values corrected for multiple comparisons was performed to assess differences between three groups. A P-value of <0.05 was considered statistically significant.
Results
Metabolic and morphometric profiles
Fasting blood glucose levels were significantly increased in STZ-induced mice compared with the control group at the age of 11 weeks (P<0.05). The blood glucose levels of STZ-induced mice did not differ significantly from those of the CQ-treated group (P>0.05; Figure 1A). The total body weight and heart weight were significantly decreased in STZ-induced mice and CQ group compared with controls (P<0.05). However, the body weight and heart weight showed no significant difference between the CQ and STZ groups (P>0.05; Figure 1B and C). The heart weight to body weight ratio was significantly decreased in the STZ and CQ groups (P<0.05; Figure 1D). The LV weight and the LV weight to body weight ratio were significantly decreased in the STZ and CQ groups (P<0.05; Figure 1E and F).
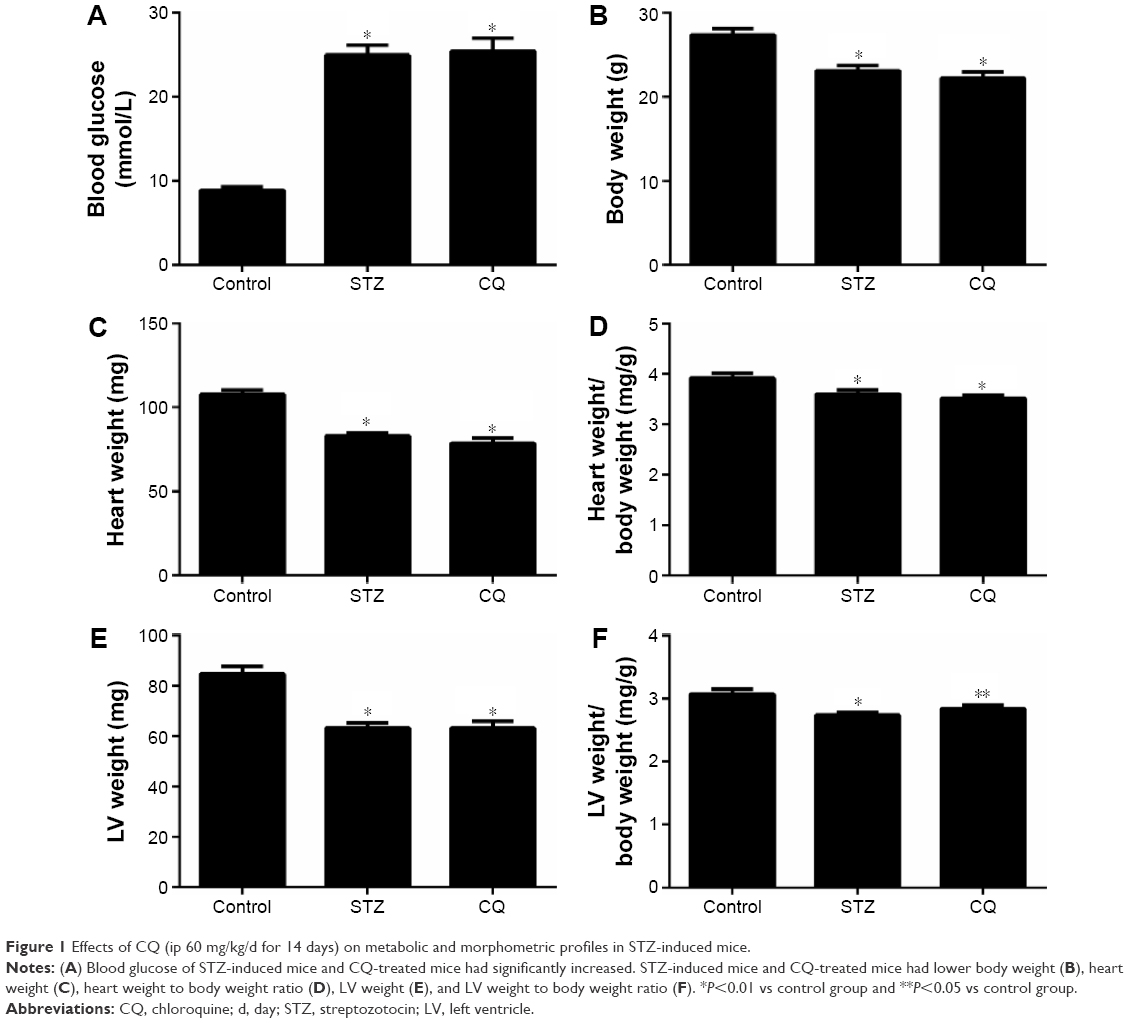 | Figure 1 Effects of CQ (ip 60 mg/kg/d for 14 days) on metabolic and morphometric profiles in STZ-induced mice. |
Systolic function and diastolic function of the heart in vivo
In STZ-induced diabetic mice and CQ-treated mice, parameters of cardiac systolic function (Figure 2A), including HR (Figure 2B), FS (Figure 2C), and EF (Figure 2D), were similar to those in the control group (P>0.05). Using pulse-wave Doppler technique, echocardiography revealed that diastolic cardiac function was significantly impaired in the STZ-induced diabetic mice (Figure 3A). Transmitral filling pattern showed inverted E/A ratio (Figure 3B) with prolongation of E-wave deceleration time (Figure 3C) in the STZ-induced diabetic mice compared with the control group (P<0.05). CQ treatment significantly improved the E/A ratio and E-wave deceleration time (P<0.05).
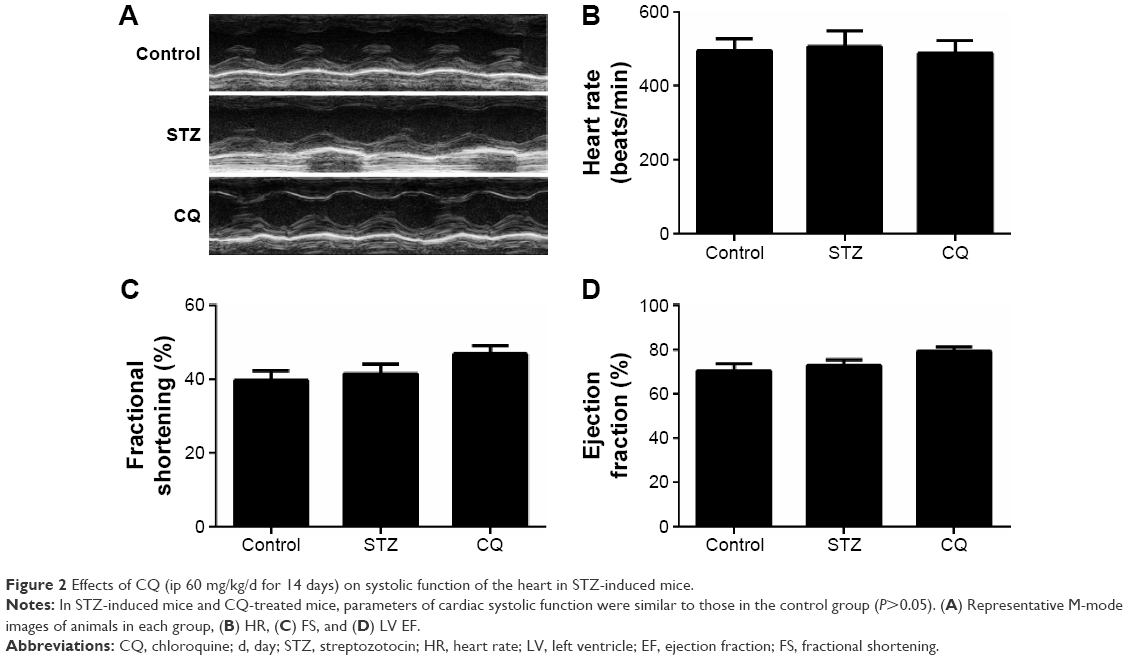 | Figure 2 Effects of CQ (ip 60 mg/kg/d for 14 days) on systolic function of the heart in STZ-induced mice. |
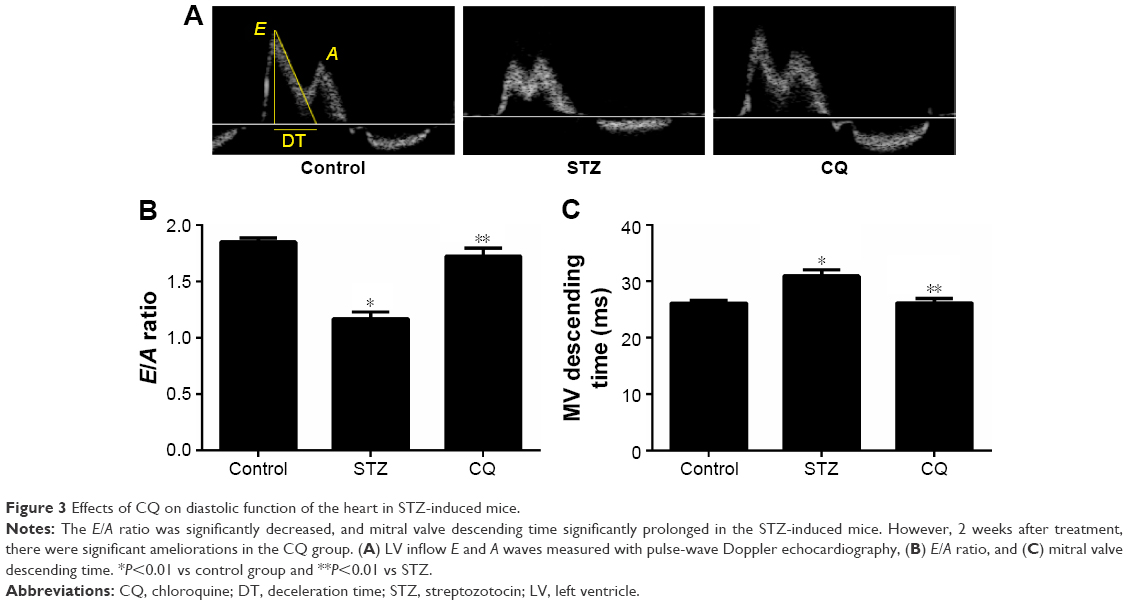 | Figure 3 Effects of CQ on diastolic function of the heart in STZ-induced mice. |
Expression of LC3, p62, and beclin 1 in myocardium
LC3, p62/SQSTM1, and beclin 1 (marker molecules of autophagic activities) in the hearts are shown in Figure 4A. LC3-II/LC3-I ratio was increased significantly in the STZ-induced mice. CQ treatment further increased LC3-II expression (Figure 4B) but decreased the LC3-II/LC3-I ratio with the increased LC3-I (Figure 4C). Compared with control, the level of p62 was increased significantly in the STZ-induced diabetic mice. The level of p62 protein was further increased significantly in the CQ-treated mice (Figure 4D). The level of beclin 1 was significantly increased in the STZ-induced diabetic mice compared with the control group. The level of beclin 1 in the CQ treated group did not differ significantly from that of the STZ-induced mice (P>0.05; Figure 4E).
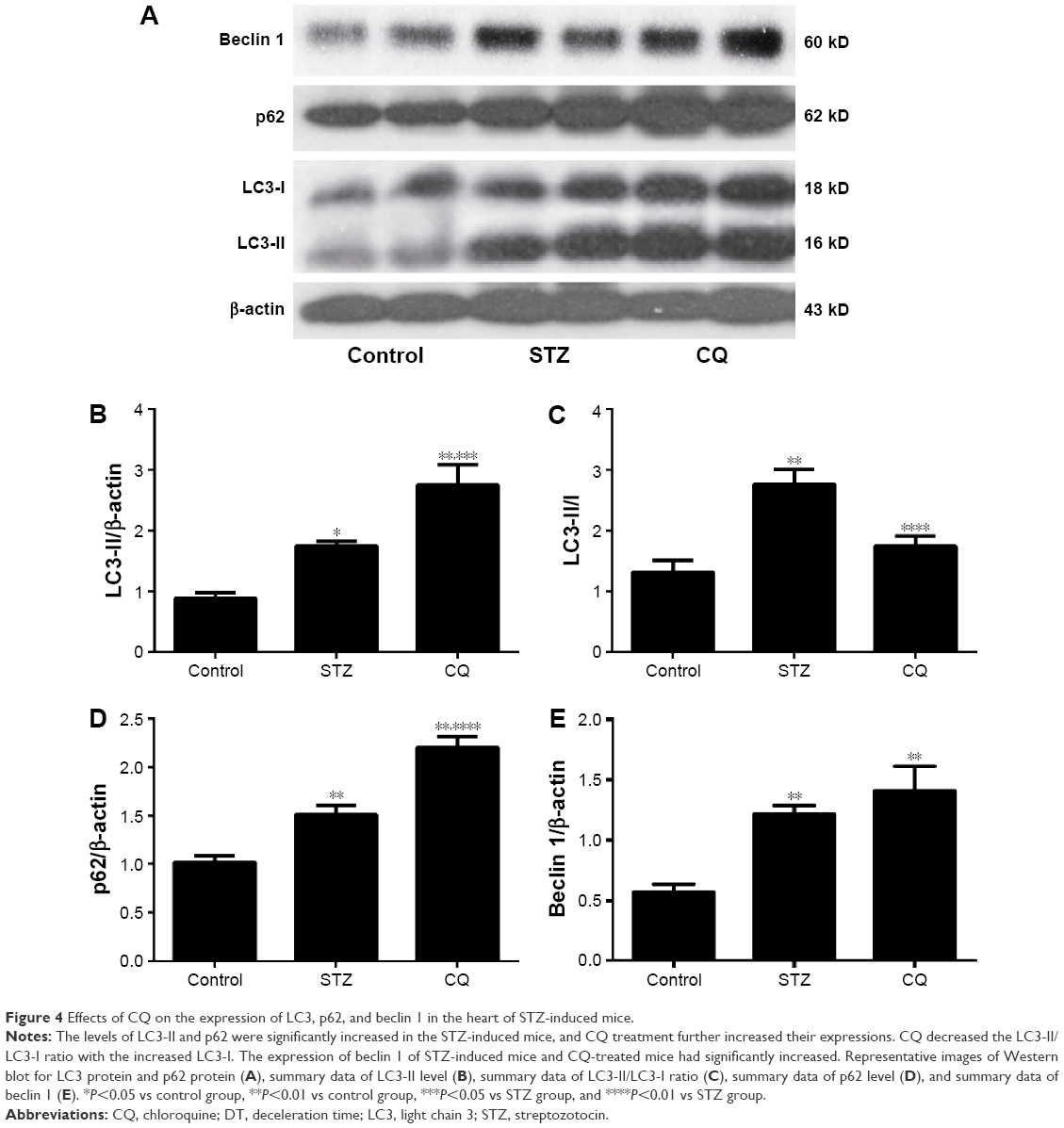 | Figure 4 Effects of CQ on the expression of LC3, p62, and beclin 1 in the heart of STZ-induced mice. |
Electron microscopy
Representative electron microscopy images are shown in Figure 5. Electron microscopy revealed that formation of the autophagosomes and the autophagolysosomes was enhanced in the hearts of STZ-induced mice. The electrographic assay showed that the autophagolysosomes were significantly decreased in the hearts of the CQ treatment group compared with the STZ-induced group (P<0.05).
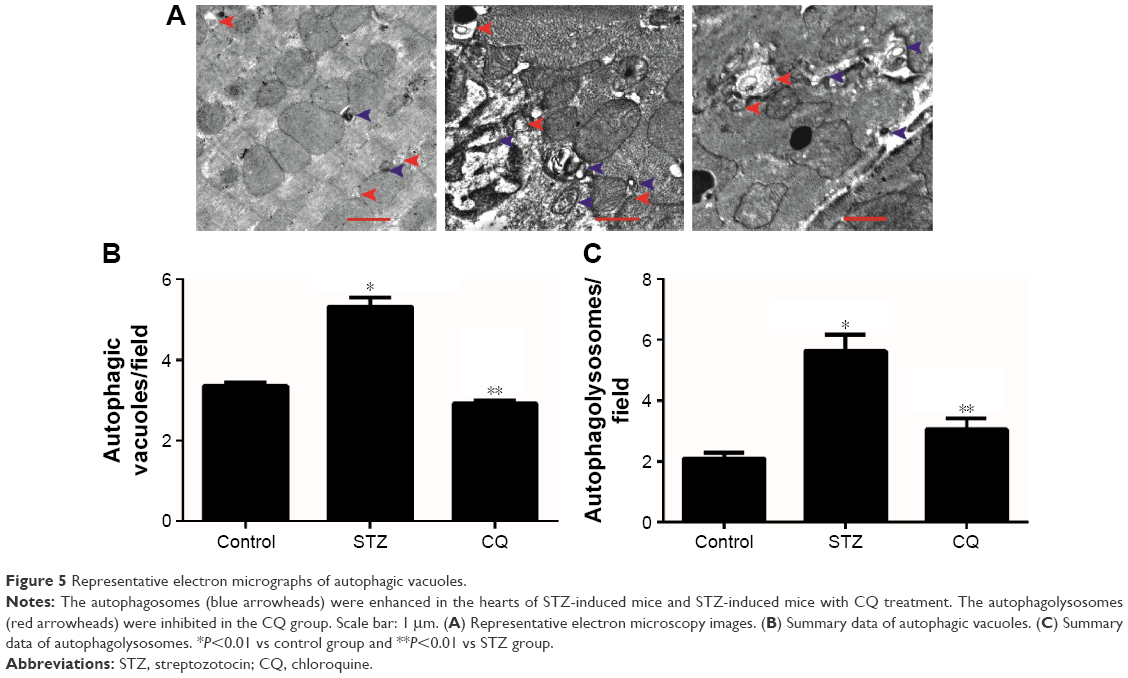 | Figure 5 Representative electron micrographs of autophagic vacuoles. |
Reactive oxygen species levels in myocardium
Myocardial reactive oxygen species (ROS) levels were quantified by DHE staining. The ROS levels were significantly increased in the STZ-induced diabetic mice (P<0.01). Treatment with CQ had no significant change in the ROS levels as compared with the untreated diabetic mice (P>0.05; Figure 6).
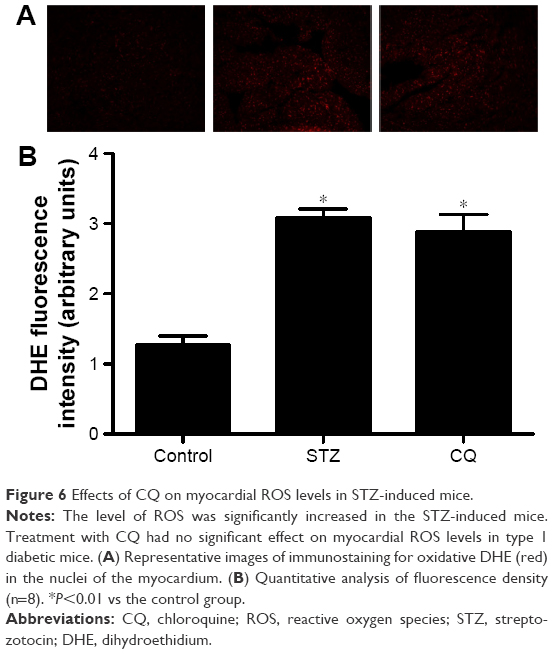 | Figure 6 Effects of CQ on myocardial ROS levels in STZ-induced mice. |
Apoptosis in myocardium
The paraffin-embedded sections of hearts from each group were stained by TUNEL to determine the cardiomyocyte apoptosis. The apoptotic cardiomyocytes were observed in STZ-induced mice, and CQ treatment reduced significantly the STZ-induced apoptosis (P<0.01; Figure 7).
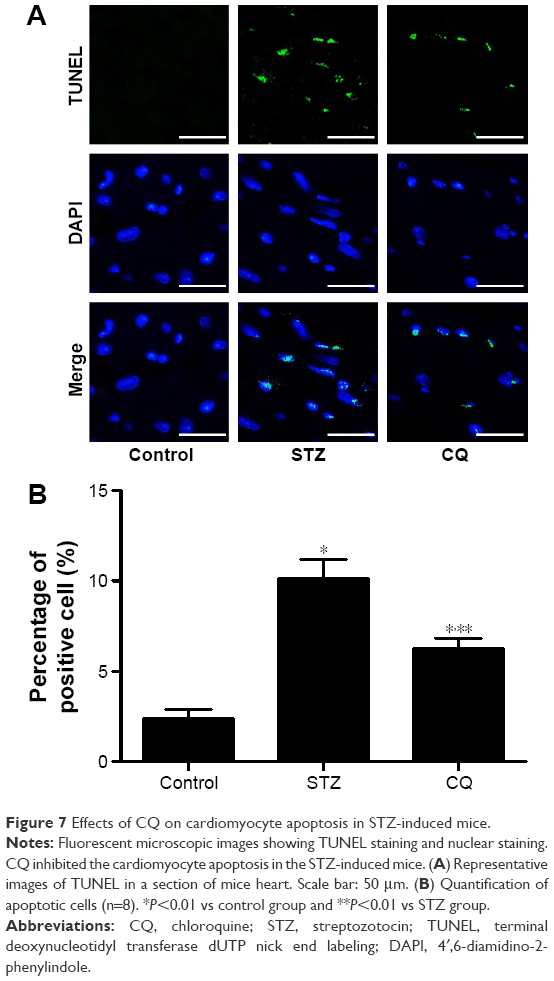 | Figure 7 Effects of CQ on cardiomyocyte apoptosis in STZ-induced mice. |
Fibrosis in myocardium
Fibrosis is an integral feature of diabetic cardiomyopathy. We performed Masson’s staining on cardiac tissue sections to determine the development of fibrosis in mice of each group. Dramatic perivascular fibrosis was observed in STZ-induced mice, which was attenuated significantly by CQ treatment (P<0.05; Figure 8).
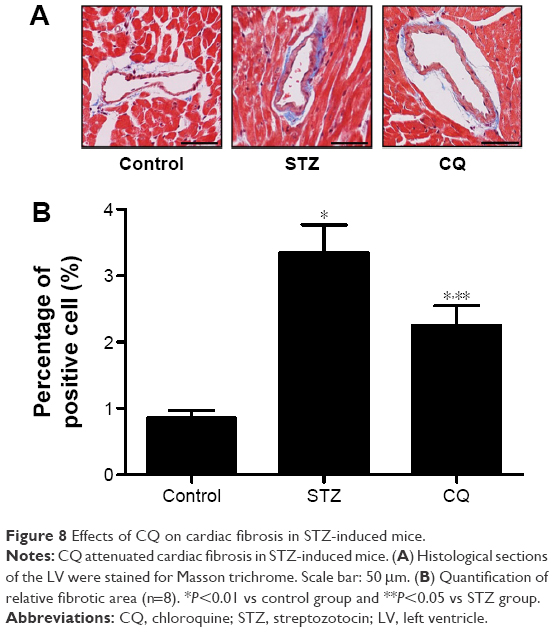 | Figure 8 Effects of CQ on cardiac fibrosis in STZ-induced mice. |
Discussion
In this study, the HFpEF model was established with STZ-induced diabetic mice. Diastolic dysfunction was found in STZ-induced diabetic mice, showing the reverse E/A ratio and increased mitral E-wave deceleration time. Additionally, the echocardiography M-mode demonstrated normal EF, FS, and stroke volume. Therefore, the STZ-induced mice model showed typical features of HF with normal EF. Echocardiography tests in type 1 diabetic mellitus patients showed that without known coronary artery disease revealed diastolic function with a reduction in early filling and increase in atrial filling.12,13 Diabetic cardiomyopathy could progress to irreversible cardiac damage; therefore, early recognition and treatment of the preclinical cardiac abnormalities are important.4 The present study showed features of metabolic syndromes with decreased body weight and increased blood glucose in STZ-induced diabetic mice. Treatment with CQ for 14 days did not lower the plasma glucose level significantly (Figure 1). The animal model provided evidence for diastolic dysfunction tested by echocardiography. The LV mitral valve blood flow showed faster relaxation and the E/A ratio back to the normal level in the CQ group, indicating that CQ treatment improved the diastolic dysfunction in the STZ-induced diabetic mice (Figures 2 and 3). These findings actually suggested that CQ, an autophagy inhibitor, might be a useful therapeutic agent for the treatment of diabetic diastolic dysfunction.
Autophagy refers to the homeostatic cellular process of sequestering organelles and long-lived proteins in a double-membrane vesicle inside the cell (autophagosome), where the contents are subsequently delivered to the lysosome for degradation.14 Under basal conditions, autophagy occurs in a healthy heart;15 however, autophagy can be activated under pathological conditions, including HF and cardiac hypertrophy.16,17 Overactivated autophagy may affect the ultrastructure of the sarcomere and cause mitochondrial structural abnormalities.18 A previous study showed that the overactivated autophagy may harm the cardiac function through affecting the titin/protein ratio.19 Insulin acts through the mTOR pathway to inhibit the autophagy. Autophagy in the heart is enhanced in type 1 diabetes, but is suppressed in type 2 diabetes. This difference provided important insight into the pathophysiology of diabetic cardiomyopathy, which was essential for the development of new treatment strategies.5,20 The present study demonstrated that autophagy was enhanced in the STZ-induced diabetic mice model (Figures 4 and 5). CQ inhibited autophagy by affecting lysosome acidification.21 CQ altered the lysosome pH, with the lysosomal neutralization inhibiting lysosome activities and can be used in assays of short-term autophagy flux.22 In addition, CQ decreased LC3-II/LC3-I protein ratio and undigested autophagosome observed by transmission electron microscopy in STZ-induced diabetic mice (Figure 5). The level of LC3-II is correlated with the extent of autophagosome formation. CQ accumulates LC3-II, a key step in autophagosome formation, by preventing the degradation of LC3-II-containing autolysosomes.23 The adaptor protein p62 (sequestosome-1) can bind directly to LC3 to facilitate degradation of ubiquitinated protein aggregated by autophagy.24 The accumulation of p62 was associated with decreased autophagy by CQ (Figure 4).
The subsequent generation of ROS and accompanying oxidative stress in diabetes are hallmarks of the molecular mechanisms underlying diabetic cardiovascular disease.25 In diabetic cardiomyopathy, the production of ROS induces inflammation, endothelial dysfunction, cell apoptosis, and myocardial remodeling.26 In the present study, the effects of CQ on oxidative stress in STZ-induced mice were analyzed. The results of the present study suggested that the autophagy inhibitor CQ was not able to decrease the ROS level in the diabetic mice, which indicated that CQ was not able to act as an antioxidant directly. Though autophagy is generally viewed as a survival mechanism, excessive autophagy has been associated with apoptosis and fibrosis.27 In alveolar macrophages, lipopolysaccharide induced autophagy and apoptosis. Blockade of the formation of autophagosomes inhibited apoptosis via the intrinsic apoptotic pathway.28 The apoptosis and fibrosis of myocardium were ameliorated in the CQ-treated diabetic mice. These results showed that inhibiting of excessive autophagy of STZ-induced diabetic mice was correlated with decreased apoptosis and fibrosis.
Conclusion
The present investigation demonstrates that overactivated autophagy in STZ-induced type 1 DM mice results in cardiac diastolic dysfunction. CQ improved LV diastolic function and preserved systolic contractility in STZ-induced type 1 diabetic mice by reducing autophagic conditions of the LV.
Acknowledgments
This work was supported by grants from the Natural Science Foundation of China (No 81000353 to Doctor G-S Zhang, No 81402928 to Doctor N Hou, No 81402926 to Doctor W-L Chen, No 81573429 to Doctor X-Q Wu, and No 81573433 to Doctor J-D Luo) and the Scientific and Technological Program of Guangzhou Education Bureau (No 2012C116 to Doctor G-S Zhang and No 2012C199 to Doctor N Hou).
Disclosure
The authors report no conflicts of interest in this work.
References
Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355(3):251–259. | ||
Takeda Y, Sakata Y, Mano T, et al. Competing risks of heart failure with preserved ejection fraction in diabetic patients. Eur J Heart Fail. 2011;13(6):664–669. | ||
Bhatia RS, Tu JV, Lee DS, et al. Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med. 2006;355(3):260–269. | ||
Felicio JS, Koury CC, Carvalho CT, et al. Present insights on cardiomyopathy in diabetic patients. Curr Diabetes Rev. Epub 2015 Sep 14. | ||
Kanamori H, Takemura G, Goto K, et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy. 2015;11(7):1146–1160. | ||
Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol. 2011;50(6):1035–1043. | ||
Zhu H, Tannous P, Johnstone JL, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117(7):1782–1793. | ||
Xu X, Kobayashi S, Chen K, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288(25):18077–18092. | ||
Liu H, Xie Q, Xin BM, et al. Inhibition of autophagy recovers cardiac dysfunction and atrophy in response to tail-suspension. Life Sci. 2015;121:1–9. | ||
Chaanine AH, Gordon RE, Nonnenmacher M, Kohlbrenner E, Benard L, Hajjar RJ. High-dose chloroquine is metabolically cardiotoxic by inducing lysosomes and mitochondria dysfunction in a rat model of pressure overload hypertrophy. Physiol Rep. 2015;3(7):ii:e12413. | ||
Xiao Q, Hou N, Wang YP, et al. Impaired sonic hedgehog pathway contributes to cardiac dysfunction in type 1 diabetic mice with myocardial infarction. Cardiovasc Res. 2012;95(4):507–516. | ||
Schannwell CM, Schneppenheim M, Perings S, Plehn G, Strauer BE. Left ventricular diastolic dysfunction as an early manifestation of diabetic cardiomyopathy. Cardiology. 2002;98(1–2):33–39. | ||
Krüger M, Babicz K, von Frieling-Salewsky M, Linke WA. Insulin signaling regulates cardiac titin properties in heart development and diabetic cardiomyopathy. J Mol Cell Cardiol. 2010;48(5):910–916. | ||
Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. | ||
Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13(5):619–624. | ||
Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell Death Differ. 2009;16(1):31–38. | ||
Cao DJ, Wang ZV, Battiprolu PK, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108(10):4123–4128. | ||
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. | ||
Gautel M. Cytoskeletal protein kinases: titin and its relations in mechanosensing. Pflugers Arch. 2011;462(1):119–134. | ||
Riahi Y, Wikstrom JD, Bachar-Wikstrom E, et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia. 2016;59(7):1480–1491. | ||
Li FJ, He CY. Acidocalcisome is required for autophagy in Trypanosoma brucei. Autophagy. 2014;10(11):1978–1988. | ||
Gurney MA, Huang C, Ramil JM, et al. Measuring cardiac autophagic flux in vitro and in vivo. Methods Mol Biol. 2015;1219:187–197. | ||
Ni HM, Bockus A, Wozniak AL, et al. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy. 2011;7(2):188–204. | ||
Nalbandian A, Llewellyn KJ, Nguyen C, Yazdi PG, Kimonis VE. Rapamycin and chloroquine: the in vitro and in vivo effects of autophagy-modifying drugs show promising results in valosin containing protein multisystem proteinopathy. PLoS One. 2015;10(4):e0122888. | ||
Kayama Y, Raaz U, Jagger A, et al. Diabetic cardiovascular disease induced by oxidative stress. Int J Mol Sci. 2015;16(10):25234–25263. | ||
Selvaraju V, Joshi M, Suresh S, Sanchez JA, Maulik N, Maulik G. Diabetes, oxidative stress, molecular mechanism, and cardiovascular disease – an overview. Toxicol Mech Methods. 2012;22(5):330–335. | ||
Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157(3):455–468. | ||
Chen S, Yuan J, Yao S, et al. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy. 2015;11(12):2346–2357. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.