Back to Journals » OncoTargets and Therapy » Volume 16
Chimeric Antigen Receptor (CAR) T-Cell Therapy for Patients with Lung Cancer: Current Perspectives
Authors Maher J ![]()
Received 16 April 2023
Accepted for publication 27 June 2023
Published 3 July 2023 Volume 2023:16 Pages 515—532
DOI https://doi.org/10.2147/OTT.S341179
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Nagashree Seetharamu
John Maher1– 3
1King’s College London, School of Cancer and Pharmaceutical Sciences, CAR Mechanics Lab, Guy’s Cancer Centre, Great Maze Pond, London, SE1 9RT, UK; 2Leucid Bio Ltd., Guy’s Hospital, Great Maze Pond, London, SE1 9RT, UK; 3Department of Immunology, Eastbourne Hospital, Kings Drive, Eastbourne, East Sussex, BN21 2UD, UK
Correspondence: John Maher, King’s College London, Guy’s Hospital, Great Maze Pond, London, SE1 9RT, UK, Tel +0044 207 188 1468, Fax +0044 207 188 0919, Email [email protected]; [email protected]
Abstract: Immunotherapy using chimeric antigen receptor (CAR)-engineered T-cells has achieved unprecedented efficacy in selected hematological cancers. However, solid tumors such as lung cancer impose several additional challenges to the attainment of clinical success using this emerging therapeutic modality. Lung cancer is the biggest cause of cancer-related mortality worldwide, accounting for approximately 1.8 million deaths worldwide each year. Obstacles to the development of CAR T-cell immunotherapy for lung cancer include the selection of safe tumor-selective targets, accounting for the large number of candidates that have been evaluated thus far. Tumor heterogeneity is also a key hurdle, meaning that single target-based approaches are susceptible to therapeutic failure through the emergence of antigen null cancers. There is also a need to enable CAR T-cells to traffic efficiently to sites of disease, to infiltrate tumor deposits and to operate within the hostile tumor microenvironment formed by solid tumors, resisting the onset of exhaustion. Multiple immune, metabolic, physical and chemical barriers operate at the core of malignant lesions, with potential for further heterogeneity and evolution in the face of selective therapeutic pressures. Although the extraordinarily adaptable nature of lung cancers has recently been unmasked, immunotherapy using immune checkpoint blockade can achieve long-term disease control in a small number of patients, establishing clinical proof of concept that immunotherapies can control advanced lung carcinomas. This review summarizes pre-clinical CAR T-cell research that is specifically focused on lung cancer in addition to published and ongoing clinical trial activity. A number of advanced engineering strategies are also described which are designed to bridge the gap to the attainment of meaningful efficacy using genetically engineered T-cells.
Keywords: immunotherapy, malignancy, engineered T-cell, pulmonary
Introduction
Lung cancer is the biggest cause of cancer deaths worldwide, accounting for over 18% of mortality attributable to malignant disease.1 It is estimated that almost 237,000 people were diagnosed with lung cancer in the United States (US) in 2022.2 Histological subtypes include (i) non-small cell lung cancer (NSCLC – approximately 85% of tumors), which includes adenocarcinoma, squamous cell, large cell and adenosquamous variants, and (ii) small cell lung cancer (SCLC – approximately 15% of tumors). Most patients present with advanced disease, significantly limiting the use of therapeutic options with curative intent. The main modalities used to treat these cancers include surgery, radiotherapy, chemotherapy, targeted therapies using small molecules, tumor-targeted monoclonal antibodies, and immune checkpoint inhibition. However, 130,000 deaths resulted from lung cancer in 2022, making this the leading cause of cancer death in the US.2 These data highlight a pressing unmet need for new treatment options for these patients.
Chimeric antigen receptors (CARs) are synthetic fusion molecules which direct the specificity of immune cells against native cell surface target molecules found on tumors and other cell types.3 In general, antigen engagement by CARs is direct rather than human leukocyte antigen (HLA)-restricted, akin to the recognition process used by an antibody. As a result, CAR T-cell immunotherapy obviates the need for HLA matching of the receptor to the target. Moreover, HLA downregulation by tumor cells (a common immune evasion mechanism in lung cancer)4–6 affords no protection to CAR T-cell recognition. CAR technology has been in development since 1987 when T-cell receptor/antibody chimeric receptors were first described.7 More than 20 years later, several large clinical centers independently described compelling clinical efficacy of CD19-targeted CAR T-cells in the treatment of patients with relapsed refractory B-cell malignancy.8 More recently, CAR T-cells specific for B-cell maturation antigen (BCMA) have proven to be highly effective in relapsed refractory multiple myeloma.9 However, solid tumors such as lung cancer remain largely refractory to this approach. Hurdles to the successful transition of this technology for common solid tumors have recently been reviewed, together with potential approaches that may be taken to mitigate these issues.10 This review sets out to consider pre-clinical efforts to develop effective CAR T-cell solutions specifically for lung cancer, in addition to ongoing and published clinical trial activity in this arena. Only primary lung cancer has been included in this review, meaning that metastatic lung cancer and malignant pleural mesothelioma are not discussed. Other T-cell-based immunotherapies for lung cancer have been considered in recent reviews and are not further covered here.11
Structurally, CARs consist of a targeting moiety, spacer domain, transmembrane region and one or more signaling units. Most commonly the targeting moiety consists of a single-chain antibody fragment (scFv) in which variable heavy and variable light chain domains are joined using a short linker. Generations of CARs that have been evaluated thus far are summarized in Figure 1. Successful application of CAR T-cell immunotherapy for blood cancers relies upon the use of so-called second generation (2G) designs in which the signaling domain consists of a co-stimulatory unit (generally either CD28 or 4–1BB), placed upstream of an activating unit (most commonly CD3 zeta). However, a range of CAR generations have been evaluated in lung cancer models as well as in the clinic.
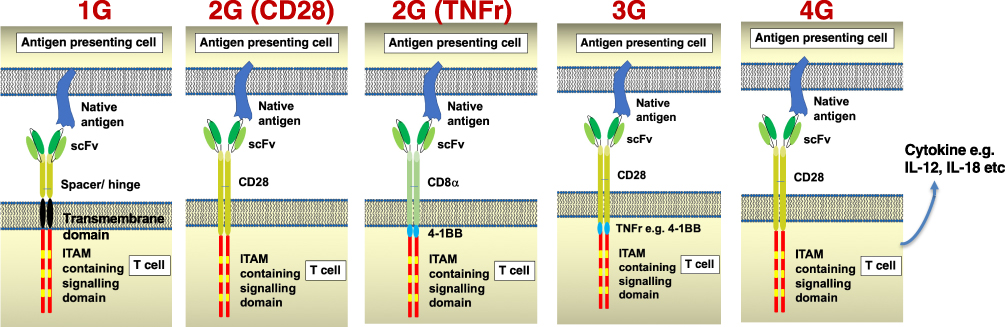 |
Figure 1 Generations of chimeric antigen receptors. Chimeric antigen receptors consist of a targeting moiety (most commonly an scFv as illustrated here), a spacer/hinge, transmembrane domain and a signaling endodomain. Antigen recognition is direct provided that a target cell (here designated as antigen-presenting cell) expresses the target antigen on the cell surface. In first generation (1G) CARs, the endodomain contains an ITAM (immunoreceptor tyrosine-based activation motif)-containing activating module. Most commonly CD3 zeta is used for this purpose since each monomer contains 3 ITAMs. Second generation (2G) CARs contain a co-stimulatory domain placed upstream of CD3 zeta and most commonly derived from CD28 or 4–1BB. Third generation (3G) CARs contain two complementary co-stimulatory units while armored CARs produce a cytokine such as IL-12 or IL-18. |
Evaluation of CAR T-Cell Immunotherapy in Models of Lung Cancer
Several pre-clinical investigations of CAR T-cell immunotherapy have been undertaken in models of lung cancer. These involve a diverse range of target molecules and are summarized in the section that follows.
Receptor Tyrosine Kinases
Epidermal Growth Factor Receptor
Many early studies focused on CAR T-cell targeting of receptor tyrosine kinases. The epidermal grown factor receptor (EGFR) is highly expressed in about 60% of NSCLC tumors.12 Zhou et al found that 2G (CD28) CAR T-cells directed against EGFR could delay disease progression in an A549 NSCLC tumor xenograft model.13 No toxicity was observed, although ability of the targeting moiety to engage murine EGFR was unclear.
A key difficulty with targeting of EGFR (like many solid tumor targets) is the fact that it is expressed at low levels in many normal tissues, most notably in the basal layers of skin epidermis.14 One approach that has been used to improve therapeutic index involves the co-expression of an appropriate chemokine receptor alongside the CAR. Using this approach, tumor cell homing of CAR T-cells is enhanced while on-target off-tumor toxicity is minimised due to reduced transit time in normal tissues.15 NSCLC tumors produce high levels of CXCL13 which has been exploited through the co-expression of CXCR5 alongside an EGFR-specific 2G (4–1BB) CAR.16 As a result, enhanced CAR T-cell migration to and destruction of A549 tumors was demonstrable, albeit only when tumor cells were engineered to over-produce CXCL13. In a related approach, 2G (4–1BB) EGFR CAR T-cells have been engineered to co-express CCR6, in combination with a PD1 blocking antibody.17 Although significant anti-tumor activity was observed, the A549 tumor model used in this study was once again engineered to overproduce the relevant chemokine ligand (in this case, CCL20).
While most CARs are targeted using an scFv, an adnectin (derived from the 10th type III domain in fibronectin) with specificity for EGFR has also been used to direct the specificity of a 2G (4–1BB) containing CAR.18 When compared to a matched cetuximab-derived scFv-based CAR, comparable anti-tumor activity was observed in a H292 lung cancer xenograft model.
Lentiviral and retroviral vectors are most commonly employed to generate CAR T-cells. In an alternative strategy, Li et al have used the piggyBac transposon system to engineer EGFR-specific 2G (4–1BB) CAR T-cells.12 Using a H460 NSCLC xenograft model, they found that co-administration of intra-tumoral and intravenous CAR T-cells was effective in mediating disease control without toxicity. Once again, however, reactivity of the scFv with mouse EGFR was not shown.
In up to 10% of NSCLC tumors, a splice variant of EGFR known as EGFRvIII is found.19 As a result, extracellular amino acids 6–273 are deleted, leading to constitutive receptor activity. A 3G (CD28 + 4–1BB) CAR with specificity for EGFRvIII demonstrated therapeutic efficacy against an established metastatic A549 xenograft in which EGFRvIII expression had been enforced.20 A difficulty with this target is the fact that expression is often heterogeneous in tumors, meaning that antigen null tumors are likely to emerge in the face of CAR T-cell-mediated selective pressure.21
c-Met Receptor
The c-Met receptor tyrosine kinase is expressed in 60–100% of NSCLC and commonly provides a mechanism by which tumors acquire EGFR inhibitor resistance.22 CARs have been engineered to recognize MET using derivatives of its natural ligand, hepatocyte growth factor23 or using an scFv.22 A 2G (4–1BB) CAR targeted against c-Met demonstrated anti-tumor activity against A549 NSCLC tumor xenografts without toxicity, although ability of this scFv-based CAR to engage the mouse ortholog of this target was unclear.22 Given the widespread expression of c-Met in normal tissues,24 clinical evaluation of CAR T-cells directed against this target was first performed using mRNA transfected T-cells, injected intratumorally in six patients with metastatic breast cancer.25 However, safety of systemically delivered CAR T-cells with specificity for c-Met remains to be determined.
Receptor Tyrosine Kinase-Like Orphan Receptor (ROR) 1
Receptor tyrosine kinase-like orphan receptor family member (ROR)1 is an onco-fetal receptor tyrosine kinase which is expressed in 93% of SCLC,26 up to 90% of lung adenocarcinomas, 12% of lung squamous cell carcinomas as well as a subset of rarer lung cancer subtypes.27,28 By contrast, normal tissue expression of ROR1 is minimal with low levels found in regenerating B-cells, parathyroid gland, pancreatic islets, gastrointestinal tract and adipose tissue.29 Moreover, 2G (4–1BB) ROR1-specific CAR T-cells have proven to be safe in primate studies.30 In vitro testing of the same CAR using 3D culture systems has revealed that they can control 3D basement membrane-supported cultures of A549 NSCLC cells,31 prompting clinical studies which are described later in this review.
Other Receptor Tyrosine Kinases
AXL is expressed in 69% of NSCLC tumors although low-level expression in a number of normal tissues has been highlighted.32 3G (CD28 + 4–1BB) AXL-specific CAR T-cells elicit anti-tumor activity against A549 NSCLC tumor xenografts.33
Vascular endothelial growth factor receptor (VEGFR)2 is expressed in tumor endothelial cells that support lung and other tumor types. VEGFR2 has been successfully targeted using a 2G (CD28) CAR in mice bearing A549 NSCLC xenografts although VEGFR2 expression was enforced in this model.34
Over 90% of NSCLC tumors are reported to express erythropoietin-producing human hepatocellular carcinoma type A receptor 2 (EphA2) with negligible expression in normal lung.35 A 2G 4–1BB containing CAR with specificity for EphA2 demonstrated anti-tumor activity in an A549 lung cancer xenograft model.35
Carcinoembryonic Antigen (CEA)
Carcinoembryonic antigen (CEA) is expressed in about 70% of NSCLC tumors.36 When 2G (CD28) CEA-specific CAR T-cells were administered to mice with an established A549 NSCLC tumor, transient disease control was noted.37 3G (CD28 + 4–1BB) CEA-specific CAR T-cells have also demonstrated anti-tumor activity in NSCLC models that were resistant to antibody-drug conjugates directed against the same target.38 Regarding healthy tissue biodistribution, CEA is found in the gastrointestinal and respiratory tract. However, in contrast to tumor cell expression, CEA is polarized at the luminal surface in normal epithelia meaning that access to CAR T-cells should theoretically be restricted at these sites. Nonetheless, Phase 1 clinical testing revealed pulmonary toxicity in subjects who received a 1G CAR with specificity for CEA, highlighting the risks associated with this and related targets that exhibit polarized epithelial cell expression.39
Glypican 3
Glypican 3 is an oncofetal heparan sulfate glycoprotein that is found in about 63% of squamous cell lung carcinomas.40 By contrast, expression in normal tissue, including lung, is negligible.40,41 Anti-tumor activity of 3G (CD28 + 4–1BB) glypican-3 CAR T-cells has been demonstrated against two squamous cell lung carcinoma tumor xenografts, although glypican-3 expression was enforced in tumor cells by lentiviral transduction in each case.40
Prostate Stem Cell Antigen (PSCA)
PSCA is also expressed in non-small cell lung cancer.42 Exploiting this, 2G (CD28) CAR T-cells have proven efficacious against patient-derived xenograft (PDX) models of NSCLC.43 When PDX co-express both PSCA and MUC1 (another potential target of interest in NSCLC), synergistic therapeutic efficacy was reported in a second PDX model.43
CD44v6 Splice Variant
The CD44v6 adhesion receptor is commonly expressed in a range of tumor types, including lung cancer. It is estimated that about 90% of squamous cell carcinomas express this splice variant while up to 50% of lung adenocarcinomas are positive.44 In pre-clinical testing, T-cells that were engineered to co-express a CD44v6-specific 2G (CD28) CAR co-expressed with a suicide gene mediated anti-tumor activity in mice engrafted with MR232 lung adenocarcinoma tumor xenografts.45 However, low-level expression of CD44v6 in skin has been documented.46 Potential significance of this finding for CAR T-cell immunotherapy is highlighted by the fact that the CD44v6-specific antibody–drug conjugate, bivatuzumab, caused fatal toxic epidermal necrolysis in one subject.47
CD56
The CD56 antigen (neural cell adhesion molecule) is expressed in many cancers that exhibit neuronal or neuroendocrine differentiation, including SCLC.48 Crossland et al used the Sleeping Beauty transposon system to generate 2G (CD28) CAR T-cells with specificity for this target.48 Therapeutic efficacy against CD56-expressing H526 SCLC xenografts was demonstrated. Although CD56 is expressed on NK cells and neurones, CAR T-cells directed against this target have been safely administered as consolidation therapy to a child with embryonal rhabdomyosarcoma following repeated treatment with surgery, radiotherapy and chemotherapy.49
Epithelial Cell Adhesion Molecule (EpCAM)
EpCAM is expressed in about 50% of NSCLC tumors.50 Similar to CEA, expression by normal cells is polarized to the luminal surface, whereas such polarity is not evident in transformed cells. Once again, however, pulmonary immunopathology has been noted in some pre-clinical models when EpCAM has been targeted using CAR T-cells.51 Using an orthotopic Lewis lung carcinoma tumor model, Xu et al demonstrated that a 2G (CD28) CAR T-cells targeted against EpCAM could delay intracranial disease progression following local but not systemic delivery, although persistence of cells was insufficient to achieve disease eradication.50 One strategy that may be used to dial down risk of toxicity associated with the targeting of self-antigens such as EpCAM entails the use of low affinity targeting moieties that can discriminate between healthy (EpCAM low) and malignant cells (EpCAM) high.52 Nonetheless, risk of antigen downregulation leading to therapeutic failure needs to be considered with this approach.
B7 Family Members
Two members of the B7 family are commonly expressed on lung cancers, namely PD-L1 and B7-H3.
Programmed Death Receptor Ligand 1 (PD-L1)
PD-L1 is widely expressed on a range of solid tumors including lung cancer. 2G (4–1BB) CAR T-cells with specificity for B7-H3 delayed disease progression in an A549 NSCLC xenograft model53 while 3G (4–1BB + Toll-like receptor 2) PD-L1-specific CAR T-cells demonstrated efficacy against an NSCLC PDX tumor. Surprisingly however, co-treatment with this and a second population of CAR T-cells targeting mesothelin (MSLN) yielded inferior anti-tumor activity to PD-L1-specific CAR T-cells alone.54 Nonetheless, PD-L1-specific CAR T-cells have been evaluated clinically in NSCLC as summarized later in this review.
B7-H3
B7-H3 (CD276) shares 30% amino acid homology with PD-L1 although its cognate receptor(s) remains unclear. Expression of B7-H3 in normal tissue is negligible while it is found in about three quarters of NSCLC tumors,53 both on tumor and vascular/stromal elements, and expression is correlated with poor prognosis.55 Clinical experience with B7-H3 targeted CAR T-cells in the treatment of other solid tumor types is summarized elsewhere.24 In pre-clinical studies, 2G (4–1BB) CAR T-cells delayed tumor progression in an A549 NSCLC xenograft model.53 B7-H3 CAR T-cell trafficking has also been directed by the co-expression of the CCR2b chemokine receptor to facilitate traversal of the blood–brain barrier in order to tackle central nervous system metastases.56
Delta-Like Ligand (DLL)3
DLL3 is an inhibitory Notch ligand that is predominantly expressed on intracellular membranes. However, due to substantial over-expression in SCLC, moderate cell surface expression is common in this tumor, of the order of 10,000 molecules per cell.57 3G (CD28 + 4–1BB) DLL3-specific CAR T-cells have been developed for application against SCLC, in which this target is selectively expressed in about 70% of tumors.58 Transient toxicity was observed in treated mice, which may have been due to cytokine release syndrome and authors found that a bispecific antibody co-targeting DLL and CD3 was more effective in similar pre-clinical models. More recently, Jaspers et al developed a panel of DLL3-specific CARs which proved efficacious in immunodeficient and immune competent mouse models of SCLC.59 On a cautionary note, however, the DLL3-specific antibody–drug conjugate rovalpituzumab tesirine (Rova)-T was unsuccessful from an efficacy perspective in Phase 3 testing in patients with SLCL.60
CD47
CD47 is commonly expressed on solid tumors including lung cancer and represents a macrophage immune checkpoint. It imparts a “don’t eat me” signal via interaction with signal regulatory protein a on macrophages.61 A 3G (CD28 + 4–1BB) CAR has demonstrated in vitro anti-tumor activity against A549 NSCLC cells, although in vivo testing was not undertaken.61
Gangliosides
Disialoganglioside D2
Disialoganglioside D2 (GD2) has been studied quite extensively as a CAR T-cell target in neuroblastoma. A recent clinical trial undertaken in Italy using a 3G (CD28 + 4–1BB) GD2-specific CAR has yielded the most compelling solid tumor CAR T-cell efficacy data yet described worldwide, with a complete response rate of 33% (9/27) and a partial response rate of 30% (8/27).62 GD2 is also expressed in 39% SCLC, 56% of squamous cell lung carcinomas and 72% of lung adenocarcinomas.63 Using both orthotopic and metastatic lung cancer models, anti-tumor activity of 2G (CD28) CAR T-cells with specificity for GD2 was demonstrated.63
Ganglioside M2
Ganglioside M2 (GM2) is also expressed in a range of solid tumors including lung cancer. Third generation (CD28 + 4–1BB) CAR T-cells directed against GM2 have been armored to co-express IL-7 and CCL19 and demonstrated compelling efficacy in a xenograft models of SCLC, contrasting with lack of efficacy of non-armored CAR T-cells in these models.64 This was accompanied by significant intra-tumoral infiltration by CAR T-cells arising from autocrine IL-7 and CCL19 stimulation, which promote increased proliferation and migration of the CAR T-cells, respectively.
Mesothelin
Mesothelin (MSLN) is a target of interest in lung cancer, in addition to mesothelioma, pancreatic and ovarian cancers. A 2G (CD28) CAR with specificity for MSLN delayed tumor progression in a Lewis lung carcinoma model in a manner that was further potentiated by delivery of a glutamine antagonist to the tumor microenvironment (TME) using anti-PD-L1 targeted nanovesicles.65 Alternatively, a novel CAR design known as a TRuC in which a MSLN-specific scFv is fused to CD3 epsilon achieved potent anti-tumor activity in a range of solid tumor models, including an A549-MSLN lung cancer xenograft.66 In the TRuC design, the scFv CD3 epsilon fusion becomes incorporated into the endogenous TCR CD3 complex, thereby providing T-cell receptor-like signaling to the host cell. When compared to a matched 2G (4–1BB) CAT, the TRuC CAR T-cells achieved more rapid tumor accumulation and disease regression.66
Intensity of co-stimulation may also influence anti-tumor activity in lung cancer models. Positioning of Dap10 downstream of CD3 zeta in a 2G (CD28) CAR with specificity for MSLN enhanced cytolytic activity, cytokine secretion, serial killing activity and therapeutic efficacy against A549 NSCLC tumor xenografts and a lung cancer PDX when compared to unmodified 2G CAR T-cells.67
Miscellaneous Targets
Several additional candidate CAR T-cell targets have been pursued pre-clinically in lung cancer models. Lung-specific (Lun)X is selectively expressed in approximately 80% of NSCLC but not healthy tissues.68,69 A 3G (CD28+4-1BB) CAR has been developed to target LunX and demonstrated efficacy in an A549 NSCLC xenograft and a patient-derived xenograft model.69
PTK7 is a non-canonical signalling Wnt family pseudokinase that is expressed in a range of solid tumors including NSCLC whereas normal tissue expression is low level in some epithelia.70 Anti-tumor activity of a 2G (4–1BB) CAR has been demonstrated in xenograft models of both NSCLC and SCLC.70 By contrast, screening of reactivity of these CAR T-cells with a limited panel of human tissues did not uncover any evidence of on-target off-tumor toxicity.
Olfactory receptor OR2H1 has recently been identified as a common solid tumor cell surface target for which normal tissue expression is restricted to testis.71 Expression has been identified in 13% of lung carcinomas and a 2G (4–1BB) CAR directed against this target demonstrated anti-tumor activity in a H2009 lung adenocarcinoma tumor xenograft model.71
Mutation of EGFR is common in NSCLC and leads to resistance to EGFR tyrosine kinase inhibitors. In this setting, CD70 has been identified as commonly expressed on these tumor cells, most notably in those that have undergone epithelial-to-mesenchymal transition.72 Moreover, expression of CD70 in NSCLC was associated with poorer survival. CAR T-cells and NK cells directed against CD70 demonstrated anti-tumor activity in a number of NSCLC models.72 Although clinical trials of CD70-targeted CAR T-cells in lung cancer were not identified, one patient with renal cell carcinoma has achieved a complete response following the infusion of CD70-targeted CAR T-cells.73
A major advantage of CAR technology is the HLA-independent nature of antigen recognition, as alluded to above. Nonetheless, this denies access to intracellular antigens, many of which are more tumor selective. One such example is MAGE-A1, a cancer testis antigen that is expressed in 44% of lung adenocarcinomas.74 CAR T-cells with anti-tumor activity against this target have been developed, although the precise HLA-restriction of the CAR is unclear.74
Multi-Targeted and Combinatorial Approaches
Several advanced CAR T-cell approaches have been described for experimental treatment of lung cancer. The first of these entails the co-administration of CAR T-cells targeted against tumor and tumor-supportive stroma. Karakla et al showed that growth of an A549 lung tumor xenograft was delayed following treatment with CAR T-cells directed against fibroblast activation protein α (FAP), which targets stromal fibroblasts, but not malignant cells.75 However, the combined administration of FAP- and EphA2-specific CAR T-cells (which target tumor cells) led to a further enhancement of therapeutic activity and survival.
Dual antigen targeted CAR T-cells represent an alternative strategy to enhance potency, mitigate risk of therapeutic failure due to antigen loss and maximize discrimination between malignant and normal cell types. Tandem CARs that co-target B7-H3 and CD70 have been shown to exert superior anti-tumor activity against a large cell lung cancer xenograft (NCI-H460) when compared to single antigen controls, meaning that lower dosing levels were sufficient for efficacy.76 Chu et al employed universal CAR T-cells with specificity for fluorescein isothiocyanate (FITC) for combined use with an NSCLC-specific FITC-conjugated ligand.77 This approach offers the potential for combined use with one or more targeting moieties, administered either simultaneously or sequentially. Improved discrimination between malignant and healthy cells may be achieved using logic gating strategies that exploit dual antigen recognition78 or use of inhibitory CARs that recognise targets on normal tissue.79,80 Multi-target recognition may also be used to boost potency within the tumor microenvironment with the use of so-called parallel CARs.81 This solution entails the co-expression of a 2G CAR (eg containing CD28) with a chimeric co-stimulatory domain that contains a complementary co-stimulatory unit (eg 4–1BB). Using this parallel approach, two or more targets may be simultaneously engaged and optimized dual co-stimulation is delivered since both signaling units are located in their natural juxtamembrane position. To address the distinct co-stimulatory requirements of different T-cell subsets, Guedan et al co-administered MSLN-specific CD4+ CAR T-cells that contained an ICOS + CD3 zeta endodomain with MSLN-specific CD8+ CAR T-cells containing a fused 4–1BB + CD3 zeta endodomain.82 Sustained persistence and anti-tumor immunity was demonstrated against subcutaneous L55 NSCLC xenografts.
A number of combinatorial strategies involving CAR T-cells and other pharmaceuticals have also been described. For example, oncolytic viruses may be used in combination with CAR T-cells since they not only exert a direct anti-tumor effect but also create a more favorable immune environment within the tumor. In the context of lung cancer, this approach was exemplified using both EGFR and HER2 CAR T-cells in A549 NSCLC xenograft models.83 Alternatively, it has been shown that that N-linked glycans hinder recognition of solid tumor cells (including lung cancer) by CAR T-cells.84 This inhibitory effect may be overcome by deletion of mannoside acetyl-glucosaminlytransferase 5 (MGAT5) in tumor cells or treatment with the N-glycan inhibitor, 2-deoxy-D-glucose.
Clinical Experience of CAR T-Cell Immunotherapy of Lung Cancer
There have been limited reports of CAR T-cell clinical trials targeted against lung cancer, although there is extensive ongoing clinical trial activity in this arena (summarized in Table 1). A summary of clinical reports published this far is presented below.
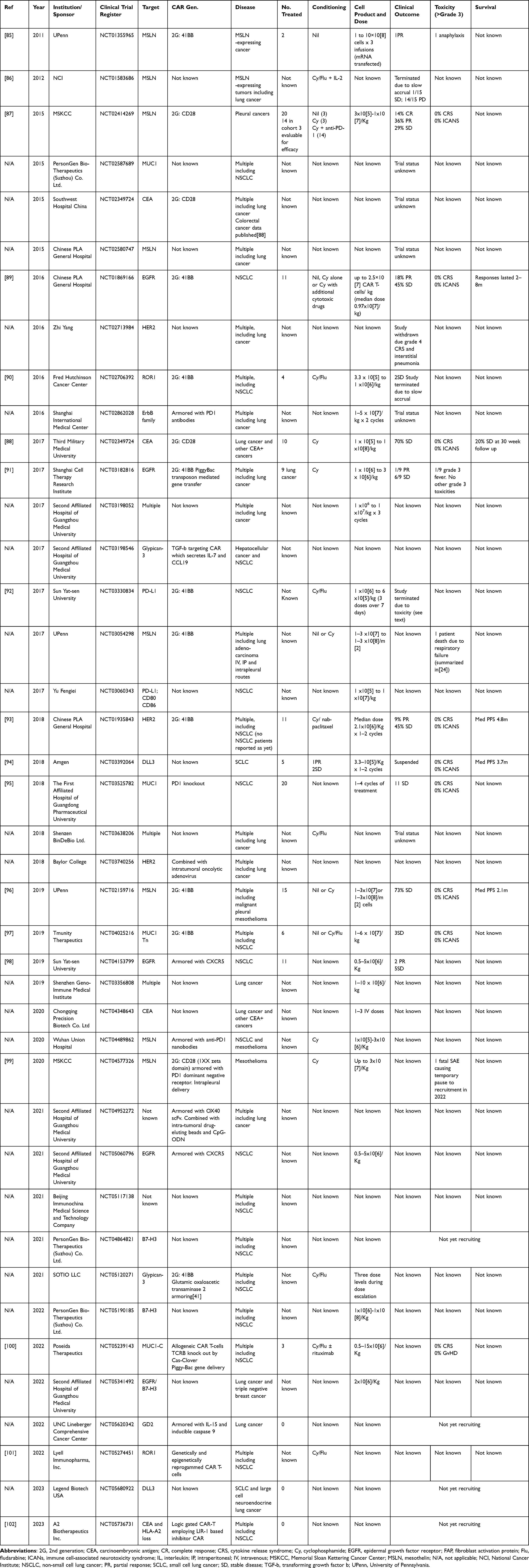 |
Table 1 Clinical Trials Evaluating CAR T-Cell Immunotherapy of Lung Cancer |
Feng et al described a clinical study in which 11 subjects with EGFR+ NSCLC were treated with 2G (4–1BB)-engineered CAR T-cells.89 Two subjects achieved a partial response, although the contributory effect of conditioning chemotherapy with cisplatin, cyclophosphamide and either pemetrexed or docetaxel remains uncertain. Five additional patients achieved stable disease in this study. Despite issues with toxicity in other EGFR-targeted clinical trials,24 therapy was well tolerated with the exception of one grade 3–4 increase in serum lipase. A more recent study in NSCLC was undertaken by a group in Shanghai using PiggyBac transposon-engineered EGFR-re-targeted CAR T-cells, administered as two doses. Once again, infusions were generally well tolerated and one durable PR that lasted 13 months was reported in a series of 9 patients.91
Given the frequent expression of ROR1 in lung cancer, a clinical trial of 2G (4–1BB) ROR1-targeted CAR T-cells was initiated in subjects with ROR1+ NSCLC and TNBC (NCT02706392).90 Following condition with cyclophosphamide and fludarabine, cells were infused intravenously but demonstrated poor trafficking to the site of disease. Two patients with NSCLC achieved a mixed response, with reduction in tumor burden observed at some metastatic sites of disease.103 Subsequent pre-clinical mouse studies demonstrated that conditioning with oxaliplatin resulted in macrophage activation and chemokine release, favoring enhanced CAR T-cell recruitment to tumors and providing a rational direction for future clinical testing.90
Mesothelin was the target of a Phase 1 clinical trial employing a 2G (CD28) CAR. One patient with lung cancer and malignant pleural disease was treated with intrapleural CAR T-cells without prior lymphodepletion and survived for approximately 12 months.104 Two mesothelioma patients treated with a similar CAR T-cell approach after cyclophosphamide conditioning and combined with pembrolizumab achieved complete metabolic responses, as detected using Positron emission tomography (PET) using18 fluoro-deoxyglucose (FDG).
PD-L1 has also been the subject of a CAR T-cell clinical trial in patients with NSCLC (clinicaltrials.gov reference NCT03330834).92 One patient was conditioned with cyclophosphamide and fludarabine and treated with a 2G (4–1BB) scFv-based CAR (scFv derived from atezolizumab) at doses of 1×105/kg on day 1, 3x105/kg on day 4 and 6×105/kg on day 8. CAR T-cell persistence at 3.3% of peripheral blood T-cells was detected on day 29. He presented on day 47 with pyrexia and adult respiratory distress syndrome, accompanied by elevated CRP and IL-6 (but not other cytokines). Investigators felt that this was atypical for cytokine release syndrome and instead most likely represented on-target off-tumor toxicity. The patient was treated with tocilizumab and methylprednisolone with rapid resolution and the study has been terminated.
Finally, MUC1 is a popular choice for clinical studies and was targeted in 20 NSCLC patients using CAR T-cells in which PD1 had been knocked out. However, stable disease was the best response observed, an outcome reported in eleven subjects.95 Similarly, stable disease (achieved in 3 of 6 treated subjects) was the best response achieved using CAR T-cells specific for the Tn glycoform of MUC1.105
Perspectives and Conclusions
Despite considerable effort, CAR T-cell immunotherapy has achieved little meaningful clinical impact against lung cancer. The main limitation has been lack of efficacy rather than unacceptable safety. Recent studies of the biology of NSCLC from the TRACERx study highlight significant issues of intra-tumoral heterogeneity, varied pathways to metastasis and the remarkable propensity of these tumors to undergo clonal evolution under selective pressure applied by chemotherapy and perhaps immune selection.106 The profound adaptability of lung cancers highlights the significant challenge in developing broadly applicable therapeutic approaches for this disease, including CAR T-cell solutions. Nonetheless, immune checkpoint blockade can achieve long-term survival in a subset of NSCLC patients, demonstrating the principle that sustained disease control can be achieved by immune effector cells even in advanced disease.107
The first key obstacle to consider is the need for efficient CAR T-cell homing towards and infiltration of solid tumors such as lung cancer. Co-expression of appropriate chemokine receptors in CAR T-cells offers one possible solution as described above, although exemplification thus far in lung cancer has involved enforced expression of cognate chemokines in tumor cells. One additional option entails the co-expression of CXCR2 in these cells15 which binds to chemokines such as IL-8 that are naturally over-expressed in NSCLC and derived cell-line models.108 Tumor infiltration by CAR T-cells may also be facilitated by the co-expression of matrix degrading enzymes such as heparanase.109
Given the heterogeneity of lung tumors described above, it is probable that single target antigens will not be uniformly expressed by all malignant cells. This highlights the need to consider combinatorial targeting strategies, as described above. Alternatively, CARs may be engineered to engage multiple targets using a single targeting moiety. One example involves the use of NKG2D-based CARs110 which bind to eight discrete ligands that are highly expressed in NSCLC.111 Alternatively, the panErbB-specific T1E28z CAR employs a promiscuous ligand, T1E, that can bind to eight discrete ErbB homo- or heterodimers.112 Although ErbB dimers are highly expressed in lung cancer,113 this CAR may be unsuited to systemic administration owing to risks of on-target off-tumor toxicity due to low-level ErbB expression in normal tissues. To mitigate risk, a hypoxia-sensing derivative has been developed whereby CAR expression is restricted to the hypoxic TME.114 These “hypoxiCAR” T-cells elicit therapeutic activity against ErbB-expressing solid tumors without toxicity in mice, despite the ability of T1E to bind mouse ErbB orthologues with high efficiency.
The tumor microenvironment (TME) is the battleground within which CAR T-cells must operate. The physical, chemical and biological hurdles which operate at that site together with generic strategies that may help to overcome these obstacles have recently been reviewed elsewhere.115–117 Additionally, a number of specific approaches have been tested to modulate the TME in lung cancer in favor of CAR T-cell activation. For example, disruption of the NSCLC tumor microenvironment using a nanozyme potentiates anti-tumor activity of B7-H3-specific CAR T-cells.118 Moreover, microwave ablation has been used to remodel the TME in order to facilitate efficacy of AXL targeted CAR T-cells against NSCLC.33
To potentiate impact, armoring strategies have also been employed to modulate inhibitory influences operative within the tumor microenvironment. High interstitial pressure within lung tumors can lead to the upregulation of the PD-L1 immune checkpoint, thereby mediating resistance to CAR T-cell immunotherapy.119 Li et al investigated whether armoring of CAR T-cells to secrete PD1-specific scFvs could enhance anti-tumor activity.120 They used a H292 lung cancer xenograft model in which expression of CD19 was enforced and observed that CD19-specific CAR T-cells that constitutively produced anti-PD1 scFvs outperformed CAR T-cells alone, or when combined with systemic PD1 blockade. Unfortunately, however, this principle was not exemplified using an endogenously expressed lung cancer target.
Cytokine armoring strategies represent another commonly used approach that has also been applied to CAR T-cells directed against lung cancer. Illustrating this, anti-tumor activity of CEA-specific 2G (CD28) CAR T-cells was potentiated by armoring with nuclear factor of activated T-cells (NFAT)-inducible IL-18, an intervention which increased M1-polarized macrophages and natural killer cells, while reducing regulatory T-cells within the tumor microenvironment.37 Similarly, DLL3-specific CAR T-cells have been armored to express constitutively active IL-18, boosting anti-tumor activity due to stimulatory actions on the CAR T-cells themselves and on myeloid cells in the TME.59 Interleukin 12 also represents an attractive armoring cytokine, albeit limited by high potential for toxicity. Immunotherapy using tumor-infiltrating lymphocytes in which IL-12 expression was directed by an NFAT (nuclear factor of activated T-cells) promoter potentiated efficacy in patients with melanoma but at the risk of severe toxicity.121 Similar considerations apply to the use of IL-18, establishing the desirability of tightly regulated control systems that restrict biological activity of these potently pro-inflammatory cytokines to the TME.
A further intriguing option to target solid tumors such as lung cancer entails the use of macrophage-targeted CARs.52,122 Macrophages have a natural tumor tropism and Sanchez-Paulete et al have shown that such cells can infiltrate orthotopic lung tumors and impede tumor progression.52 Clinical efficacy data using this novel approach are awaited.
In conclusion, lung cancer presents a substantial challenge to the development of effective CAR-based immunotherapies. Nonetheless, advanced genetic engineering technologies that allow precise delivery of cells to the site of disease, recognition of a desired target portfolio, re-education of the immunosuppressive TME and also the stimulation of endogenous immune reactivity and epitope spreading123–127 together offer hope for future success in this daunting quest.
Disclosure
J.M. is CSO, scientific founder and shareholder of Leucid Bio, is a member of the scientific advisory board of Arovella Therapeutics Ltd and has undertaken consultancy work for Bristol-Meyers-Squibb, Juno, Celgene, Poolbeg Pharma, Ellipses Pharma and Biotest. The author reports no other conflicts of interest in this work.
References
1. Kocarnik JM, Compton K, Dean FE, et al.; Global Burden of Disease Cancer C. Cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life years for 29 cancer groups from 2010 to 2019: a systematic analysis for the global burden of disease study 2019. JAMA Oncol. 2022;8(3):420–444.
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33.
3. Maher J. Immunotherapy of malignant disease using chimeric antigen receptor engrafted T cells. ISRN Oncol. 2012;2012:278093.
4. Koukourakis IM, Giatromanolaki A, Mitrakas A, Koukourakis MI. Loss of HLA-class-I expression in non-small-cell lung cancer: association with prognosis and anaerobic metabolism. Cell Immunol. 2022;373:104495.
5. Ichinokawa K, Nakanishi Y, Hida Y, et al. Downregulated expression of human leukocyte antigen class I heavy chain is associated with poor prognosis in non-small-cell lung cancer. Oncol Lett. 2019;18(1):117–126.
6. Gettinger S, Choi J, Hastings K, et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017;7(12):1420–1435.
7. Kuwana Y, Asakura Y, Utsunomiya N, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149(3):960–968.
8. Halim L, Maher J. CAR T-cell immunotherapy of B-cell malignancy: the story so far. Ther Adv Vaccines Immunother. 2020;8:2515135520927164.
9. Biltibo E, Berdeja JG. SOHO State-of-the-Art Updates and Next Questions | BCMA-Directed CAR T-Cells: early Results and Future Directions. Clin Lymphoma Myeloma Leuk. 2023:45.
10. Maher J. Solid tumours: building bridges to CAR-T success. Clin Translational Discovery. 2023;3:e179.
11. Want MY, Bashir Z, Najar RA. T cell based immunotherapy for cancer: approaches and strategies. Vaccines. 2023;11(4):56.
12. Li H, Huang Y, Jiang DQ, et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Dis. 2018;9(2):177.
13. Zhou X, Li J, Wang Z, et al. Cellular immunotherapy for carcinoma using genetically modified EGFR-specific T lymphocytes. Neoplasia. 2013;15(5):544–553.
14. Tran QT, Kennedy LH, Leon Carrion S, et al. EGFR regulation of epidermal barrier function. Physiol Genomics. 2012;44(8):455–469.
15. Whilding LM, Halim L, Draper B, et al. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers. 2019;11(5):674.
16. Li G, Guo J, Zheng Y, et al. CXCR5 guides migration and tumor eradication of anti-EGFR chimeric antigen receptor T cells. Mol Ther Oncolytics. 2021;22:507–517.
17. Wang J, Wang Y, Pan H, et al. Chemokine Receptors CCR6 and PD1 Blocking scFv E27 Enhances Anti-EGFR CAR-T Therapeutic Efficacy in a Preclinical Model of Human Non-Small Cell Lung Carcinoma. Int J Mol Sci. 2023;24(6):5424.
18. Han X, Cinay GE, Zhao Y, Guo Y, Zhang X, Wang P. Adnectin-Based Design of Chimeric Antigen Receptor for T Cell Engineering. Mol Ther. 2017;25(11):2466–2476.
19. Okamoto I, Kenyon LC, Emlet DR, et al. Expression of constitutively activated EGFRvIII in non-small cell lung cancer. Cancer Sci. 2003;94(1):50–56.
20. Zhang Z, Jiang J, Wu X, et al. Chimeric antigen receptor T cell targeting EGFRvIII for metastatic lung cancer therapy. Front Med. 2019;13(1):57–68.
21. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984.
22. Min J, Long C, Zhang L, et al. c-Met specific CAR-T cells as a targeted therapy for non-small cell lung cancer cell A549. Bioengineered. 2022;13(4):9216–9232.
23. Thayaparan T, Petrovic RM, Achkova DY, et al. CAR T-cell immunotherapy of MET-expressing malignant mesothelioma. Oncoimmunology. 2017;6(12):e1363137.
24. Maher J, Davies DM. CAR Based Immunotherapy of Solid Tumours-A Clinically Based Review of Target Antigens. Biology. 2023;12(2):287.
25. Tchou J, Zhao Y, Levine BL, et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol Res. 2017;5(12):1152–1161.
26. Wang WZ, Shilo K, Amann JM, et al. Predicting ROR1/BCL2 combination targeted therapy of small cell carcinoma of the lung. Cell Death Dis. 2021;12(6):577.
27. Baldwin ET, Gotte M, Tchesnokov EP, et al. Human endogenous retrovirus-K (HERV-K) reverse transcriptase (RT) structure and biochemistry reveals remarkable similarities to HIV-1 RT and opportunities for HERV-K-specific inhibition. Proc Natl Acad Sci U S A. 2022;119(27):e2200260119.
28. Balakrishnan A, Goodpaster T, Randolph-Habecker J, et al. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clin Cancer Res. 2017;23(12):3061–3071.
29. Kipps TJ. ROR1: an orphan becomes apparent. Blood. 2022;140(14):1583–1591.
30. Berger C, Sommermeyer D, Hudecek M, et al. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol Res. 2015;3(2):206–216.
31. Wallstabe L, Gottlich C, Nelke LC, et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight. 2019;4(18):8754.
32. Maher J, Davies DM. CAR-Based Immunotherapy of Solid Tumours-A Survey of the Emerging Targets. Cancers. 2023;15(4):1171.
33. Cao B, Liu M, Wang L, et al. Remodelling of tumour microenvironment by microwave ablation potentiates immunotherapy of AXL-specific CAR T cells against non-small cell lung cancer. Nat Commun. 2022;13(1):6203.
34. Zhong M, Chalbatani GM, Deng M, et al. Functional characterization and development of novel human kinase insert domain receptor chimeric antigen receptor T-cells for immunotherapy of non-small cell lung cancer. Eur J Pharm Sci. 2023;180:106331.
35. Li N, Liu S, Sun M, et al. Chimeric Antigen Receptor-Modified T Cells Redirected to EphA2 for the Immunotherapy of Non-Small Cell Lung Cancer. Transl Oncol. 2018;11(1):11–17.
36. Grunnet M, Sorensen JB. Carcinoembryonic antigen (CEA) as tumor marker in lung cancer. Lung Cancer. 2012;76(2):138–143.
37. Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017;21(11):3205–3219.
38. Kim YJ, Li W, Zhelev DV, Mellors JW, Dimitrov DS, Baek DS. Chimeric antigen receptor-T cells are effective against CEACAM5 expressing non-small cell lung cancer cells resistant to antibody-drug conjugates. Front Oncol. 2023;13:1124039.
39. Thistlethwaite FGD, Guest R, Rothwell D, et al. The clinical efficacy of first‑generation carcinoembryonic antigen (CEACAM5)‑specific CAR T cells is limited by poor persistence and transient pre‑conditioning‑dependent respiratory toxicity. Cancer Immunol Immunother. 2017;66:1425–1436.
40. Li K, Pan X, Bi Y, et al. Adoptive immunotherapy using T lymphocytes redirected to glypican-3 for the treatment of lung squamous cell carcinoma. Oncotarget. 2016;7(3):2496–2507.
41. Hickman TL, Choi E, Whiteman KR, et al. BOXR1030, an anti-GPC3 CAR with exogenous GOT2 expression, shows enhanced T cell metabolism and improved anti-cell line derived tumor xenograft activity. PLoS One. 2022;17(5):e0266980.
42. Kawaguchi T, Sho M, Tojo T, et al. Clinical significance of prostate stem cell antigen expression in non-small cell lung cancer. Jpn J Clin Oncol. 2010;40(4):319–326.
43. Wei X, Lai Y, Li J, et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology. 2017;6(3):e1284722.
44. Heider KH, Kuthan H, Stehle G, Munzert G. CD44v6: a target for antibody-based cancer therapy. Cancer Immunol Immunother. 2004;53(7):567–579.
45. Porcellini S, Asperti C, Corna S, et al. CAR T Cells Redirected to CD44v6 Control Tumor Growth in Lung and Ovary Adenocarcinoma Bearing Mice. Front Immunol. 2020;11:99.
46. Casucci M, Nicolis Di Robilant B, Falcone L, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122(20):3461–3472.
47. Tijink BM, Buter J, de Bree R, et al. A Phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin Cancer Res. 2006;12(20 Pt 1):6064–6072.
48. Crossland DL, Denning WL, Ang S, et al. Antitumor activity of CD56-chimeric antigen receptor T cells in neuroblastoma and SCLC models. Oncogene. 2018;37(27):3686–3697.
49. Jiang C, Zhao W, Qin M, Jin M, Chang L, Ma X. CD56-chimeric antigen receptor T-cell therapy for refractory/recurrent rhabdomyosarcoma: a 3.5-year follow-up case report. Medicine. 2019;98(43):e17572.
50. Xu T, Karschnia P, Cadilha BL, et al. In vivo dynamics and anti-tumor effects of EpCAM-directed CAR T-cells against brain metastases from lung cancer. Oncoimmunology. 2023;12(1):2163781.
51. Qin D, Li D, Zhang B, et al. Potential lung attack and lethality generated by EpCAM-specific CAR-T cells in immunocompetent mouse models. Oncoimmunology. 2020;9(1):1806009.
52. Sanchez-Paulete AR, Mateus-Tique J, Mollaoglu G, et al. Targeting Macrophages with CAR T Cells Delays Solid Tumor Progression and Enhances Antitumor Immunity. Cancer Immunol Res. 2022;10(11):1354–1369.
53. Liu J, Yang S, Cao B, et al. Targeting B7-H3 via chimeric antigen receptor T cells and bispecific killer cell engagers augments antitumor response of cytotoxic lymphocytes. J Hematol Oncol. 2021;14(1):21.
54. Qin L, Zhao R, Chen D, et al. Chimeric antigen receptor T cells targeting PD-L1 suppress tumor growth. Biomark Res. 2020;8:19.
55. Yim J, Koh J, Kim S, et al. Effects of B7-H3 expression on tumour-infiltrating immune cells and clinicopathological characteristics in non-small-cell lung cancer. Eur J Cancer. 2020;133:74–85.
56. Li H, Harrison EB, Li H, et al. Targeting brain lesions of non-small cell lung cancer by enhancing CCL2-mediated CAR-T cell migration. Nat Commun. 2022;13(1):2154.
57. Sharma SK, Pourat J, Abdel-Atti D, et al. Noninvasive Interrogation of DLL3 Expression in Metastatic Small Cell Lung Cancer. Cancer Res. 2017;77(14):3931–3941.
58. Chen X, Amar N, Zhu Y, et al. Combined DLL3-targeted bispecific antibody with PD-1 inhibition is efficient to suppress small cell lung cancer growth. J Immunother Cancer. 2020;8(1):46.
59. Jaspers JE, Khan JF, Godfrey WD, et al. IL-18-secreting CAR T cells targeting DLL3 are highly effective in small cell lung cancer models. J Clin Invest. 2023:64.
60. Blackhall F, Jao K, Greillier L, et al. Efficacy and Safety of Rovalpituzumab Tesirine Compared With Topotecan as Second-Line Therapy in DLL3-High SCLC: results From the Phase 3 TAHOE Study. J Thorac Oncol. 2021;16(9):1547–1558.
61. La HT, Tran DBT, Tran HM, Nguyen LT. Third-Generation Anti-CD47-Specific CAR-T Cells Effectively Kill Cancer Cells and Reduce the Genes Expression in Lung Cancer Cell Metastasis. J Immunol Res. 2021;2021:5575260.
62. Del Bufalo F, De Angelis B, Caruana I, et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N Engl J Med. 2023;388(14):1284–1295.
63. Reppel L, Tsahouridis O, Akulian J, et al. Targeting disialoganglioside GD2 with chimeric antigen receptor-redirected T cells in lung cancer. J Immunother Cancer. 2022;10(1):54.
64. Sasaki T, Sakoda Y, Adachi K, Tokunaga Y, Tamada K. Therapeutic effects of anti-GM2 CAR-T cells expressing IL-7 and CCL19 for GM2-positive solid cancer in xenograft model. Cancer Med. 2023:23.
65. Li X, Zhu T, Wang R, et al. Genetically Programmable Vesicles for Enhancing CAR-T Therapy against Solid Tumors. Adv Mater. 2023;e2211138.
66. Ding J, Guyette S, Schrand B, et al. Mesothelin-targeting T cells bearing a novel T cell receptor fusion construct (TRuC) exhibit potent antitumor efficacy against solid tumors. Oncoimmunology. 2023;12(1):2182058.
67. Zhao R, Cheng L, Jiang Z, et al. DNAX-activating protein 10 co-stimulation enhances the anti-tumor efficacy of chimeric antigen receptor T cells. Oncoimmunology. 2019;8(1):e1509173.
68. Iwao K, Watanabe T, Fujiwara Y, et al. Isolation of a novel human lung-specific gene, LUNX, a potential molecular marker for detection of micrometastasis in non-small-cell lung cancer. Int J Cancer. 2001;91(4):433–437.
69. Hu Z, Zheng X, Jiao D, et al. LunX-CAR T Cells as a Targeted Therapy for Non-Small Cell Lung Cancer. Mol Ther Oncolytics. 2020;17:361–370.
70. Jie Y, Liu G, Feng L, et al. PTK7-Targeting CAR T-Cells for the Treatment of Lung Cancer and Other Malignancies. Front Immunol. 2021;12:665970.
71. Martin AL, Anadon CM, Biswas S, et al. Olfactory Receptor OR2H1 Is an Effective Target for CAR T Cells in Human Epithelial Tumors. Mol Cancer Ther. 2022;21(7):1184–1194.
72. Nilsson MB, Yang Y, Heeke S, et al. CD70 is a therapeutic target upregulated in EMT-associated EGFR tyrosine kinase inhibitor resistance. Cancer Cell. 2023;41(2):340–355 e346.
73. Pal S, Tran B, Haanen J, et al. 558 CTX130 allogeneic CRISPR-Cas9–engineered chimeric antigen receptor (CAR) T cells in patients with advanced clear cell renal cell carcinoma: results from the phase 1 COBALT-RCC study. J ImmunoTherapy Cancer. 2022;10(Suppl 2):A584–A584.
74. Mao Y, Fan W, Hu H, et al. MAGE-A1 in lung adenocarcinoma as a promising target of chimeric antigen receptor T cells. J Hematol Oncol. 2019;12(1):106.
75. Kakarla S, Chow KK, Mata M, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21(8):1611–1620.
76. Yang M, Tang X, Zhang Z, et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics. 2020;10(17):7622–7634.
77. Chu W, Zhou Y, Tang Q, et al. Bi-specific ligand-controlled chimeric antigen receptor T-cell therapy for non-small cell lung cancer. Biosci Trends. 2018;12(3):298–308.
78. Wilkie S, van Schalkwyk MC, Hobbs S, et al. Dual Targeting of ErbB2 and MUC1 in Breast Cancer Using Chimeric Antigen Receptors Engineered to Provide Complementary Signaling. J Clin Immunol. 2012;32(5):1059–1070.
79. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172.
80. Tokatlian T, Asuelime GE, Mock JY, et al. Mesothelin-specific CAR-T cell therapy that incorporates an HLA-gated safety mechanism selectively kills tumor cells. J Immunother Cancer. 2022;10(1).
81. Muliaditan T, Halim L, Whilding LM, et al. Synergistic T cell signaling by 41BB and CD28 is optimally achieved by membrane proximal positioning within parallel chimeric antigen receptors. Cell Rep Med. 2021;2(12):100457.
82. Guedan S, Posey AD, Shaw C, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. 2018;3(1):546.
83. Sonzogni O, Zak DE, Sasso MS, et al. T-SIGn tumor reengineering therapy and CAR T cells synergize in combination therapy to clear human lung tumor xenografts and lung metastases in NSG mice. Oncoimmunology. 2022;11(1):2029070.
84. Greco B, Malacarne V, De Girardi F, et al. Disrupting N-glycan expression on tumor cells boosts chimeric antigen receptor T cell efficacy against solid malignancies. Sci Transl Med. 2022;14(628):eabg3072.
85. Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2(2):112–120.
86. Castelletti L, Yeo D, van Zandwijk N, Rasko JEJ. Anti-Mesothelin CAR T cell therapy for malignant mesothelioma. Biomark Res. 2021;9(1):11.
87. Adusumilli PS, Zauderer MG, Rusch VW, et al. Regional delivery of mesothelin-targeted CAR T cells for pleural cancers: safety and preliminary efficacy in combination with anti-PD-1 agent. J Clin Oncol. 2019:53.
88. Zhang C, Wang Z, Yang Z, et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA(+) Metastatic Colorectal Cancers. Mol Ther. 2017;25(5):1248–1258.
89. Feng K, Guo Y, Dai H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci. 2016;59(5):468–479.
90. Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy when Combined with Checkpoint Blockade. Cancer Cell. 2021;39(2):193–208 e110.
91. Zhang Y, Zhang Z, Ding Y, et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J Cancer Res Clin Oncol. 2021;147(12):3725–3734.
92. Liu H, Ma Y, Yang C, et al. Severe delayed pulmonary toxicity following PD-L1-specific CAR-T cell therapy for non-small cell lung cancer. Clin Transl Immunol. 2020;9(10):e1154.
93. Feng K, Liu Y, Guo Y, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018;9(10):838–847.
94. Byers L, Heymach J, Gibbons D, et al. 697 A phase 1 study of AMG 119, a DLL3-targeting, chimeric antigen receptor (CAR) T cell therapy, in relapsed/refractory small cell lung cancer (SCLC). J ImmunoTherapy Cancer. 2022;10(Suppl 2):A728–A728.
95. Lin Y, Chen S, Zhong S, An H, Yin H, McGowan E. Phase 1 clinical trial of PD1 knockout anti-MUC1 CAR-T cells in the treatment of patients with non-small cell lung cancer. Ann Oncol. 2019;30:xi12–xi15.
96. Haas AR, Tanyi JL, O’Hara MH, et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified t cells recognizing mesothelin in advanced solid cancers. Mol Ther. 2019;27(11):1919–1929.
97. Gutierrez R, Shah PD, Hamid O, et al. Phase I experience with first in class TnMUC1 targeted chimeric antigen receptor T-cells in patients with advanced TnMUC1 positive solid tumors. J Clin Oncol. 2021;39(15_suppl):e14513–e14513.
98. Qu J, Mei Q, Chen L, Zhou J. Chimeric antigen receptor (CAR)-T-cell therapy in non-small-cell lung cancer (NSCLC): current status and future perspectives. Cancer Immunol Immunother. 2021;70(3):619–631.
99. Adusumilli PS, Amador AM, Chintala N, et al. Promoting functional persistence in solid tumor CAR T-cell therapy: mesothelin-targeted CAR (M28z1XXPD1DNR) with T-cell intrinsic PD1 dominant negative receptor. Ann Oncol. 2021;32:S1392–S1393.
100. Henry J, Oh D, Eskew J, et al. 728 Phase 1 study of P-MUC1C-ALLO1 allogeneic CAR-T cells in patients with epithelial-derived cancers. J ImmunoTherapy Cancer. 2022;10(Suppl 2):A761–A761.
101. Spigel DR, Murthy H, Chumsri S, et al. Phase I study of LYL797, a ROR1-targeted CAR T-cell therapy with genetic and epigenetic reprogramming for the treatment of advanced solid tumors. Ann Oncol. 2022;33.
102. DiAndreth B, Hamburger AE, Xu H, Kamb A. The Tmod cellular logic gate as a solution for tumor-selective immunotherapy. Clin Immunol. 2022;241:109030.
103. Specht JM, Lee S, Turtle CJ, et al. Abstract CT131: a phase I study of adoptive immunotherapy for advanced ROR1+ malignancies with defined subsets of autologous T cells expressing a ROR1-specific chimeric antigen receptor (ROR1-CAR). Cancer Res. 2018;78:CT131.
104. Adusumilli PS, Zauderer MG, Riviere I, et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021;11(11):2748–2763.
105. Gutierrez R, Shah PD, Hamid O, et al. Phase I experience with first in class TnMUC1 targeted chimeric antigen receptor T-cells in patients with advanced TnMUC1 positive solid tumors. J Clin Oncol. 2021;39(15S):e14513.
106. Hayes TK, Meyerson M. Molecular portraits of lung cancer evolution. Nature. 2023.
107. Antonia SJ, Borghaei H, Ramalingam SS, et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: a pooled analysis. Lancet Oncol. 2019;20(10):1395–1408.
108. Yan X, Han L, Zhao R, Fatima S, Zhao L, Gao F. Prognosis value of IL-6, IL-8, and IL-1beta in serum of patients with lung cancer: a fresh look at interleukins as a biomarker. Heliyon. 2022;8(8):e09953.
109. Caruana I, Savoldo B, Hoyos V, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21(5):524–529.
110. Obajdin J, Davies DM, Maher J. Engineering of chimeric natural killer cell receptors to develop precision adoptive immunotherapies for cancer. Clin Exp Immunol. 2020;202(1):11–27.
111. Okita R, Maeda A, Shimizu K, Nojima Y, Saisho S, Nakata M. Clinicopathological relevance of tumor expression of NK group 2 member D ligands in resected non-small cell lung cancer. Oncotarget. 2019;10(63):6805–6815.
112. Davies DM, Foster J, Van Der Stegen SJ, et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol Med. 2012;18:565–576.
113. Del Re M, Cucchiara F, Petrini I, et al. erbB in NSCLC as a molecular target: current evidences and future directions. ESMO Open. 2020;5(4):54.
114. Kosti P, Opzoomer JW, Larios-Martinez KI, et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep Med. 2021;2(4):100227.
115. Glover M, Avraamides S, Maher J. How Can We Engineer CAR T Cells to Overcome Resistance? Biologics. 2021;15:175–198.
116. Hull CM, Maher J. Approaches for refining and furthering the development of CAR-based T cell therapies for solid malignancies. Expert Opin Drug Discov. 2021;16(10):1105–1117.
117. Shen L, Xiao Y, Tian J, Lu Z. Remodeling metabolic fitness: strategies for improving the efficacy of chimeric antigen receptor T cell therapy. Cancer Lett. 2022;529:139–152.
118. Zhu L, Liu J, Zhou G, et al. Remodeling of Tumor Microenvironment by Tumor-Targeting Nanozymes Enhances Immune Activation of CAR T Cells for Combination Therapy. Small. 2021;17(43):e2102624.
119. Ou Z, Dou X, Tang N, Liu G. Pressure increases PD-L1 expression in A549 lung adenocarcinoma cells and causes resistance to anti-ROR1 CAR T cell-mediated cytotoxicity. Sci Rep. 2022;12(1):6919.
120. Li S, Siriwon N, Zhang X, et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin Cancer Res. 2017;23(22):6982–6992.
121. Zhang L, Morgan RA, Beane JD, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. 2015;21(10):2278–2288.
122. Achkova DY, Beatson RE, Maher J. CAR T-Cell Targeting of Macrophage Colony-Stimulating Factor Receptor. Cells. 2022;11(14):2190.
123. Conde E, Vercher E, Soria-Castellano M, et al. Epitope spreading driven by the joint action of CART cells and pharmacological STING stimulation counteracts tumor escape via antigen-loss variants. J Immunother Cancer. 2021;9(11):687.
124. Lai J, Mardiana S, House IG, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol. 2020;21(8):914–926.
125. Tatsumi T, Gambotto A, Robbins PD, Storkus WJ. Interleukin 18 gene transfer expands the repertoire of antitumor Th1-type immunity elicited by dendritic cell-based vaccines in association with enhanced therapeutic efficacy. Cancer Res. 2002;62(20):5853–5858.
126. Bhatia V, Kamat NV, Pariva TE, et al. Targeting advanced prostate cancer with STEAP1 chimeric antigen receptor T cell and tumor-localized IL-12 immunotherapy. Nat Commun. 2023;14(1):2041.
127. Jin C, Ma J, Ramachandran M, Yu D, Essand M. CAR T cells expressing a bacterial virulence factor trigger potent bystander antitumour responses in solid cancers. Nat Biomed Eng. 2022;6(7):830–841.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Immune-Checkpoint Inhibitors in Lung Neuroendocrine Tumors – A Systematic Review and Meta-Analysis
Pichel RC, Benini L, Romelli M, Gandini S, Gervaso L, Valente M, De Sousa MJ, Araújo A, Araújo A, Di Giacomo AM, Fazio N
OncoTargets and Therapy 2025, 18:833-843
Published Date: 31 July 2025