Back to Journals » Cancer Management and Research » Volume 11
Chimeric antigen receptor (CAR) T-cell therapy as a treatment option for patients with B-cell lymphomas: perspectives on the therapeutic potential of Axicabtagene ciloleucel
Authors Viardot A, Wais V, Sala E, Koerper S
Received 18 December 2018
Accepted for publication 28 February 2019
Published 25 March 2019 Volume 2019:11 Pages 2393—2404
DOI https://doi.org/10.2147/CMAR.S163225
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Andreas Viardot,1 Verena Wais,1 Elisa Sala,1 Sixten Koerper2,3
1Department of Internal Medicine III, University Hospital Ulm, Ulm, Germany; 2Institute of Clinical Transfusion Medicine and Immunogenetics Ulm, German Red Cross Blood Transfusion Service Baden-Württemberg-Hessen and University Hospital Ulm, Ulm, Germany; 3Institute of Transfusion Medicine, University of Ulm, Ulm, Germany
Abstract: Axicabtagene lisoleucel (Axi-cel) is the second approved gene-alterating cancer treatment and the first in aggressive lymphoma using the “chimeric antigen receptor” (CAR) technology. T-cells from patients were transfected with CARs and reinfused after a lymphodepleting chemotherapy. CAR T-cells are “living drugs” with the ability to persist and expand after a single infusion. Axi-cel is a “second generation” CAR product characterized by the use of a retroviral gene vector transfer and by CD28 as costimulatory domain. In a phase II trial with heavily pretreated patients with aggressive B-cell lymphoma, the overall response rate was 82% with an ongoing complete response rate of 40% after 6 months – with expectations of long-term remissions and cure, even though follow-up data are still limited. There are some prominent side effects like cytokine release syndrome (Grade 3–5: 13%) and neurotoxicity (Grade 3–5: 28%). Novel strategies for prediction, prevention and treatment of these critical side effects are warranted. There are new concepts to enhance the efficacy and prevent resistance in lymphomas. CAR T-cells represent an extremely evolving field with an inestimable potential in general and particularly in aggressive lymphoma. However, we are still learning how to use Axi-cel and other CAR-T cells compounds effectively to optimize the long-term results.
Keywords: chimeric antigen receptor, CAR T-cells, Axicabtagene ciloleucel, diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma, transformed follicular lymphoma, cytokine release syndrome, neurotoxicity, CAR related encephalopathy syndrome
Background
Axicabtagene ciloleucel (Axi-cel; KTE-C19) was approved by the United States (US) Food and Drug Administration (FDA) in October 2017 and by the European Medical Agency (EMA) in August 2018 for the treatment of refractory and relapsed (r/r) aggressive B-cell lymphoma (diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma and transformed follicular lymphoma) after two prior therapies. It represents the first-in-class approval in lymphoma. Two months before, in September 2017, Tisagenlecleucel was approved by FDA for the treatment of r/r B precursor acute lymphoblastic leukemia (ALL) in children and young adults. Meanwhile, both compounds are approved by FDA and EMA for aggressive lymphoma, and a third compound – Lisocabtagene maraleucel (Liso-cel, JCAR017) – has received the FDA breakthrough designation and the EMA PRIME status for fast-track approval for aggressive lymphoma. Cancer treatment with chimeric antigen receptor transfected T-cells will shift the paradigms of immunotherapy and influence the therapeutic field in lymphoma and other neoplasia. However, the complexity of dealing with genetically modified and “living” drugs produced from autologous lymphocytes challenges manufacturers, logisticians, regulatory authorities, health insurances, and medical teams. This review will focus on the experience of Axi-cel as a paradigm of a new generation of immunotherapeutics and will summarize the first experiences after the US marketing authorization.
Treatment options in aggressive lymphomas
Diffuse large B-cell lymphoma (DLBCL) represents the most frequent lymphoma entity in the world.1 Regardless of intensive efforts to establish new treatment standards, the 40-year-old combination of cyclophosphamide, doxorubicin, vincristine and prednisolone (CHOP), including the monoclonal anti-CD20 antibody rituximab (R-CHOP), is still the basis of the first-line treatment. Using R-CHOP and R-CHOP-like regimens, two thirds of the patients will achieve long-term remission, unless they have clinical or molecular high risk features.2,3 On the other side, the prognosis of r/r patients is still poor. If intensive treatment is not possible due to patient´s age and comorbidity, the disease is not curable in most of the cases.4 For patients without comorbidity, high-dose chemotherapy followed by autologous stem cell transplantation (ASCT) is an accepted standard of care. However, the rate of long-term remissions is about 20% in more recently published clinical trials.5–7 Relapses after ASCT have a median overall survival of 6.1 months.8 In the case of refractory disease, the overall survival is usually not longer than 6–7 months.8 Considering, that the majority of DLBCL patients is older than 65 years and unfit for intensive treatment, experts estimate that not more than 10% of relapsed or refractory patients can be cured.9
Allogeneic stem cell transplant (Allo-SCT) represents an alternative approach in some countries for younger and highly selected patients in order to achieve disease control by immunotherapy. In prospective and registry trials,10,11 up to 40% of patients are shown to have long-term remission. Allo-SCT has still some open issues, like the availability of a suitable matched donor and the morbidity and mortality rates associated with complications, like uncontrolled infections or graft-versus-host disease. For these reasons, patients should by highly selected with regard to age and comorbidity. Furthermore, considering that the graft-versus-lymphoma effect needs up to 3 months after transplant in order to exploit its function, this option is suitable only in the case of chemo- or radiotherapy-controlled disease at the time of transplant.
Novel drugs with the potential to improve the prognosis are still limited. The antibody-drug conjugate Polatuzumab vedotin achieved a breakthrough designation of the FDA and the Prime designation of the EMA in 2017 based on a randomized phase-II trial with significant prolongation of progression free survival (PFS) and overall survival (OS) in the combination with rituximab and bendamustin compared with them alone.12 The combination of the anti-CD19 antibody MOR208 with lenalidomide shows a promising prolongation of the PFS in a still recruiting phase II trial13 and received a FDA breakthrough designation in 2017. Most other compounds tested in phase-I or II trials show a response rate of approximately 30% and a PFS of only 6 months.4 The unmet medical need is obviously high.
Axi-cel is also approved for r/r primary mediastinal B-cell lymphoma (PMBCL) and transformed follicular lymphoma (tFL). PMBCL is an aggressive Non-Hodgkin lymphoma with some Hodgkin-lymphoma-like features (young patients, mediastinal involvement). The prognosis was poor until the introduction of rituximab. Nowadays, long-term remission are achieved with R-CHOP-like regimens in the vast majority of patients.14 However, r/r patients still have a poor outcome. In a phase-II trial, the checkpoint inhibitor pembrolizumab showed an overall response rate of 47% leading to a FDA approval in 2018.15
De novo tFL may have an acceptable prognosis – without pretreatment of the follicular lymphoma part. In the r/r setting, the outcome is comparable to r/r DLBCL.16
With the approval of two commercial CAR T-cell products (Axi-cel, Tisagenlecleucel) and the FDA breakthrough designation of a third construct (Liso-cel), there is a real chance for game-changing improvements in the field of aggressive B-cell lymphoma.
History of CAR T-cell therapy and Axi-cel
The first description of a chimeric antigen receptor composed of the T-cell receptor domains fused to the antibody’s variable domains comes from Zelig Esshar from Weizmann Institute in Rehovot, Israel, in 1989.17 This simple construct represents the “first generation” of CAR.18 The first clinical trials in humans with solid tumors using these “first generation” CARs were published in 2006 and showed disappointing results without significant tumor regression19,20 The main reason was the lack of costimulatory domains that could elicit the T-cell activation. However, the first clinical report of a patient with metastatic colon carcinoma using a “third generation CAR” (anti-ERBB2-CD28-4-1BB-CD3ζ) ended in a fatal cytokine storm.21 The first success of a second generation CAR was reported on an anti-CD19 CAR (4.1BB; CD3ζ) constructed by the University of Pennsylvania in three patients with chronic lymphocytic leukemia (CLL)22 followed by publications in pediatric ALL.23 At the National Cancer Institute (NCI), several first and second generation CARs were developed.24,25 An anti-CD19 CAR (CD28; CD3ζ) compound went into further clinical evaluation for lymphoma patients. The first proof of principle of a similar construct was published in 2012 in patients with indolent lymphoma.26 In the same year, the NCI entered into cooperation with Kite Pharma for development and commercialization of cancer therapeutics including a CAR product named KTE-C19 or Axi-cel. Based on the data of a large phase II-trial (ZUMA-1),27 Axi-cel was approved for the treatment of patients with two relapses with DLBCL, PMBCL and tFL in October 2017 (FDA) and August 2018 (EMA). As assessed in December 2018, six clinical trials using Axi-cel are registered in clinicaltrials.gov.28
Review of pharmacology, mode of action, preparation and pharmacokinetics of Axicabtagene ciloleucel
Axi-cel is generated from autologous lymphocytes expressing a so-called second generation CAR consisting of a peptide sequence from the variable region of a monoclonal antibody against CD19 (FCM63, the same as in Tisagenlecleucel and Liso-cel), the ζ chain of the T-cell receptor and CD28 as a costimulatory molecule.29 CD19 is a transmembrane glycoprotein which is highly expressed in all the stages of differentiation of normal and pathological B-cells. CD19 directed therapies were effective in both ALL and most B-cell Non-Hodgkin lymphomas. The T-cell stimulation by the ζ chain of the T-cell receptor is weak as demonstrated by in vivo and in vitro trials of the first generation CARs.24 A costimulatory domain is necessary in order to obtain an enhanced activation of T-cells. CD28-based CAR T-cell constructs are thought to have a greater peak expansion whereas 4-1BB-based constructs show a longer persistence.29 On the other hand, 4-1BB-based CAR T-cells can induce early exhaustion limiting the efficacy.30 However, no direct in vivo comparisons of different CAR compounds were performed in order to prove these preclinical observations.
The manufacturing process begins with the collection of the starting material by leukapheresis. The target cell count in the ZUMA-1 trial was 5–10×109 mononuclear cells.31 T-cells were selected through magnetic beads and activated eg, by anti-CD3 antibodies and Interleukin-2, subsequently transfected with CAR genes by a gamma retroviral viral vector, followed by expansion and preparation of the final product.32 For an extended clinical use, the production process was optimized, eg, omitting the preparation with magnetic beads and a shortening of the expansion period.33
Viral vectors guide RNA to reverse-transcribe into DNA and permanently integrate into the genome of the autologous T-cells. Subsequently, the viral vector will be washed out. Lentiviral vectors, which are used in other commercially available products (eg, Tisagenlecleucel), had the theoretical advantage of a safer integration site profile.34 However, there is so far no report about secondary malignancies or other genotoxic events in the follow-up of treated patients.
Axi-cel is administered as a single-dose infusion containing a suspension of 2×106 CAR T-cells per kilogram body weight up to a maximum of 2×108 cells.35 The rapid expansion of these cells has a peak within 7–14 days and is accompanied with peak levels of interleukins secreted from activated immune cells.27 In the ZUMA-1 trial, CAR T-cells were still detectable in most patients at 180 days after infusion.28 The extent of expansion, eg, measured by the area under the curve within 28 days was associated with a response but also with acute side effects.27 The prognostic relevance of CAR T-cell persistence remains unclear: in the first clinical trials from NCI with lymphoma36 and ALL,37 the loss of CAR T-cell persistence or recovery of normal B-cells was associated with a poor prognosis. This is in contrast to the long-term follow-up data of the ZUMA-1 trial, where B-cell recovery was observed in over half the patients with ongoing remission at 12 months.38
Efficacy studies, including any comparative studies and relevant case reports
The preclinical and early clinical development of Axi-cel started at the NCI, including the first clinical experience in humans using an anti-CD19-CD28-CD3ζ CAR. Axi-cel was then further developed by Kite Pharma (now owned by Gilead) towards the approval of the FDA and EMA.
In 2012, the first clinical trial in eight patients with indolent B-cell malignancies (four chronic lymphocytic leukemia (CLL), three follicular lymphoma (FL) and one splenic marginal zone lymphoma (MZL)) was published25 using the NCI anti-CD19 CAR (CD28; CD3ζ) compound. In this trial, a single agent lymphodepleting course with fludarabine was used for conditioning and administration of interleukin-2 after reinfusion was mandatory. Six of eight patients achieved objective remissions (three CLL, two FL, one MZL) and four out of eight patients had long-term depletion of normal polyclonal CD19 B-cells. Four out of eight patients experienced significant toxicity in terms of cytokine-mediated side effects which resolved completely except for one patient who died from influenza.
In a subsequent clinical trial, 15 patients with B-cell malignancies (nine DLBCL, two indolent lymphoma, and four CLL) were treated with fludarabine/cyclophosphamide as a preparation regimen, without the addition of interleukin-2.38 Out of 15 patients, eight achieved a complete remission (CR) including four out of seven patients with refractory DLBCL. Thirteen out of 15 patients experienced grade 3 or 4 toxicities including hypotension (n=4) and neurological side effects (n=6).
Given the promising results particularly in DLBCL, the following trial focused on aggressive lymphoma. Twenty-two patients (19 with DLBCL, two with FL and one with mantle cell lymphoma) were treated with anti CD19 CAR T-cells.39 There was a 73% remission rate with 55% of patients achieving a CR. Eleven out of 12 CR patients presented an ongoing response with a median duration of 12.5 months without any further treatment. The most predictive factors for achieving a CR were a higher peak of blood CAR T-cells and an elevated serum level of IL-15.40 All patients experienced grade 3 or 4 toxicities which are in line with the subsequently recognized CAR-related cytokine release syndrome (CRS). Furthermore, 12 patients experienced grade 3 or 4 neurological events. Grade 3 and 4 neurologic toxicities were associated with increased blood CAR T-cell counts. CAR T-cells were also detected in the cerebrospinal fluid.
The ZUMA-1 trial27 – the first trial with Axi-cel or KTE-019 in cooperation with Kite pharma – consisted of a phase I and phase II part. In the phase I,41 seven patients were treated resulting in an overall response rate of five out of seven and CR rate of four out of seven (three patients with ongoing CR over 12 months). In the subsequent multicenter phase II trial (ZUMA-1),27 111 patients with DLBCL, PMBCL and tFL refractory to last treatment or relapsing after autologous stem cell transplantation were included. Since the majority of sites were located in the US (21 sites, one site in Israel), the time from leukapheresis to transfusion was short (17 days). Only one patient was excluded due to unsuccessful manufacturing, seven patients were excluded due to adverse events and/or tumor progression and two patients due to non-measurable disease. The rate of effectively treated patients (101/111: 91%) is higher than in the JULIET trial with Tisagenlecleucel42 or the TRANSCEND trial with Lisocaptagene maraleucel.43 In contrast to these both trials, the time between inclusion and treatment and particularly between leukapheresis and reinfusion (“vein to vein” time) was exceptionally short (median: 17 days). Bridging chemotherapy was not allowed. During the commercial phase, the “vein to vein” time will be probably prolonged, especially for countries outside the US, with the need of cryopreserving the cell products at least for a transitional period.
Among the 101 patients receiving Axi-cel, the response rate was 82% with a 54% CR.27 The median duration of response was 8.1 months. At an updated analysis with a median follow-up of 15.4 months, 40% of patients remained in CR. The overall survival at 18 months was 52%.
Grade 3–5 cytokine release syndrome (CRS) occurred in 13% of patients and grade 3–5 neurologic events in 28%.27 Three patients died during treatment, one patient from hemophagocytic lymphohistiocytosis and another patient from cardiac arrest in association with a CRS, while a third patient died from unrelated pulmonary embolism.
One year after the approval of Axi-cel, “real world data” were presented at the annual meeting of the American Society of Hematology (ASH) 2018 for the first time (Table 1).44–47 One of the most remarkable observations was that a significant proportion (up to 50%) of patients did not fulfill the original inclusion criteria of the ZUMA-1 trial (eg, due to reduced performance status, low platelets, low glomerular filtration rate, history of central nervous system (CNS) lymphoma, previous Allo-SCT etc.)44 this fact obviously does not affect the outcome in the largest series.44 The time from leukapheresis to reinfusion was longer (21–27 days),44,47 and bridging chemotherapy was often necessary. Furthermore, histological diagnoses other than DLCBL were included more frequently as compared with the ZUMA-1. Nevertheless, the best response rate was comparable or slightly decreased compared to the ZUMA-1 t trial. The toxicity profile is similar to the ZUMA-1 trial. The reported incidence of severe CRS appears lower, which can be partially explained by a higher usage of tocilizumab. In the retrospective analysis of a single center experience, safety and efficacy of Axi-cel therapy were comparable in the two subsets of elderly and younger patients (≥ and <65 years).46
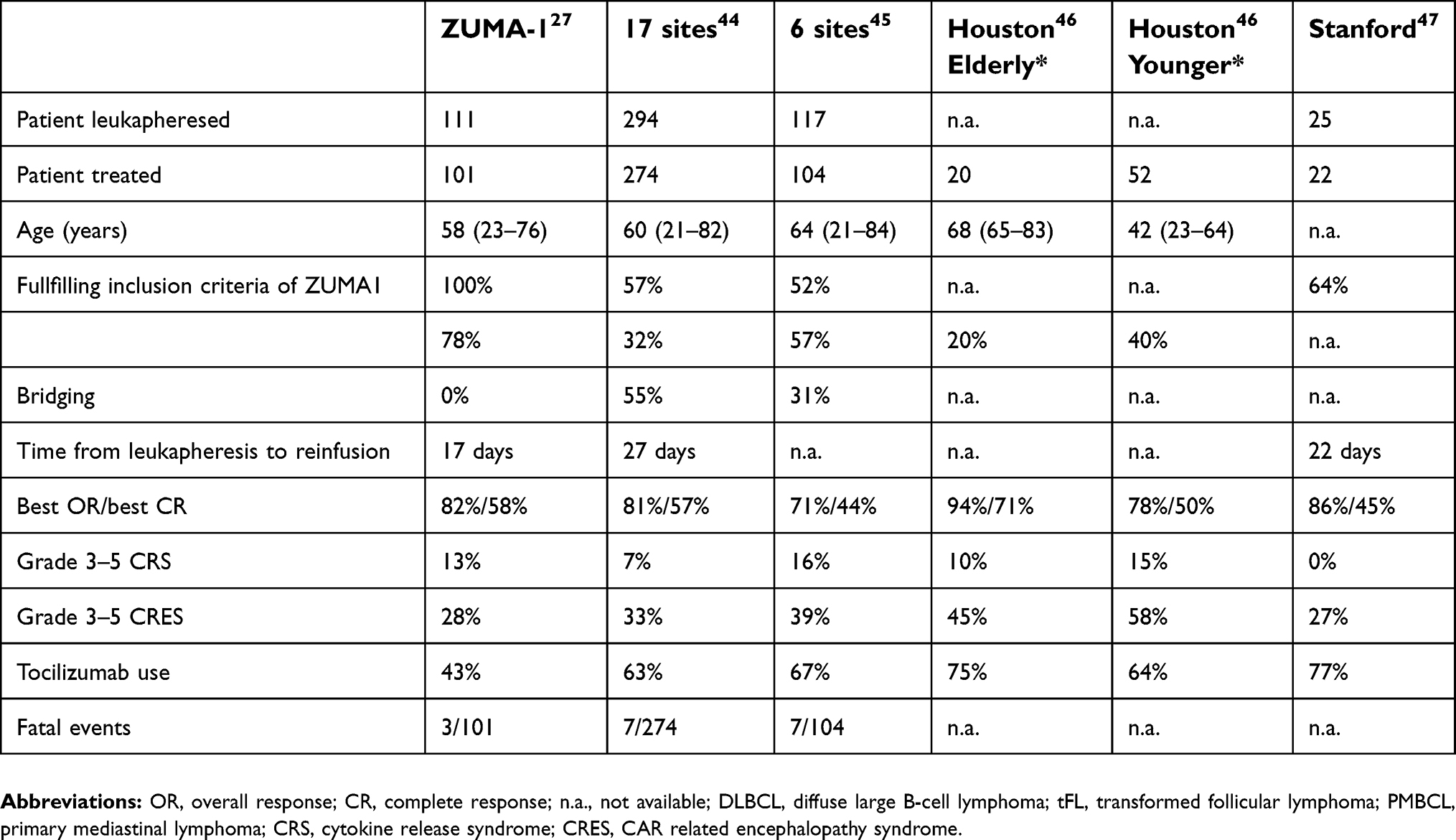 | Table 1 “Real world” data with Axi-cel from US sites, as presented at the ASH meeting 2018 |
Currently, potential markers for efficacy and response are under study. Recent works pointed out that a low day 0 C-reactive protein (CRP) together with a high absolute leucocyte count (ALC) during the leukapheresis represent potential predictors of response,45 as well as a low day 0 CRP, low levels of ferritin, IL-6 and TNF-α could be associated with a better outcome at day 30 after the reinfusion. High serum cytokine levels of IL-6 and angiopoietin 2/angiopoietin 1 ratio at day 1 were correlated with severe cytokine release syndrome.48
Safety and tolerability
The reinfusion of CAR T-cells is associated with several acute but usually self-limiting toxicities. The most prominent side effects are the CRS and the neurotoxicity, recently described as CAR-related encephalopathy syndrome (CRES).49 Ongoing cytopenia is frequently found in trials of ALL50 Late effects are rare; however, the B-cell depletion is ongoing and may lead to a clinically significant hypogammaglobinemia.50
CRS is caused by the activation of lymphocytes and myeloid cells with the subsequent release of inflammatory cytokines, eg, IL-2, IL6 and GM-CSF deriving from activated T-cells and IL-1RA, IL-10, IL-6, IFNα from monocytes and macrophages.51 The clinical symptoms are highly variable, starting from mild constitutional symptoms until life threatening multiorgan dysfunction including the hemophagocytic lymphohistiocytosis (HLH). After CAR T-cell reinfusion and during their expansion, an increase of serum level of several cytokines as IFN-γ, Interleukin-6 and TNF-α was documented and correlates with clinical symptoms. Since the increment of T-cells starts after reinfusion and the maximum peak of T-cell expansion is about day 8, CRS-associated symptoms are expected between day 2 and 14. In most cases, CRS is fully reversible within 2–3 weeks. In context of CAR T-cell treatment and other immunotherapies, a new grading system was developed and published by Lee.48 For the comparison of different CAR T-cell trials, it is worth mentioning that grading scores may differ, eg, in trials with Tisagenlecleucel.
In the ZUMA-1 trial,27 the incidence of higher grade neurologic events was significantly associated with the expansion of CAR T-cells. In an analysis of 44 serum biomarkers, elevated serum levels of Interleukin-2, GM-CSF and ferritin were associated particularly with the occurrence of neurotoxicity, but not with CRS.
Tocilizumab, an anti-IL-6 receptor antibody initially approved for treatment of several rheumatic diseases, was widely used in the treatment of CRS. The therapeutic effect was described for the first time in a series of pediatric ALL patients treated with Tisagenlecleucel.23 Usually, the symptoms of CRS were mitigated within a few hours. Tocilizumab is now approved by FDA and EMA for the treatment of CRS. After its administration, IL-6 levels typically rise transiently suggesting theoretically, that the neurotoxicity can be triggered or enhanced. However, clinical data provide no significant differences in the neurotoxicity in tocilizumab-treated patients. Siltuximab, an anti-IL-6 antibody approved for the treatment of Multicentric Castleman disease, has the theoretical advantage in rapid reduction of the IL-6 levels. Siltuximab is used as a reserve medication in case of tocilizumab failure. Therefore, the experience is still limited.
In the real world experience of Axi-cel,44–47 collected data show that there might be a decrease of the incidence of CRS due to the implementation of risk management systems and broad use of Interleukin-6 receptor antagonists. On the other side, the neurotoxicity – or CRES – remains an unmet problem. Clinical symptoms may vary from mild tremor, headache or impaired handwriting to disorientation, aphasia, somnolence, seizures and cerebral edema in the severe forms.49–51 The pathophysiology is not completely understood. The passive diffusion of peak level interleukins as well as the trafficking of CAR T-cells through the brain blood barrier may contribute to these symptoms. However, severe neurotoxicity seems to be a “class effect” of anti-CD19 redirected T-cell therapies, also observed in Blinatumomab and other CD19-directed bispecific antibodies.52 In other CAR and bispecific antibody trials with alternative targets, neurologic symptoms are often mild despite high interleukin peaks. Since CD19 antigen is usually not detectable in brain tissue, this aspect remains unexplained. Some investigators observed a biphasic course of CRS,50 with a first phase within 5 days after reinfusion, with is reversible by anti-IL-6 treatment and a second phase after 5 days, which is generally not responsive to anti-IL-6 treatment, but partially to high dose corticosteroids.
Surveillance and early diagnosis are essential in the management of CRES. For the grading, the three times daily neurological assessment method CARTOX is recommended,51 which includes a simple 10-point neurological assessment. A new experimental approach for the treatment of CRES is provided by mouse experiments, suggesting that IL-1 receptor antagonist anakinra may protect mice from CRS and CRES in contrast to tocilizumab.53 However, clinical experience is limited at the time of this publication.
CAR T-cells “late effects” is an emerging field analyzing the complications occurring and/or persisting beyond 90 days after the reinfusion. Among late effects one of the most frequent is prolonged cytopenia, in some cases requiring platelets or red blood cell transfusions and/or hematopoietic growth factors. The B-cell depletion occurring after CAR T-cells reinfusion could lead to a clinically significant hypogammaglobinemia, with a consequently increased risk of late infections.50 Secondary malignancies were also observed: therapy-related myelodysplastic syndromes, non-melanoma skin cancer, non-invasive bladder cancer. However, given the intensive pretreatment of the patients, the incidence of secondary malignancies is within the expectation. Late immune-related and neurologic events, like, respectively, granulomatous disease and transient ischemic attack, were also detected.54 Finally in a small series of patients receiving CART-cells after failure of Allo-SCT (time from transplant to CAR T-cells was 46.3 months), the onset of a late acute graft versus host disease was documented. Most of those late events were mild, suggesting the long-term safety of this therapeutic strategy.54
Patient’s perspective
In the ZUMA-1 trial, Axi-cel met the primary endpoint of improving the response rate in comparison to a prespecified response rate of 20% according to historical results in the setting of r/r DLBCL.27 Considering the adjusted comparison of ZUMA-1 and SCHOLAR-1, Axi-cel seems to prolong significantly the survival in patients with a mostly incurable status of disease.55 However, the fast track approval of FDA and EMA is based on a phase II trial including 101 patients with a median follow-up of only 15.1 months. There should be an awareness about the immaturity of these data and the lack of randomized comparisons. Whereas there is a detailed knowledge about management of acute side effects, data about long-term side effects and quality of life is limited. Persistent B-cell aplasia and immunoglobulin deficit is the only well-documented long-term side effect manageable with intravenous immunoglobulin replacement. There are potential long-term side effects at least theoretically predictable, eg, secondary malignancies by insertional oncogenesis of the viral vectors, development or exacerbation of neurologic disorders, development or exacerbation of autoimmune disorders.56 Furthermore, there is no systematic evaluation of quality of life in any of the commercially tested CAR T-cell products.
An example of a more critical view to the new technologies is the appraisal of the United Kingdom National Institute of Health and Care Excellence (NICE) which did not recommend the treatment with Axi-cel within the market authorization.57 This was justified by the fact that the “exact size of the benefit … is unknown” without a valid comparator, and that “the cost-effectiveness estimates are above the range…”. With a similar rationale, there is no recommendation for Tisagenlecleucel in DLBCL.
Comparison to other commercial CAR T-cell products
In 2018, another anti-CD19 CAR T-cell product, Tisagenlecleucel, was approved by FDA and EMA for treatment of DLBCL and tFL.42 Furthermore, Lisocabtagene maraleucel (Liso-cel or JCAR017) achieved the FDA breakthrough and EMA PRIME designation in 2017.43 Compared with Axi-cel, both compounds use a lentiviral vector and a 4-1BB instead of CD28 as costimulatory domain. In contrast to Tisagenlecleucel, Liso-cel is a product with precisely defined flat doses of transfected CD4 and CD8 cells (Table 2). For both products, data from phase-II trials with more than 100 patients are available (Tisagenlecleucel: JULIET, Liso-cel: TRANSCEND). The time from inclusion to treatment and from leukapheresis to reinfusion was variable and longer than in the ZUMA-1 trial. Bridging therapy was allowed and used in the majority of patients. A larger proportion of patients did not receive the reinfusion due to medical reasons such as disease progression or technical reasons like nonconforming products (JULIET42 54 out of 165 patients; TRANSCEND43 32 out of 134). As a result of different strategies in the recruitment phase and considering the various clinical background, it is difficult to compare the three different trials, especially with regard to efficacy data. The best overall response rate in JULIET was 52% (CR 40%), and in TRANSCEND 75% (CR 55%). After 6 months, 30% or 34% of patients remain in CR, respectively. The toxicity profile appeared to be similar between the three compounds, although, as previously mentioned, a direct comparison is nearly impossible, not only due to imbalances in patient characteristics, but also considering the different scoring systems and management strategies (eg, Tocilizumab use) adopted for the occurrence of CRS and CRES. Grade 3 and 4 CRES might be higher in Axi-cel (28%, JULIET: 12%; TRANSCEND: 12%), and grade 3 and 4 CRS lower in Liso-cel (1%, JULIET: 23%, ZUMA-1: 13%). Differences between the commercial products will crystallize more in future, while up-to-date technical and logistic matters might influence the choice of the compound more than medical reasons alone.
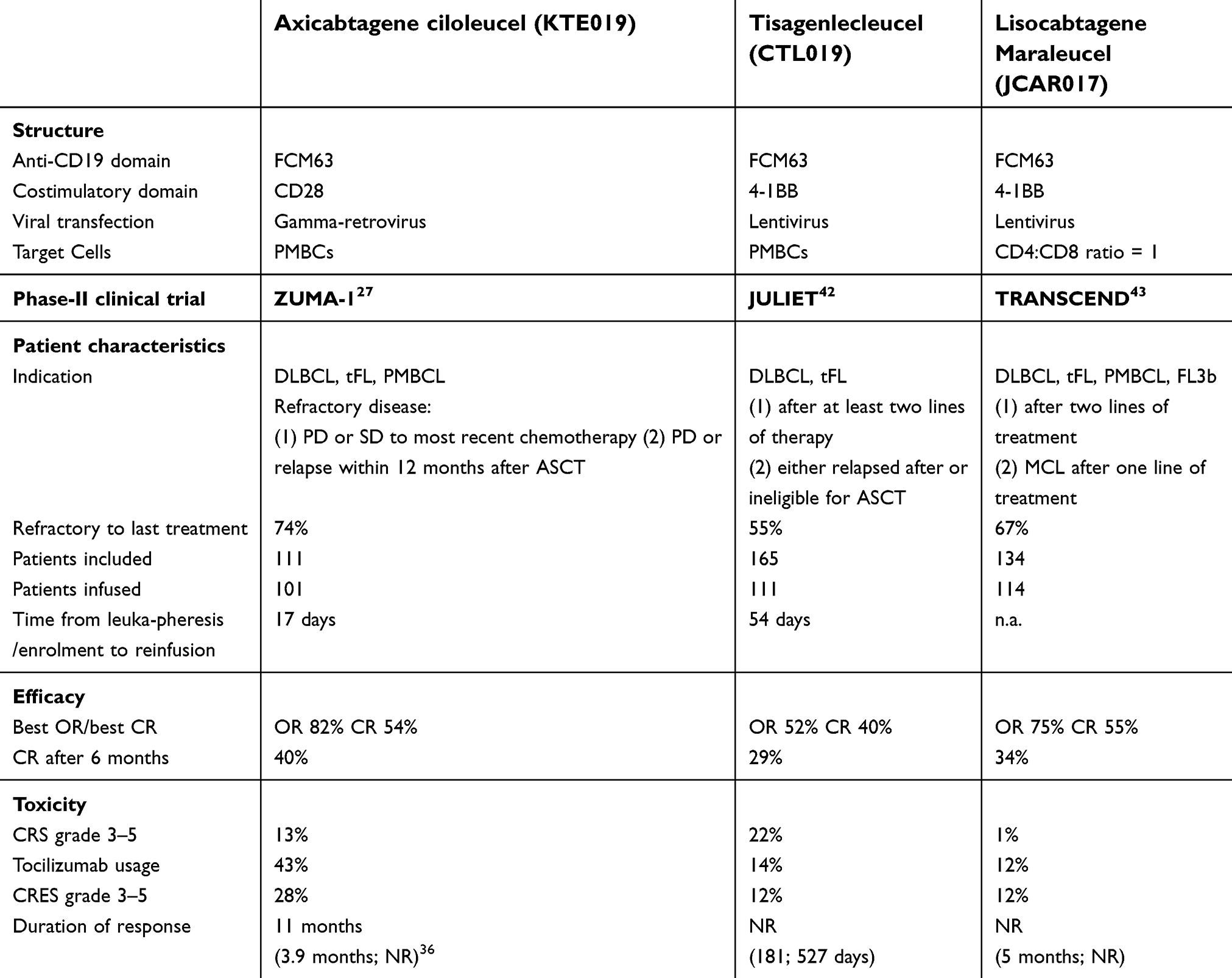 | Table 2 Comparison of commercially developed anti-CD19 CAR T-cell compounds |
Mechanism of resistance and new strategies
In the ZUMA-1 trial, 82% of patients initially responded.27 However, many patients lost their response within 6–9 months after treatment. Therefore, it is important to evaluate the mechanisms of primary and secondary resistance.
CD19 loss is a prominent mechanism of resistance in CD19 redirected T-cell therapies observed with bispecific antibodies as well as CARs. In ZUMA-1, 3 out of 11 (27%) patients lost their CD19 expression at time of disease progression.58 In an additional report including patients treated with commercially available Axi-cel, five out of eight patients with progressive disease had a CD19 loss.59
A more relevant mechanism of resistance derives from the immunosuppressive environment of the lymphoma. Inhibitory checkpoint regulators such as PD-1 and PD-L1 have been shown to be upregulated in CAR-T cells and in the microenvironment and may contribute to resistance.60,61 In the ZUMA-1, eight out of ten evaluable patients experiencing disease progression presented PD-L1 positivity (80%).58 PD-L1 positivity is also observed in many other trials with CD19 CARs, particularly in ALL, and in CD19 redirected bispecific antibodies.62 This suggests that Axi-cel activity could be augmented by incorporating PD-L1 blockade into the treatment strategy. In a case report presented at the ASH congress in 2017,63 one patient with DLBCL experiencing disease progression after treatment with Axi-cel with a high expression level of PD-L1 received nivolumab as a further salvage strategy and proved to get a secondary expansion of CART-cells and a subsequent clinical response. In the ZUMA-6 trial,64 the PD-L1 antagonist atezolizumab was given in addition to Axi-cel starting at different time points (day 21, 14 and 1 in different cohorts). In a phase-I trial including 12 patients, the combination showed an acceptable safety profile and high response rates, opening the phase II part of this trial.
How to place Axi-cell in the future treatment of DLBCL
In the SCHOLAR-1 analysis of refractory DLBCL from two large randomized trials and two academic databases,7 the overall survival was limited to 6–7 months. In a comparison of SCHOLAR-1 and ZUMA-1 adjusted for imbalances in key prognostic covariates, the CR rate of Axi-cel is tenfold higher than after standard-of-care treatment.55 The risk of death decreased at 77%. Considering that a relevant proportion of patients included in the SCHOLAR-1 registry would not fulfill the ZUMA-1 inclusion criteria, this analysis cannot replace a randomized comparison. Given the fact, that only 10–20% of patients with relapsed or refractory patients are curable by ASCT or conventional chemotherapy,9 a vast majority of those patients would be theoretically candidates for CAR T cell therapy.65 So far, the limited availability of certified sites, the manufacturing capacity of the companies and the economic burden challenge the broad implementation of CAR T-cells in the treatment of r/r DLBCL.
CAR T-cell therapy will also challenge the use of allo-SCT in selected patients. Although evaluated particularly in phase-II trials10 and in registries,11 allo-SCT is a valid option for younger patients with relapsed and refractory diseases. The use of allo-SCT is limited by several factors: 1) the availability of a matched donor (maybe enhanced by the use of haploidentical donors); 2) the time for donor identification and selection; 3) the time to establish a competent host immunity and consequently a graft-versus-lymphoma effect after transplant; and finally 4) the significant mortality and morbidity by graft-versus-host disease and opportunistic infections. However, in phase-II trials of highly selected patients with completely HLA matched donors, the survival rates are promising. Whether CAR T-cells will present an alternative therapy for selected young patients with available completely matched donors, is still a matter of debate.
Given the unsatisfactory results of salvage treatment after first relapse,5–7 an earlier placement of CAR-T cell therapy in first relapse is under discussion and will be addressed in several phase III trials. ZUMA-7 is a phase III randomized trial of Axi-cel versus standard-of-care (R-DHAP, R-ICE, R-ESHAP followed by ASCT in responding patients) in patients with r/r DLBCL.66 Comparable concepts of other CAR-T cell compounds are underway, eg, the BELINDA trial (NCT03570892) with Tisagenlecleucel or the TRANSFORM trial with liso-cel (NCT03575351).
Future directions
Beyond Axi-cel, a confusing amount of new developments will push into clinical practice. Given the class effect of neurotoxicity in CD19-directed immunotherapies and the frequent secondary loss of CD19, alternative targets in B-cell lymphoma, like CD20, CD22 and HLA-DR are warranted. CD20 is well-established, but has the theoretical disadvantage of loss of efficacy in anti-CD20 antibody pretreated patients. However, first clinical results suggest efficacy without evidence of neurotoxicity.67 CD22 was successfully used for the treatment of ALL.68 CD22 CARs may also be a valuable option in B-cell lymphoma, partially since no commercial CD22 antibody pretreatment may decrease the efficacy.69 Finally, thanks to an initial desensitization of the chimeric receptor with a consequent increase of the avidity for the target, HLA-DR CARs could improve CAR T-cells selectivity while sparing the normal B cells.70
Multitarget CAR approaches comes into clinical investigation. Particularly in ALL patients, where resistance by CD19 loss is more frequent than in lymphoma patients, CD19 and CD22 can be simultaneously attacked eg, using a CAR “cocktail”, a bicistronic CAR (two CARs in one retroviral vector) or a bispecific CAR.71–73
The strong lymphocyte activation by CD19-CD28-CD3ζ CAR constructs is responsible for side effects and might lead to early exhaustion and shortened persistence. Replacing the hinge and transmembrane domain from CD28 (as usual in Axi-cel) to eg, CD8-alpha lowers the cytokine levels in vitro and the side-effects in first clinical experience.74 In another model, CD28 “null” mutations can decrease the CAR T-cell exhaustion in vitro.75 By reducing the immunoreceptor tyrosine-based activation motifs (ITAMs) of the CAR molecule from three to one, strong activation and early exhaustion can be prevented, calibrating the activation potential and efficacy of CARs.76
An alternative strategy for prevention of T-cell exhaustion is the modulation of the primary transfected cells by cell selection. In the setting of Axi-cel, peripheral mononuclear cells (PMCs) were transfected after limited modifications, in contrast to Liso-cel, where defined amounts of CD4- and CD8-cells were transfected independently. However, there is no evidence that differences in CD4/CD8 composition may impact the outcome.27 As an alternative strategy, autologous central memory-enriched T-cells (Tcm) can be enriched ex-vivo and transfused in the setting of ASCT.77 In a phase-I trial with 17 patients, the PFS after ASCT was promising (36.1 months).78 There are at least theoretical considerations about pretreatment before harvesting in order to improve the function of CAR T-cells. At least in CLL patients, side effects can diminished and expansion can be enhanced by the use of ibrutinib pretreatment during leukapheresis and in the first weeks after CART reinfusion.79,80
Conclusions
Since the first successes of CAR-T-cells in ALL and aggressive lymphoma, we are experiencing a new era of immunotherapy in cancer. To date, the treatment landscape of aggressive lymphoma and pediatric ALL is widely changing – at this time mainly limited by the establishment of highly complex manufacturing and distribution pathways and the high costs of the products. We are learning rapidly how to expand this principle also to elderly and unfit patients, to earlier stages of aggressive lymphomas, and to other non-Hodgkin lymphomas such as mantle cell lymphoma (ZUMA-2; NCT02601313) or indolent lymphoma (ZUMA-5; NCT03105336). Axi-cel treatment could be further improved 1) by optimizing the logistic aspects like the application (short “vein to vein” time, fresh application of the product); 2) by making progress in predicting, preventing and treating CRS and CRES (eg, by alternative cytokine inhibitors like anakinra or siltuximab); and 3) by enhancing the expansion, persistence and efficacy of CAR T-cells (eg, immune checkpoint blockade, ibrutinib to enhance CAR T-cell expansion, other immunomodulating drugs). The most exciting field is the development of new CAR constructs like interleukin armoured (fourth generation) CARs, bispecific CARs to avoid resistance by antigen loss or “on-off” CARs for regulating toxic effects.81,82 The development of allogeneic “off the shelf” CAR-Tcells might help to shorten the time to transfusion. With the selection of suitable tumor antigens, the CAR principle could be extended to many fields of cancer therapy.
Disclosure
AV received honoraria from Amgen, Pfizer, Roche and BMS; travel grants from Abbvie, Roche, Celgene and BMS; and participated in advisory boards from Amgen, Kite-Gilead, Roche, Pfizer. VW, ES and SK have no further disclosures. Dr Andreas Viardot reports personal fees, non-financial support from Kite/Gilead, personal fees, non-financial support from Roche, personal fees from Amgen, personal fees from BMS, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1.
2. Cunningham D, Hawkes EA, Jack A, et al. Rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone in patients with newly diagnosed diffuse large B-cell non-Hodgkin lymphoma: a phase 3 comparison of dose intensification with 14-day versus 21-day cycles. Lancet. 2013;381(9880):1817–1826. doi:10.1016/S0140-6736(13)60313-X
3. Vitolo U, Trněný M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol. 2017;35(31):3529–3537. doi:10.1200/JCO.2017.73.3402
4. Sarkozy C, Sehn LH. Management of relapsed/refractory DLBCL. Best Pract Res Clin Haematol. 2018;31(3):209–216. doi:10.1016/j.beha.2018.07.014
5. Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28(27):4184–4190. doi:10.1200/JCO.2010.28.1618
6. Crump M, Kuruvilla J, Couban S, et al. Randomized comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine, and cisplatin chemotherapy before autologous stem-cell transplantation for relapsed and refractory aggressive lymphomas: NCIC-CTG LY.12. J Clin Oncol. 2014;32(31):3490–3496. doi:10.1200/JCO.2013.53.9593
7. van Imhoff GW, McMillan A, Matasar MJ, et al. Ofatumumab versus rituximab salvage chemoimmunotherapy in relapsed or refractory diffuse large B-cell lymphoma: the ORCHARRD study. J Clin Oncol. 2017;35(5):544–551. doi:10.1200/JCO.2016.69.0198
8. Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800–1808. doi:10.1182/blood-2017-03-769620
9. Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. Hematology Am Soc Hematol Educ Program. 2011;498–505. doi:10.1182/asheducation-2011.1.498
10. Glass B, Hasenkamp J, Wulf G, et al. Rituximab after lymphoma-directed conditioning and allogeneic stem-cell transplantation for relapsed and refractory aggressive non-Hodgkin lymphoma (DSHNHL R3): an open-label, randomised, phase 2 trial. Lancet Oncol. 2014;15(7):757–766. doi:10.1016/S1470-2045(14)70161-5
11. Fenske TS, Ahn KW, Graff TM, et al. Allogeneic transplantation provides durable remission in a subset of DLBCL patients relapsing after autologous transplantation. Br J Haematol. 2016;174(2):235–248. doi:10.1111/bjh.14046
12. Sehn LH, Kamdar M, Herrera AF, et al. Randomized phase 2 trial of polatuzumab vedotin (pola) with bendamustine and rituximab (BR) in relapsed/refractory (r/r) FL and DLBCL. J Clin Oncol. 2018;36(15_suppl):7507. doi:10.1200/JCO.2018.36.15_suppl.7507
13. Salles GA, Duell J, González-Barca E, et al. Single-arm phase II study of MOR208 combined with Lenalidomide in patients with relapsed or refractory diffuse large B-cell lymphoma: L-mind. Blood. 2018;131(abstract):227.
14. Dunleavy K, Pittaluga S, Maeda LS, et al. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med. 2013;368(15):1408–1416. doi:10.1056/NEJMoa1214561
15. Zinzani PL, Ribrag V, Moskowitz CH, et al. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood. 2017;130(3):267–270. doi:10.1182/blood-2016-12-758383
16. Link BK, Maurer MJ, Nowakowski GS, et al. Rates and outcomes of follicular lymphoma transformation in the immunochemotherapy era: a report from the University of Iowa/MayoClinic specialized program of research excellence molecular epidemiology resource. J Clin Oncol. 2013;31(26):3272–3278. doi:10.1200/JCO.2012.48.3990
17. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. 1989;86:10024–10028.
18. Eshhar Z, Waks T, Gross G, Schindler DG Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993;90:720–724.
19. Lamers CH, Sleijfer S, Vulto AG, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20–2. doi:10.1200/JCO.2006.05.9964
20. Park JR, Digiusto DL, Slovak M, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15(4):825–833. doi:10.1038/sj.mt.6300104
21. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi:10.1038/mt.2010.24
22. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi:10.1056/NEJMoa1103849
23. Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi:10.1056/NEJMoa1215134
24. Kochenderfer JN, Feldman SA, Zhao Y, et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;32:689–702. doi:10.1097/CJI.0b013e3181ac6138
25. Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116(19):3875–3886. doi:10.1182/blood-2010-01-265041
26. Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi:10.1182/blood-2011-10-384388
27. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;37:2531–2544. doi:10.1056/NEJMoa1707447
28. US national Library of Medicine [Internet]. Available from:
29. van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499–509. doi:10.1038/nrd4597
30. Long AH, Haso WM, Shern JF, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. doi:10.1038/nm.3838
31. Better M, Chiruvolu V, Oliver J, et al. Production of KTE-C19 (Anti-CD19 CAR T cells) for ZUMA-1: A phase 1/2 multi-center study evaluating safety and efficacy in subjects with refractory aggressive Non-Hodgkin Lymphoma (NHL). Mol Ther. 2016;24(Supplement 1). doi:10.1016/S1525-0016(16)33096-9.
32. Wang X, Riviere I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncol. 2016;3:16015. doi:10.1038/mto.2016.15
33. Jain MD, Bachmeier CA, Phuoc VH, Chavez JC. Axicabtagene ciloleucel (KTE-C19), an anti-CD19 CAR T therapy for the treatment of relapsed/refractory aggressive B-cell non-Hodgkin’s lymphoma. Ther Clin Risk Manag. 2018;14:1007–1017. doi:10.2147/TCRM.S145039
34. McGarrity GJ, Hoyah G, Winemiller A, et al. Patient monitoring and follow-up in lentiviral clinical trials. J Gene Med. 2013;15(2):78–82. doi:10.1002/jgm.2691
35. Yescarta (Axicabtagene ciloleucel) [package insert]. Santa Monica, CA, USA: Kite Pharma, Inc; 2017.
36. Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33:540–549. doi:10.1200/JCO.2014.56.2025
37. Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi:10.1016/S0140-6736(14)61403-3
38. Neelapu SS, Ghobadi A, Jacobson CA, et al. 2-year follow-up and high-risk subset analysis of Zuma-1, the pivotal study of Axicabtagene Ciloleucel (Axi-Cel) in patients with refractory large B cell Lymphoma. Blood. 2018;131:2967(abstract).
39. Kochenderfer JN, Somerville RPT, Lu T, et al. Long-duration complete remissions of diffuse large B cell lymphoma after anti-CD19 chimeric antigen receptor T cell therapy. Mol Ther. 2017;25:2245–2253. doi:10.1016/j.ymthe.2017.07.004
40. Kochenderfer JN, Somerville RPT, Lu T, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol. 2017;35:1803–1813. doi:10.1200/JCO.2016.71.3024
41. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 results of ZUMA-1: A multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285–295. doi:10.1016/j.ymthe.2016.10.020
42. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2018. doi:10.1056/NEJMoa1804980
43. J Abramson, Gordon LI, Palomba ML, et al. Updated safety and Long term clinical outcomes in TRANSCEND NHL 001, pivotal Trial of lisocabtagene maraleucel (JCAR017) in r/r aggressive NHL. J Clin Oncol. 2018; 36(suppl):7505 (abstract) . doi: 10.1200/JCO.2018.36.15_suppl.7505
44. Nastoupil LJ, Jain MD, Spiegel JY, et al. Axicabtagene ciloleucel (Axi-cel) CD19 Chimeric Antigen Receptor (CAR) T-cell therapy for relapsed/refractory large B-cell lymphoma: real world experience. Blood. 2018;131:91 (abstract).
45. Jacobson CA, Hunter B, Armand P, et al. Axicabtagene ciloleucel in the real world: outcomes and predictors of response, resistance and toxicity. Blood. 2018;131:92 (abstract).
46. Sano D, Nastoupil LJ, Fowler NH, et al. Safety of Axicabtagene ciloleucel CD19 CAR T-cell therapy in elderly patients with relapsed or refractory large B-cell lymphoma. Blood. 2018;131:96 (abstract).
47. Spiegel JY, Sahaf B, Hossain N, et al. Elevated Axicabtagene ciloleucel (CAR-19) expansion by immunophenotyping is associated with toxicity in diffuse large B-cell lymphoma. Blood. 2018;131:576 (abstract).
48. Faramand R, Kotani H, Morrissey D, et al. Prediction of CAR T-related toxicities in R/R DLBCL patients treated with axicabtagene ciloleucel using point of care cytokine measurements. Blood. 2018;131:95 (abstract).
49. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. doi:10.1182/blood-2014-05-552729
50. Park JH, Rivière I, Gonen M, et al. Long-term follow-up of CD19 CAR Therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–459. doi:10.1056/NEJMoa1709919
51. Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy – assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62. doi:10.1038/nrclinonc.2017.148
52. Viardot A, Bargou R. Bispecific antibodies in haematological malignancies. Cancer Treat Rev. 2018;65:87–95. doi:10.1016/j.ctrv.2018.04.002
53. Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–748. doi:10.1038/s41591-018-0036-4
54. Cordeiro A, Bezerra E, Hill JA, et al. Late effects of CD19- targeted CAR-T cell therapy. Blood. 2018;131:576 (abstract 223).
55. Neelapu SS, Locke FL, Bartlett NL, et al. A comparison of one year outcomes in ZUMA-1 (Axicabtagene ciloleucel) and SCHOLAR-1 in patients with refractory, aggressive Non-Hodgkin lymphoma (NHL). Blood. 2017;130:579 (abstract).
56. Zheng PP, Kros JM, Li J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov Today. 2018;23(6):1175–1182. doi:10.1016/j.drudis.2018.02.012
57.
58. Neelapu SS, Locke FL, Bartlett NL, et al. Long-term follow-up ZUMA-1: A pivotal trial of Axicabtagene ciloleucel (Axi-cel; KTE-C19) in patients with refractory aggressive Non-Hodgkin Lymphoma (NHL). Blood. 2017;130:578 (abstract).
59. Oak J, Spiegel JY, Sahaf B, et al. Target Antigen downregulation and other mechanisms of failure after axicabtagene ciloleucel (CAR19) therapy. Blood. 2018;131:4656 (abstract).
60. Vranic S, Ghosh N, Kimbrough J, et al. PD-L1 status in refractory lymphoma. PLoS One. 2016;11(11):e0166266. doi:10.1371/journal.pone.0166266
61. Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130–3144. doi:10.1172/JCI83092
62. Feucht J, Kayser S, Gorodezki D, et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget. 2016;7(47):76902–76919. doi:10.18632/oncotarget.12357
63. Hill BT, Roberts ZJ, Rossi JM, Smith MR. Marked re-expansion of chimeric antigen receptor (CAR) T cells and tumor regression following nivolumab treatment in a patient treated with Axicabtagene ciloleucel (Axi-cel; KTE-C19) for refractory Diffuse Large B cell Lymphoma (DLBCL). Blood. 2017;130:2825 (abstract).
64. Jacobson CA, Locke FL, Miklos DB, et al. End of phase 1 results from zuma-6: axicabtagene ciloleucel (Axi-cel) in combination with atezolizumab for the treatment of patients with refractory diffuse large B cell lymphoma. Blood. 2018;131:4192 (abstract).
65. Chow VA, Shadman M, Gopal AK. Translating anti-CD19 CAR T-cell therapy into clinical practice for relapsed/refractory diffuse large B-cell lymphoma. Blood. 2018;132(8):777–781. doi:10.1182/blood-2018-04-839217
66. Oluwole OO, Bishop MR, Gisselbrecht C, et al. ZUMA-7: A phase 3 randomized trial of Axicabtagene ciloleucel (Axi-cel) versus standard-of-care (SOC) therapy in patients with relapsed/refractory diffuse large B cell lymphoma (R/R DLBCL). J Clin Oncol. 2018;36(15_suppl). abstract. doi:10.1200/JCO.2018.36.15_suppl.TPS7585
67. Zhang WY, Wang Y, Guo YL, et al. Treatment of CD20-directed chimeric antigen receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an early phase IIa trial report. Signal Transduct Target Ther. 2016;1:16002. doi:10.1038/sigtrans.2016.2
68. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–28. doi:10.1038/nm.4441
69. Yates B, Shalabi H, Salem D, et al. Sequential CD22 targeting impacts CD22 CAR-T cell response. Blood. 2018;131:282 (abstract).
70. Han C, Sim SJ, Kim SH, et al. Desensitized chimeric antigen receptor T cells selectively recognize target cells with enhanced antigen expression. Nat Commun. 2018;9(1):468. doi:10.1038/s41467-018-02912-x
71. Yang J, Li J, Zhang X, et al. A feasibility and safety study of CD19 and CD22 chimeric antigen receptors-modified T cell cocktail for therapy of B cell acute lymphoblastic leukemia. Blood. 2018;131:277 (abstract).
72. Amrolia PJ, Wynn R, Hough R, et al. Simultaneous targeting of CD19 and CD22: phase I Study of AUTO3, a bicistronic Chimeric Antigen Receptor (CAR) T-cell therapy, in pediatric patients with relapsed/refractory B-cell Acute Lymphoblastic Leukemia (r/r B-ALL): amelia study. Blood. 2018;131:279 (abstract).
73. Schultz LM, Davis KL, Baggott C, et al. Phase 1 study of CD19/CD22 bispecific Chimeric Antigen Receptor (CAR) therapy in children and young adults with B cell Acute Lymphoblastic Leukemia (ALL). Blood. 2018;131:898 (abstract).
74. Brudno J, Hartman S, Lam N, et al. Low levels of neurologic toxicity with retained anti-Lymphoma activity in a phase I clinical trial of T cells expressing a novel anti-CD19 CAR. Blood. 2018;131:898 (abstract).
75. Boucher JC, Li G, Shrestha B, et al. Mutation of the CD28 costimulatory domain confers decreased CAR T cell exhaustion. Blood. 2018;131:966 (abstract).
76. Feucht J, Sun J, Eyquem J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 2019;25:82–88. doi:10.1038/s41591-018-0290-5
77. Wang X, Popplewell LL, Wagner JR. Phase 1 studies of central memory–derived CD19 CAR T–cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127:2980–2990. doi:10.1182/blood-2015-12-686725
78. Popplewell LL, Wang X, Blanchard S, et al. CD19-CAR therapy using naive/memory or central memory T cells integrated into the autologous stem cell transplant regimen for patients with B-NHL. Blood. 2018;131:610 (abstract).
79. Fraietta JA, Beckwith KA, Patel PR, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127:1117–1127. doi:10.1182/blood-2015-11-679134
80. Gauthier J, Hirayama AV, Hay KA, et al. Comparison of efficacy and toxicity of CD19-specific chimeric antigen receptor T-cells alone or in combination with ibrutinib for relapsed and/or refractory CLL. Blood. 2018;131:299 (abstract).
81. Guedan S, Calderon H, Posey AD
82. Lin C, Zhang J. Reformation in chimeric antigen receptor based cancer immunotherapy: redirecting natural killer cell. Biochim Biophys Acta Rev Cancer. 2018;1869(2):200–215. doi:10.1016/j.bbcan.2018.01.005
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.