")
Back to Journals » OncoTargets and Therapy » Volume 16
Chidamide and Oxaliplatin Synergistically Inhibit Colorectal Cancer Growth by Regulating the RPS27A-MDM2-P53 Axis
Authors Li Z , Bu D , Wang X, Zhu L, Lei D, Tang F , Sun X , Chen C, Ji X , Bai S
Received 12 April 2023
Accepted for publication 13 August 2023
Published 30 August 2023 Volume 2023:16 Pages 703—721
DOI https://doi.org/10.2147/OTT.S416824
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Zhaopeng Li,1,* Deyong Bu,1,* Xiaobin Wang,1 Lin Zhu,2 Daoyan Lei,3 Fengling Tang,1 Xianghua Sun,1 Cheng Chen,4 Xiang Ji,5 Song Bai1
1Department of Geriatric General Surgery, the First Affiliated Hospital of Kunming Medical University, Kunming, Yunnan, 650000, People’s Republic of China; 2Department of Ultrasound, the Affiliated Nanchong Central Hospital of North Sichuan Medical College, Nanchong, Sichuan, 637000, People’s Republic of China; 3Department of Ultrasound, Jiangchuan District People’s Hospital, Yuxi, Yunnan, 652600, People’s Republic of China; 4Department of Breast Surgery, the First Affiliated Hospital of Kunming Medical University, Kunming, Yunnan, 650000, People’s Republic of China; 5Department of Day Surgery, the First Affiliated Hospital of Kunming Medical University, Kunming, Yunnan, 650000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Deyong Bu; Song Bai, Department of Geriatric General Surgery, the First Affiliated Hospital of Kunming Medical University, Kunming, Yunnan, 650000, People’s Republic of China, Tel/Fax +86-0871-5324-888, Email [email protected]; [email protected]
Purpose: The present study explored the anti-tumor effects of chidamide plus oxaliplatin on colorectal cancer (CRC) and examined its underlying mechanism.
Material and Methods: First, the Combination Index (CI) of chidamide and oxaliplatin was evaluated via CCK-8 assay. Second, the effects of chidamide and oxaliplatin monotherapy and the combined treatment on cell proliferation, invasion, migration, and apoptosis were detected. Third, whole-transcriptome RNA sequencing (RNA-seq) was performed to seek the potential targeted gene by which chidamide plus oxaliplatin exerted anti-tumor effects. Fourth, the validation of the targeted gene and the signal pathway it regulated were performed. Finally, the anti-tumor effect of chidamide plus oxaliplatin on mice xenograft was examined.
Results: Chidamide and oxaliplatin acted synergistically to inhibit CRC growth in vitro and in vivo (CI< 1). Besides, compared with oxaliplatin monotherapy, chidamide could significantly enhance oxaliplatin-induced inhibition in cell proliferation, invasion, and migration, and promotion in HCT-116 and RKO cell apoptosis (P< 0.05). The RNA-seq displayed that, compared to oxaliplatin monotherapy, RPS27A mRNA was evidently decreased in HCT-116 cells treated with chidamide plus oxaliplatin (P< 0.001). Then, we found RPS27A was highly expressed in CRC tissues and CRC cell lines (P< 0.001). Silence of RPS27A attenuated proliferation and induced apoptosis in HCT-116 and RKO cells via downregulation of MDM2 expression and upregulation of P53. Next, RPS27A overexpression could partially reverse chidamide plus oxaliplatin induced growth inhibition and apoptosis in HCT-116 and RKO cells (P< 0.01). RPS27A overexpression could promote the upregulation of MDM2 and downregulation of P53 after the combined treatment of chidamide with oxaliplatin.
Conclusion: Chidamide and oxaliplatin acted synergistically to suppress CRC growth by the inhibition of the RPS27A-MDM2-p53 axis.
Keywords: chidamide, oxaliplatin, colorectal cancer, synergism
Introduction
CRC is the third malignant tumor in terms of morbidity, ranking second in the cause of cancer-related death worldwide.1 With the early screening techniques being widely used, the detection rate of early-stage CRC increases, while, a large proportion of patients are in the middle or late stages of CRC illness when diagnosed.2 So, chemotherapy is critical for them. Oxaliplatin is the frontline chemotherapeutic drug for treating high-risk stage II, stage III, and stage IV CRC, functioning through the binding of the platinum atom to DNA, which makes the DNA strands broken and ultimately interferes with the replication of DNA.3,4 Although dose intensification of oxaliplatin might enhance the anti-tumor effect of CRC, it is associated with seriously accumulative peripheral neurotoxicity5,6 and acquired chemoresistance.7 Therefore, it urgently demanded a more effective and less toxic regimen to treat CRC.
Epigenetic abnormalities being independent of DNA sequence alterations, such as DNA hypermethylation and histone hypoacetylation, play significant roles in modulating cellular processes and gene expression.8 Histone deacetylases (HDACs) and histone acetylases (HATs) are participating in maintaining the dynamic equilibrium of histone acetylation.9 It is well established that HDACs are over-expressed in several types of tumors, including lymphoma,10 CRC,11 lung cancer,12 breast cancer,13 and hepatic cancer.14 High expression of HDACs in cancer cells usually correlates with poor prognosis.15,16 Recently, inhibition of HDACs has become an emerging and attractive therapy for malignancies. HDAC inhibitors (HDACis) are involved in the depression of the removal of the acetyl groups in the certain N-terminal lysine of histone proteins by targeting HDACs, resulting in histone proteins hyperacetylation, which supports the formation of open chromatin configuration and re-expression of silent genes.17,18 The more opened chromatin is expected to facilitate not only the transcription factors binding to the DNA, but also the DNA damage drugs’ access to it easily.
Chidamide is an oral and benzamide-type HDACi, selectively targeting HDACs 1–3 belonging to class I and HDAC 10 subdivided into class IIB. Compared with MS-275, a kind of benzamide-based HDACi, chidamide not only has improved efficacy,19 but also has less toxicity, better tolerability, and a longer half-life.20 HDACs 1–3 are highly expressed in CRC and overexpression of HDAC2 is correlated with poor outcomes.11 Chidamide has elaborated promising properties against colon cancer in vitro.20 However, the single chidamide towards solid tumors in clinical intervention appears to be unsatisfactory. Thus, the combination strategy involving chidamide and conventional agents warrants the investigation in solid cancers. Early studies have demonstrated that chidamide could improve the efficacy of conventional chemotherapeutic agents,21 immune checkpoint inhibitors,22,23 targeted drugs,24,25 and reverse drug resistance26 in human cancers. Chidamide combined 5-Fu displayed synergistic antitumor efficacy on colon cancer in vivo.27 In the treatment of non-small-cell lung cancers, the combination of chidamide with carboplatin displayed good synergism in inducing apoptosis and inhibition growth, as well as DNA damage response.28 However, whether chidamide could synergistically enhance the cytotoxicity of oxaliplatin in CRC remains unclear. This study was the first time to examine the effects of chidamide in combination with oxaliplatin on CRC, simultaneously defined by what mechanisms these two drugs exerted the anticancer effect on CRC.
Material and Methods
Reagents and Antibodies
Chidamide (purity >95%) powder was supplied by Chipscreen Biosciences Ltd. (Shenzhen, Guangdong Province, China) and solubilized in dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St Louis, MO, USA) to achieve the stock concentration of 32 mM. Oxaliplatin (purity >95%) powder was supplied by Beijing Suo Laibao Biotechnology Co., Ltd (Beijing, China) and solubilized in DMSO, which was stored in 80 mM. The concentration of DMSO in all experiments did not exceed 0.1%. All the above stock solutions were kept at −80°C. It was diluted to the designated concentrations with culture medium in subsequent experiments.
The reagent TSnanofect-V1 was purchased from Tsingke Biotechnology Co., Ltd (Beijing, China). The TRIGen reagent (GenStarⓇ, Beijing, China) and First-Strand cDNA Synthesis kit (GenStarⓇ, Beijing, China) were utilized to isolate total RNA and perform cDNA synthesis according to the instruction manual, respectively. Cell Counting Kit-8 (CCK-8) assay kit was supplied by Abbkine Scientific Co., Ltd (Wuhan, Hubei Province, China). Annexin V/PI assay kit was bought from US Everbright® Inc (Suzhou, Jiangsu Province, China). The protein concentration was quantified with a bicinchoninic acid (BCA) Kit, which was purchased from Shanghai Beyotime Biotech Inc (Shanghai, China). Electrochemiluminescence (ECL) reagents were purchased from Abbkine Scientific Co., Ltd (Wuhan, Hubei Province, China). Anti-β-actin and horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibodies were supplied by Boster Biological Technology Co., Ltd (Wuhan, Hubei Province, China). Antibodies recognizing RPS27A, MDM2, P53 were obtained from Abcam® (Cambridge, MA, USA). RPS27A overexpressing plasmid and empty vector, siRNA of RPS27A and scramble siRNA were constructed by Tsingke Biotechnology Co., Ltd (Beijing, China).
Human Samples and CRC Cell Lines
A total of 30 CRC tissues and matched normal colorectal tissues were obtained from the Department of Gastrointestinal Surgery in The First Affiliated Hospital of Kunming Medical University, and before surgery, written informed consent was taken from all the CRC participants. The CRC patients had no history of neoadjuvant therapy. All the samples were immediately put into liquid nitrogen once detached ex vivo. The human CRC cell lines HCT-116 and RKO and normal colonic epithelial cell NCM-460 were purchased from the Cell Bank in Chinese Academy of Sciences. HCT-116, RKO, and NCM-460 cells were grown in RPMI 1640 medium (Gibco, USA) with 10% fetal bovine serum (FBS, Gibco, USA) in a 5% CO2 incubator at 37°C.
Cell Viability Assay
The cytotoxic effects of oxaliplatin and chidamide in monotherapy and in combination on RKO and HCT-116 cells were monitored by CCK-8 assay. RKO (7×103 cells/well) and HCT-116 (7×103 cells/well) were incubated in 96-well plates with 100 ul RPMI 1640 overnight, then they were treated with designated concentrations of oxaliplatin and chidamide alone and in combination for 48 h. Next, 10 ul CCK-8 solution per well was put into the medium and continued to culture at 37°C for an additional 1–2 h. Lastly, the optical density of each well at 450 nm was measured by Cytocube Auto (Guangzhou, China). The combination index (CI) values representing the degree of the two drugs’ interaction was calculated via CompuSyn version 1.0 software. CI<1, CI=1, and CI>1 meant synergistic, additive, and antagonistic effects, respectively.29
Colony Formation Assay
Different impacts of chidamide and oxaliplatin single agent and in combination on cell proliferation of HCT-116 and RKO were determined by colony formation assay. HCT-116 (700 cells/well) and RKO (700 cells/well) cells were plated in 6-well plates and incubated in the culture medium overnight. Then, HCT-116 and RKO cells were treated with chidamide and oxaliplatin alone and in combination for 48 h, respectively. The medium without DMSO or drugs was replaced every three days until visible clones formed. Next, methyl alcohol and 0.1% crystal violet were utilized to fix and dye the different drug-treated cells, respectively. Finally, colonies stained with crystal violet were calculated via ImageJ software.
Wound-Healing Assay
To investigate whether chidamide could further enhance the inhibition of migration capability induced by oxaliplatin, Wound-healing assays were conducted to test the migratory ability of different drug-treated HCT-116 and RKO cells. Cells (6×105 cells/well) were plated in 6-well plates and treated with chidamide and oxaliplatin single agent, and in combination for 48 h, respectively. Then, the primary culture medium was removed until 90% confluency was reached and a straight line was scratched via a 200-ul tip in the middle of each cell layer. The medium without serum was added to each Petri well for 24 h incubation. Images at 0 h, 6 h, 12 h, and 24 h following the scratch were captured with the microscope. Next, wound closure rates in the different experimental groups were calculated with ImageJ software.
Transwell Assay
Cell invasion and migration alteration after chidamide and oxaliplatin in monotherapy and in combination for 48 h were examined by Transwell assays. Transwell chambers coated with Matrigel were employed to detect cell invasiveness. HCT-116 and RKO cells were treated with chidamide and oxaliplatin single agent and in combination for 48 h. The cells with different treatments were collected, respectively. Then, a total of 200 ul of treated cells in upper chambers were resuspended with serum-free RPMI 1640 medium. A total of 800 ul RPMI 1640 medium with 10% FBS was added into the lower chambers. Next, the cells in Transwell chambers were cultivated for 24 h. After washing cells with PBS, fixing cells with methanol, and staining cells with crystal violet, five randomly selected visual fields were imaged (×100 magnification) and counted via ImageJ software. Transwell chambers without Matrigel were employed to detect cell migration.
Annexin V /PI Apoptosis Assay
To investigate whether chidamide could enhance the cell apoptosis induced by oxaliplatin, the dual-labeled with Annexin V-APC/PI reagent was utilized to dye the differently treated cells. First of all, the differently treated cells were gathered, washed, and diluted in an appropriate concentration. Then, the suspension with 8×104 cells was added into EP tubes. Next, 5 ul Annexin V solution and 5 ul propidium iodide (PI) solution were added to the cells and then cultured in dark at room temperature for 15 min according to the kit protocols. Further, the stained cells were analyzed via flow cytometry (FACS Fortessa, BD Bioscience). The early apoptotic cells were referred to those with Annexin V positive but PI negative, and late apoptotic cells were considered when both Annexin V and PI were positive. The apoptotic population was the total of early and late apoptotic cells.
Quantitative Real-Time Polymerase Chain Reactions (RT-qPCR)
Total RNA was isolated in strict accordance with the TRIGene reagent instructions and was reversely transcribed to cDNA according to the protocols of the First-Strand cDNA Synthesis kit. All the sequences of primers (Table 1) were designed via the NCBI primer design tool to amplify the cDNA. The RT-qPCR was performed in accordance with the instructions of the supplier under the following conditions: denaturation at 95°C for 20s, annealing at 60°C for 30s, and extension at 72°C for 30s, respectively. The cycling conditions for RT-qPCR were repeated 40 times. The targeted gene transcripts were normalized to the housekeeping gene Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression. Each sample was tested in triplicate. The RT-qPCR results were quantified using the 2−∆∆Ct method.
 |
Table 1 Primers Sequences |
Whole-Transcriptome RNA Sequencing and Bioinformatics Analysis
HCT-116 cells were gathered after pretreatment with indicated drug administrations, and total RNA was extracted as mentioned above. Then, RNA sequencing (RNA-seq) was conducted. Firstly, total mRNA was fragmented into small pieces and synthesized cDNA. Secondly, PCR amplification was carried out to enrich and purify double-stranded DNA. Thirdly, the amplified products were sequenced using Illumina Novaseq. Fourthly, genes with altered expression among the different treatment groups were detected using the DESeq2 from the SARTools package. The differentially expressed genes (DEGs) should meet both of the following criteria, including (a) at least 2-fold upregulation or downregulation after the drug treatment; (b) The adjusted p value less than 0.05 were used to select DEGs. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was performed to clarify how the DEGs played roles in the different treatments. Finally, the protein–protein interaction (PPI) network of DEGs was constructed and visualized via the STRING website and Cytoscape software, respectively.
RPS27A Gene Overexpression and Knockdown
When HCT-116 and RKO cells in 6-well plates proliferated to a confluence of 50%, they were transiently transfected with 1 ug RPS27A overexpressing plasmid and empty vector using TSnanofect-V1 according to the product instruction. Meanwhile, RPS27A siRNA and scramble siRNA sequence as control were transfected into HCT-116 and RKO cells utilizing TSnanofect-V1 in accordance with the kit protocol. The cells were collected at 48 h post-transfection, then RT-qPCR and Western blot were used to verify the RPS27A expression. Next, CCK-8 assays and flow cytometry were utilized to evaluate the alteration of cell proliferation and apoptosis of transfected cells.
Western Blotting (WB) Analysis
The HCT-116 and RKO cells were harvested and then lysed on ice for 30 min in radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, Shanghai, China). Next, the cell lysates were centrifuged at 14,000 rpm for 5 min at 4°C, and the supernatant was collected. The total cytoplasmic protein was quantified via a BCA assay kit. Subsequently, protein lysates were resolved via SDS-PAGE and further electrotransferred onto the polyvinylidene difluoride (PVDF) membranes. The 5% skimmed milk was utilized to block PVDF membranes for 2 h. The PVDF membranes were incubated with primary antibodies overnight at 4°C. Thereafter, the membranes were incubated with the appropriate HRP-conjugated goat anti-rabbit secondary antibodies for another hour at room temperature to detect the designated proteins. Finally, the enhanced ELC detection solution was utilized to visualize the blots and the β-actin was considered as the internal reference.
Immunohistochemical (IHC) Staining
Tissues were put into 10% formalin to be fixed and then they were embedded in paraffin. The paraffin-embedded tissues were sectioned into slides with 4 mm thickness. The slides were deparaffinized and incubated with primary antibodies in TBS overnight at 37°C. Subsequently, they were incubated with HRP-conjugated goat anti-rabbit secondary antibodies for 30 min at room temperature. Next, sections were visualized with DAB chromogenic solution, then dehydrated and mounted.
Xenograft Tumor in Mice
The five-week-old Balb/c athymic nude female mice (18–22g) were purchased from the Animal Center of Kunming Medical University. All mice were raised in the condition without pathogens. Then, 100 ul cell suspension containing 5×106 HCT-116 cells was subcutaneously inoculated into the right flank of each mouse to establish the CRC xenograft model. Mice were randomly assigned to different treatment groups when the volume of xenograft reached about 50–100 mm3. The mice with approximately uniform tumor size in different groups were treated with vehicle (intraperitoneal injection, 5% glucose solution), chidamide (gastric gavage, 3 mg/kg), oxaliplatin (intraperitoneal injection, 5 mg/kg,), chidamide plus oxaliplatin (gastric gavage, 3 mg/kg, chidamide, intraperitoneal injection, 5 mg/kg, oxaliplatin), respectively. The mice in each group were treated three times a week and were administered consecutively for three weeks. During the period, the body weight of the mouse and xenograft volume were recorded every three days. The xenograft volume (V) was calculated via the formula: V = (L×W2)/2, L refers to the largest longitudinal diameter of the tumor, and W is the largest transverse diameter. CI representing the interaction of chidamide and oxaliplatin in vivo was calculated according to the formula: CI =[(cA + cB) – cA × cB] / cAB.30 cA is the tumor inhibition rate of chidamide monotherapy, cB is the tumor inhibition rate of oxaliplatin monotherapy, and cAB is the tumor inhibition rate of the combination treatment of chidamide with oxaliplatin. CI<1, CI=1, and CI>1 meant synergistic, additive, and antagonistic effects, respectively. The mice were sacrificed on day 22 of treatment. The tumor was wholly extracted from each mouse to measure its weight.
Statistical Analysis
The measurement data were displayed as the mean ± standard deviation and were calculated by the student’s test or one-way analysis of variance (ANOVA). The Categorical data between the two groups were calculated via the χ2-test statistic. All statistical analyses were performed using GraphPad Prism 8 software. All the results were representative of at least three separate experiments. A two-sided P value less than 0.05 was regarded as statistically significant.
Results
Chidamide Synergistically Enhanced Oxaliplatin-Induced Cytotoxicity in CRC Cells
To investigate the cytotoxicity of chidamide and oxaliplatin in a single agent, CCK-8 assays were performed after the indicated concentrations drug treated HCT-116 and RKO cells for 48 h. Either oxaliplatin or chidamide could inhibit cell viability of HCT-116 and RKO cells in a dose-dependent manner, as shown in Figure 1A. To further explore whether chidamide could increase the cytotoxicity induced by oxaliplatin on CRC, we tested the effects that the indicated concentrations of oxaliplatin in combination with different dosages of chidamide had on HCT-116 and RKO cells viability via CCK-8 assays. The addition of 4 μM, 8 μM, 16 μM, and 32 μM chidamide to oxaliplatin at indicated concentrations (8 μM, 16 μM, and 32 μM) could obviously reduce both HCT-116 and RKO cells viability separately, as shown in Figure 1B. Then we calculated CI values of different combinations of chidamide with oxaliplatin by the Chou-Talalay equation via CompuSyn software to evaluate whether there was synergism in the combined treatment for CRC. The 4 to 32 μM chidamide combined with different concentrations of oxaliplatin exhibited synergy, while chidamide at relatively low concentrations, such as 0.5 μM, 1 μM, and 2 μM, combined with oxaliplatin displayed antagonism in HCT-116 and RKO cells, as shown in Figure 1C. The relatively low concentration combination of chidamide at 4 μM and oxaliplatin at 8 μM with good synergy was selected to do the flowing experiments.
 |
Figure 1 Chidamide synergistically enhanced oxaliplatin-induced cytotoxicity in HCT-116 and RKO cells. (A) Cell viability curves of HCT-116 and RKO treated with 0.5 to 32 μM of chidamide or oxaliplatin for 48 h. (B) Cell viability curves of HCT-116 and RKO treated with different concentrations of chidamide (0 to 32 μM) combined with indicated concentrations of oxaliplatin (4 μM, 8 μM, and 32 μM) for 48 h, respectively. (C) The combination index (CI) values of different concentrations of chidamide with fixed concentrations of oxaliplatin were calculated via CompuSyn. |
Chidamide Enhanced Oxaliplatin-Induced Proliferation Inhibition and Apoptosis in CRC Cells
In order to verify whether chidamide could enhance oxaliplatin-induced cell proliferation inhibition, colony formation assay and CCK-8 assay were operated in HCT-116 and RKO cell lines. The effects of the colony formation assay exhibited that, compared with chidamide and oxaliplatin in monotherapy for 48 h, a significant decrease in the number of cell clones could observe in HCT-116 and RKO cells after the combined treatment, as shown in Figure 2A. The CCK-8 assay revealed that, compared with chidamide and oxaliplatin in monotherapy, cell viability of HCT-116 and RKO reduced significantly after the combined treatment for 48 h, as shown in Figure 2B. To further explore the impact of the two agents alone and in combination on cell apoptosis, we subsequently conducted flow cytometry analyses. Annexin V/PI staining displayed chidamide plus oxaliplatin significantly increased the percentage of HCT-116 and RKO apoptotic cells versus oxaliplatin or chidamide single agent, as shown in Figure 2C. Compared with the apoptosis induction by oxaliplatin in HCT-116 and RKO cells, we also found chidamide alone did not dramatically increase the proportion of apoptotic CRC cells. All these results displayed that chidamide could significantly enhance oxaliplatin-induced proliferation inhibition and apoptosis in CRC cells.
 |
Figure 2 Chidamide enhanced oxaliplatin-induced proliferation inhibition and apoptosis in HCT-116 and RKO cells. (A) Colony formation analyzed the effects of chidamide and oxaliplatin monotherapy and the combined treatment on HCT-116 and RKO cell proliferation. (B) CCK-8 analyzed the effects of chidamide and oxaliplatin monotherapy and the combined treatment on HCT-116 and RKO cell viability. (C) Flow cytometry analyzed the effects of chidamide and oxaliplatin monotherapy and the combined treatment on HCT-116 and RKO cell apoptosis. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Abbreviations: NC, negative control (0.1% DMSO); CHI, 4 μM chidamide; OXA, 8 μM oxaliplatin; CHI+OXA, 4 μM chidamide + 8 μM oxaliplatin. |
Chidamide Enhanced Oxaliplatin-Induced Migration and Invasion Inhibition in CRC Cells
To determine the influence the combined treatment had on CRC metastasis, Transwell and Wound-healing assays were conducted to assess the alteration of HCT-116 and RKO cell invasion and migration capacity after the treatment of oxaliplatin plus chidamide for 48 h. As shown in Figure 3A, compared with chidamide and oxaliplatin treatment alone, the cell number of HCT-116 migration and invasion in the lower chamber decreased dramatically after the combined treatment (p < 0.0001). For RKO cells, the migrated and invaded RKO cells in the oxaliplatin plus chidamide group were also significantly reduced compared with the single agent (p < 0.0001), shown in Figure 3B, compared with chidamide and oxaliplatin monotherapy, the rate of wound closure significantly decreased after the combined treatment of chidamide with oxaliplatin, as shown in Figure 3C. These results displayed Chidamide could enhance oxaliplatin-induced inhibition in migration and invasion of HCT-116 and RKO cells.
 |
Figure 3 Chidamide significantly enhanced oxaliplatin-induced migratory and invasive abilities inhibition of HCT-116 and RKO cells. (A) Transwell assays analyzed the effects of chidamide and oxaliplatin monotherapy and the combined treatment on HCT-116 cell migration and invasion (×100 magnification). (B) Transwell assays analyzed the effects of chidamide and oxaliplatin monotherapy and the combined treatment on RKO cell migration and invasion (×100 magnification). (C) Wound-healing assays analyzed the effects of chidamide and oxaliplatin monotherapy and the combined treatment on HCT-116 and RKO cell migration. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Abbreviations: NC, negative control (0.1% DMSO); CHI, 4 μM chidamide; OXA, 8 μM oxaliplatin; CHI+OXA, 4 μM chidamide + 8 μM oxaliplatin. |
Chidamide Plus Oxaliplatin Downregulated RPS27A Expression in CRC Cells
Anti-tumor activity of both HDACi and DNA damage agents involves in alteration of gene transcription. To further elucidate the mechanism of chidamide synergistically enhancing the cytotoxic effect of oxaliplatin on CRC, the RNA-seq was performed to explicit the profile of altered genes transcription in HCT-116 cells after the different drugs treatment for 48 h. There were 4919 genes with differential expression between the oxaliplatin treatment group and the combined treatment group. Compared to the oxaliplatin treatment group, upregulation of 3068 DEGs and downregulation of 1851 DEGs were detected in the combined treatment group, shown in the Heatmap and Volcano plot (Figure 4A). The KEGG analysis was conducted to illustrate potential biological functions of 4919 DEGs between the two groups. KEGG analysis exhibited that DEGs were enriched in Focal adhesion, Ribosome, Pathways in cancer, apoptosis, and so on, as shown in Bubble Plot and Bar Plot (Figure 4B). The PPI network was composed of the top ten core DEGs and they were UBA52, RPS27A, FBXO15, SOCS1, RBX1, LNX1, CUL1, RNF7, CUL7, FZR1 (Figure 4C). UBA52 and RPS27A in the PPI network ranked first and second, respectively, and both of them were ribosomal proteins. Therefore, we further examined the expression of relevant ribosome mRNAs, and the results revealed that compared to oxaliplatin monotherapy, the vast majority of genes implicated in ribosome assembly were downregulated in the combined group (Figure 4D). To date, only a few studies focused on RPS27A in colon cancer, and these studies solely clarified that RPS27A was overexpressed in colon carcinoma tissues31 and RPS27A knockdown could attenuate apolipoprotein M overexpression-induced CRC cell proliferation.32 However, no literature reported how RPS27A regulated cell proliferation and apoptosis in CRC, so, we got more intense on RPS27A in this study. To further validate the results of RNA-seq, RT-qPCR and WB techniques were implemented to measure the expression of RPS27A in HCT-116 and RKO cells with different drugs treatment. Consistent with the RNA-seq results, exposure to chidamide plus oxaliplatin resulted in an evident decrease of RPS27A expression in both mRNA and protein levels, relative to oxaliplatin monotherapy (Figure 4E and F).
 |
Figure 4 Transcriptomic sequencing of HCT-116 cells after different treatments and validation of the target gene. (A) The heatmap and Volcano map exhibited the DEGs in HCT-116 cells treated with oxaliplatin alone and chidamide plus oxaliplatin for 48 h. 116-C represents HCT-116 cells treated with 8 uM oxaliplatin. 116-D means HCT-116 cells treated with 4 uM chidamide plus 8 uM oxaliplatin. (B) The bubble plot and bar plot showed enriched pathways of DEGs between oxaliplatin monotherapy and the combined treatment via KEGG analysis. (C) The PPI network is composed of the top ten hub DEGs between oxaliplatin monotherapy and the combined treatment. The red gene ranked first and was followed by the Orange and yellow genes in order. (D) DEGs enriched in ribosome pathway. Red represented upregulation and green represented downregulation expression. (E) The RPS27A mRNA levels were quantified by RT-qPCR in HCT-116 and RKO cells treated with chidamide and oxaliplatin single agent and the combined treatment for 48 h. (F) The RPS27A protein levels were determined by WB in HCT-116 and RKO cells treated with chidamide and oxaliplatin single agent and the combined treatment for 48 h. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Abbreviations: NC, negative control (0.1% DMSO); CHI, 4 μM chidamide; OXA, 8 μM oxaliplatin; CHI+OXA, 4 μM chidamide + 8 μM oxaliplatin. |
High RPS27A Expression in CRC
The above observations displayed that chidamide plus oxaliplatin could downregulate the RPS27A expression in both mRNA and protein levels. Then, RT-qPCR and WB were utilized to determine RPS27A expression in CRC cells and normal colorectal epithelial cells. The RT-qPCR and WB results revealed that RPS27A was higher expressed in CRC cells than that in normal colorectal epithelial cells (Figure 5A and B). Then, RPS27A expression in 30 human CRC tissues and matched normal colorectal epithelial tissues were determined by IHC. The IHC results revealed that RPS27A expression was significantly higher in CRC tissues than that in normal colorectal epithelial counterparts (Figure 5C).
 |
Figure 5 RPS27A was highly expressed in both CRC tissues and CRC cells. (A) RPS27A expression in HCT-116, RKO, and NCM-460 cells was quantified via RT-qPCR. (B) RPS27A expression in HCT-116, RKO, and NCM-460 cells was quantified via WB. (C) RPS27A expression in 30 CRC tissues and corresponding normal colorectal tissues were quantified via IHC. ***P<0.001, ****P<0.0001. |
RPS27A Silence Inhibited Proliferation and Promoted Apoptosis in CRC Cells via the MDM2-P53 Pathway
Now that RPS27A was highly expressed in both CRC tissues and CRC cell lines, RPS27A silence was utilized to investigate its biological function in HCT-116 and RKO cells. First of all, RT-qPCR and WB were utilized to quantify the efficiency of siRNA-mediated RPS27A silence. The results of RT-qPCR showed siRNA-2 had a better inhibition efficiency with respect to siRNA-1 and siRNA-3 in HCT-116 and RKO cells (Figure 6A). Then, the results of WB also verified that siRNA-2 could significantly suppress RPS27A protein expression (Figure 6B). Next, CCK-8 assays and flow cytometry were utilized to quantify the alteration of cell proliferation and apoptosis in HCT-116 and RKO cells when RPS27A was knockdown. The CCK-8 results showed that cells’ proliferative ability was significantly inhibited when RPS27A was silenced (Figure 6C). Flow cytometry analysis demonstrated that the apoptotic cell rate increased when RPS27A was silenced (Figure 6D). These results suggested RPS27A might play the role of an oncogene in CRC development and progression. Recently, several studies verified that RPS27A could regulate cancer development via the MDM2-P53 axis.33,34 Next, we investigated whether RPS27A silence could promote HCT-116 and RKO cell apoptosis and inhibit cell proliferation via the MDM2-P53 axis. RT-qPCR and WB were utilized to determine the expression of MDM2 and P53 when RPS27A was silenced. A significant decrease of MDM2 in both mRNA and protein levels was observed, which was in agreement with decreased RPS27A expression, While, the P53 expression in both mRNA and protein levels was upregulated when RPS27A was silenced (Figure 6E and F). All these results exhibited that RPS27A knockdown could inhibit HCT-116 and RKO cell proliferation and promote apoptosis via the MDM2-P53 pathway.
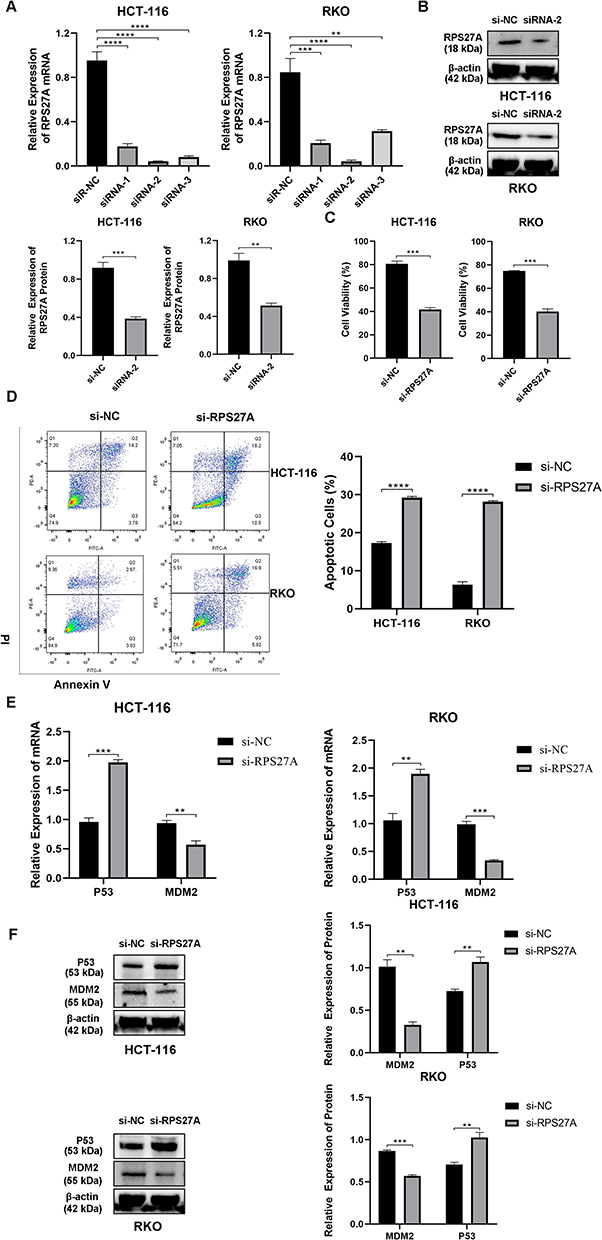 |
Figure 6 RPS27A knockdown inhibited CRC cell proliferation and promoted apoptosis via the MDM2-P53 pathway. (A) RPS27A mRNA expression was quantified by RT-qPCR in HCT-116 and RKO cells transfected with siRNAs targeted RPS27A. (B) RPS27A protein expression was determined by WB in HCT-116 and RKO cells transfected with siRNA-2 targeted RPS27A. (C) CCK-8 displayed the influence that RPS27A silence had on HCT-116 and RKO cell viability. (D) flow cytometry displayed the influence that RPS27A silence had on HCT-116 and RKO cell apoptosis. (E) MDM2 and P53 expression were quantified by RT-qPCR in HCT-116 and RKO cells transfected with siRNA which targeted RPS27A. (F) MDM2 and P53 expression was quantified by WB in HCT-116 and RKO cells transfected with siRNA which targeted RPS27A **P<0.01, ***P<0.001, ****P<0.0001. |
RPS27A Overexpression Partially Reversed Cell Proliferation Inhibition and Apoptosis Induction Resulting from the Combined Treatment of Chidamide with Oxaliplatin via the MDM2-P53 Pathway
Since RPS27A might play a role of an oncogene in CRC development and it was downregulated in the combined treatment of chidamide with oxaliplatin, we wondered whether chidamide plus oxaliplatin exerted the anti-cancer effect through targeting RPS27A in CRC cells. First, the RT-qPCR and WB were used to determine the RPS27A overexpression rate when RPS27A overexpressing plasmid transfected into both HCT-116 and RKO cells. The RT-qPCR and WB results displayed that RPS27A was significantly highly expressed when HCT-116 and RKO cells were transfected with RPS27A overexpressing plasmid (Figure 7A and B). Then, CCK-8 assays and flow cytometry were utilized to evaluate the alteration of cell proliferation and apoptosis when HCT-116 and RKO cells with RPS27A overexpressing plasmid treated with chidamide and oxaliplatin. We found RPS27A overexpression could partially reverse that chidamide-oxaliplatin induced the suppression of cell proliferation and apoptosis (Figure 7C and D). Now that we had verified that RPS27A knockdown could inhibit HCT-116 and RKO cells proliferation and promote apoptosis via the MDM2-P53 axis, and validated that chidamide in combination with oxaliplatin could induce CRC cells apoptosis via downregulation of RPS27A expression, we then analyzed the effect of RPS27A overexpression on MDM2-P53 axis by RT-qPCR and WB. The RT-qPCR results manifested that MDM2 and P53 expression upregulated when HCT-116 and RKO cells with RPS27A overexpressing plasmid treated with chidamide and oxaliplatin (Figure 7E). While, the WB results showed a decrease in MDM2 expression and an increase in P53 expression occurred when RPS27A was overexpressed in HCT-116 and RKO cells which were treated with chidamide plus oxaliplatin (Figure 7F). All the observations supported that chidamide could enhance oxaliplatin-induced anti-tumor effects by downregulating RPS27A expression, and RPS27A downregulation further inhibited the MDM2-P53 signaling axis, which ultimately exerted cell proliferation inhibition and apoptosis induction.
 |
Figure 7 RPS27A overexpression partially reversed chidamide plus oxaliplatin exerted cell proliferation inhibition and apoptosis induction in HCT-116 and RKO cells. (A) RPS27A expression was quantified by RT-qPCR when HCT-116 and RKO cells were transfected with RPS27A overexpression plasmid. (B) RPS27A expression was quantified by WB when HCT-116 and RKO cells were transfected with RPS27A overexpression plasmid. (C) CCK-8 analyzed the influence that RPS27A overexpression had on HCT-116 and RKO cell viability when they were treated with chidamide plus oxaliplatin. (D) flow cytometry analyzed the influence that RPS27A overexpression had on HCT-116 and RKO cell apoptosis when they were treated with chidamide plus oxaliplatin. (E) MDM2 and P53 expression determined by RT-qPCR in HCT-116 and RKO cells transfected with RPS27A overexpression plasmid after the treatment of chidamide plus oxaliplatin for 48 h. (F) MDM2 and P53 expression determined by WB in HCT-116 and RKO cells transfected with RPS27A overexpression plasmid after the treatment of chidamide plus oxaliplatin for 48 h. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
Chidamide and Oxaliplatin Synergistically Inhibited Tumor Growth in vivo
Since chidamide plus oxaliplatin exerted significant growth suppression in HCT-116 and RKO cells in vitro, the anti-tumor activity of the combined treatment in vivo was examined and assessed. Compared to the negative control, chidamide or oxaliplatin alone had a relatively weak suppression on the tumor volume of mice xenograft, however, the significantly decreased growth of tumors could be observed in the oxaliplatin plus chidamide group (Figure 8A). Besides, by the end of drug administration, similar to the tumor volume shrinking, the tumor weight inhibition rate of the combined administration was significantly higher than that of the single agent treatment (Figure 8B). Meanwhile, the calculated CI values of tumor volume and weight alteration after chidamide-oxaliplatin cotreatment were 0.804 and 0.714, respectively, less than 1, which indicated that the combined treatment of chidamide with oxaliplatin exhibited an ideal synergistic anti-CRC effect in vivo. Although the body mass of mice rapidly decreased in the primary stage of the combined treatment, the body weight gain could be detected in subsequent treatment (Figure 8C). All these observations demonstrated that chidamide plus oxaliplatin could synergistically treat CRC in vivo with good tolerance to some extent.
 |
Figure 8 Chidamide and oxaliplatin synergistically inhibited tumor growth in vivo. (A) The influence of chidamide and oxaliplatin monotherapy and the combined treatment had on tumor volume of mice xenograft. (B) The influence of chidamide and oxaliplatin monotherapy and the combined treatment had on tumor weight of mice xenograft. (C) The effects of chidamide and oxaliplatin monotherapy and the combined treatment on mice weight. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Abbreviations: NC, negative control (5% glucose); CHI, 3 mg/kg chidamide; OXA, 5 mg/kg oxaliplatin; CHI+OXA, 3mg/kg chidamide + 5 mg/kg oxaliplatin. |
Discussion
CRC is a kind of common malignant tumor with high incidence and lethality. A majority of CRC patients have been already in the middle-advanced stage at first diagnosis,35 surgical resection is difficult to cure this disease, so, chemotherapy is essential for these individuals. Although oxaliplatin as the first-line chemotherapeutic agent in combination with 5-Fu prolongs the survival period of CRC patients, the serious side effects6 and acquired chemoresistance36 stemming from long-term accumulation of oxaliplatin limit its utilization and lead to treatment discontinuation, which may induce neoplastic progression and relapse. Therefore, it is imperative to develop new regimens against CRC. Chidamide, a novel HDACi, was approved in December 2014 for refractory peripheral T-cell lymphoma in China.37 Chidamide has been discovered to be a potential therapeutic agent for CRC in vitro and in vivo. This research has for the first time identified the synergistic antitumor activity of chidamide plus oxaliplatin on CRC.
Class I HDACs are highly expressed in several cancers and are associated with poor prognosis38 and chemoresistance.39 Besides, inhibition of class I HDACs could lead tumor cells, but not normal cells, to apoptosis.40 It is worth noting that HDAC1,41 HDAC2,42 and HDAC343 are overexpressed in CRC, therefore HDACis targeted Class I HDACs show great interest as candidate therapeutic agents for CRC. HDACis targeted class I HDACs have obtained excellent therapeutic effects in CRC.44,45 It is reported that HDAC10 inhibition could impede the efflux of chemotherapy reagents and enhance DNA damage.46 So, HDACis targeted HDACs 1,2,3,10 may exhibit better synergism with DNA damage drugs, such as platinum drugs, in the treatment of CRC. Coincidentally, chidamide mainly targets for HDACs 1,2,3,10, which prompts us to speculate that coadministration of chidamide and oxaliplatin can enhance the antineoplastic efficacy in CRC. The observations in this research demonstrated that chidamide and oxaliplatin alone could reduce cell activity in a dose-dependent manner and just small doses of chidamide in combination with small doses of oxaliplatin could achieve desired efficacy in vitro and in vivo, the CI value of chidamide and oxaliplatin less than 1, which showed that chidamide plus oxaliplatin had good synergism. Chidamide and radiotherapy synergistically suppress tumor growth in lung cancer.47 Chidamide synergistically enhances cytotoxicity induced by gemcitabine in pancreatic cancer.48 Besides, the results in this study suggested that chidamide could significantly enhance oxaliplatin-induced suppression of cell proliferation, invasion, and migration, and induction of cell apoptosis as well.
In order to clarify how chidamide enhanced oxaliplatin-induced HCT-116 and RKO cell proliferation inhibition and apoptosis, RNA-seq was employed to search for candidate genes. We found that compared with oxaliplatin monotherapy, RPS27A was significantly downregulated in the combined treatment. RPS27A is a ribosomal protein, and it is the component of 40S ribosomal submit.49 RPS27A plays critical roles in tumor progression in addition to its ribosomal functions. RPS27A is overexpressed in several cancers, such as CRC,50 cervical cancer,51 lung adenocarcinoma52 and breast cancer.53 In this research, we also verified RPS27A was highly expressed in CRC tissues and CRC cell lines via IHC, RT-qPCR, and WB experimental techniques. RPS27A often regulates cancer development through the MDM2-P53 axis. RPS27A translocating from the nucleolus to the nucleoplasm could lead to a reduction in MDM2 phosphorylation, which further suppressed MDM2-mediated P53 degradation in P-3F-treated HeLa cells.54 Knockdown of RPS27A induced A549 cell viability inhibition, cell cycle arrest, and apoptosis, and the specific mechanism it actioned is that RPS27A knockdown enhanced the binding between RPL11 and MDM2, which inhibited MDM2-mediated p53 degradation.52 In the present study, RPS27A silenced could inhibit HCT-116 and RKO cell proliferation and promote cell apoptosis, and by mechanism analysis, we found that MDM2 protein decreased and P53 protein increased when RPS27A was knocked down. These observations mentioned above concluded RPS27A, as an oncogene, could regulate CRC development via MDM2-P53 axis.
To further investigate whether chidamide plus oxaliplatin inhibited cell proliferation and induced apoptosis via decreasing RPS27A protein expression, RPS27A overexpression plasmids were transfected into HCT-116 and RKO cells treated with chidamide plus oxaliplatin, then CCK-8 and flow cytometry were carried out to analyze the alteration in proliferation and apoptosis of CRC cells. We found that RPS27A overexpression could partially reverse chidamide plus oxaliplatin induced inhibition of HCT-116 and RKO cell proliferation and promotion of cell apoptosis. Next, we explored whether the combined treatment of chidamide with oxaliplatin inhibited CRC cell proliferation and induced apoptosis via the regulation of the MDM2-P53 pathway. The mRNA of both MDM2 and P53 were up-regulated when RPS27A was highly expressed in the combined treatment. While, the level of MDM2 protein was in line with RPS27A protein, and P53 protein was in contrast to the expression of RPS27A protein. These observations indicated that RPS27A could regulate MDM2 expression at the posttranscriptional level rather than the transcriptional level. All these results showed that chidamide plus oxaliplatin could suppress CRC cell proliferation and promote cell apoptosis by the suppression of the RPS27A-MDM2-P53 pathway. What we all know is that not all CRC patients can benefit from every regimen, even though the frontline treatment options, so we should stratify these patients and search for the ones who can get more benefits from the administration of chidamide plus oxaliplatin. According to our compelling data in vitro, we speculated that CRC patients with high expression of RPS27A and wild-type p53 might get more benefits from the combined treatment of chidamide and oxaliplatin to CRC in future clinical trials.
Nevertheless, there are some limitations in this study. Firstly, we utilized subcutaneous models to prove that chidamide could synergistically enhance oxaliplatin-induced inhibition of CRC growth in vivo, however, the ideal orthotopic models were neglected by us, which were appropriate in the translational setting. Secondly, our study only investigated the mechanism of relatively high concentration of chidamide synergistically enhancing oxaliplatin-induced suppression of CRC cell proliferation and induction of cell apoptosis, while we did not explore the mechanism of relatively low concentration of chidamide antagonistically oxaliplatin-induced inhibiting on the growth of CRC cells. Finally, we did not evaluate the therapeutic efficacy of sequential administration of chidamide and oxaliplatin on CRC cells, but explored the effects that chidamide plus oxaliplatin had on it in vitro. In view of the shortcomings in this study, our team members will explore the anti-tumor effects of chidamide plus oxaliplatin on CRC orthotopic models and the therapeutic effect of sequential administration of chidamide and oxaliplatin on CRC cells in the future.
Conclusions
Chidamide and oxaliplatin acted synergistically to suppress CRC growth in vitro and in vivo. Chidamide could enhance oxaliplatin-induced inhibition in cell invasion and migration, and promotion of cell apoptosis. The whole-transcriptome RNA sequencing displayed that RPS27A was significantly downregulated in HCT-116 cells treated with chidamide and oxaliplatin. RPS27A was highly expressed in CRC and RPS27A knockdown could suppress cell proliferation and induce apoptosis via MDM2-P53 axis. RPS27A overexpression could partially reverse chidamide plus oxaliplatin induced inhibition in cell proliferation and promotion of cell apoptosis by the inhibition of the RPS27A-MDM2-P53 pathway. This research provided theoretical support for clinical practice of chidamide plus oxaliplatin against CRC.
Ethics Approval and Informed Consent
The procedures related to human tissues from CRC patients in this research was approved by the Medical Ethics Committee of The First Affiliated Hospital of Kunming Medical University (approval No. 2022. L. 56). All procedures involving human individuals in our study were performed in line with the Declaration of Helsinki. All procedures on mice were operated in accordance with the National Institutes of Health (NIH) regulations related to the use and care of experimental animals, which were granted by the Animal Care Ethics Committee of Kunming Medical University (No. KMMU20220887).
Consent for Publication
All authors of the present study have carefully read the final manuscript and agreed to publish it in your journal.
Acknowledgments
Zhaopeng Li and Deyong Bu are co-first authors for this study. We are extremely thankful to the teachers of Department of Gastrointestinal Surgery who provided histological samples from CRC patients for us. We also highly acknowledge the Transcriptomic Platform providing by Kunming Institute of zoology, Chinese academy of sciences for cDNA libraries sequencing.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Clinical Research Center for Geriatric Diseases of Yunnan Province - diagnosis and treatment of geriatric comorbidity and clinical translational research. (No. 202102AA310069).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Muller P, Woods L, Walters S. Temporal and geographic changes in stage at diagnosis in England during 2008-2013: a population-based study of colorectal, lung and ovarian cancers. Cancer Epidemiol. 2020;67:101743. doi:10.1016/j.canep.2020.101743
3. Arango D, Wilson AJ, Shi Q, et al. Molecular mechanisms of action and prediction of response to oxaliplatin in colorectal cancer cells. Br J Cancer. 2004;91(11):1931–1946. doi:10.1038/sj.bjc.6602215
4. Martinez-Balibrea E, Martinez-Cardus A, Gines A, et al. Tumor-related molecular mechanisms of oxaliplatin resistance. Mol Cancer Ther. 2015;14(8):1767–1776. doi:10.1158/1535-7163.MCT-14-0636
5. Prutianu I, Alexa-Stratulat T, Cristea EO, et al. Oxaliplatin-induced neuropathy and colo-rectal cancer patient’s quality of life: practical lessons from a prospective cross-sectional, real-world study. World J Clin Cases. 2022;10(10):3101–3112. doi:10.12998/wjcc.v10.i10.3101
6. Wei G, Gu Z, Gu J, et al. Platinum accumulation in oxaliplatin-induced peripheral neuropathy. J Peripher Nerv Syst. 2021;26(1):35–42. doi:10.1111/jns.12432
7. Noordhuis P, Laan AC, van de Born K, Honeywell RJ, Peters GJ. Coexisting molecular determinants of acquired oxaliplatin resistance in human colorectal and ovarian cancer cell lines. Int J Mol Sci. 2019;20(15):5676.
8. Almeida LO, Neto PC, Sousa M. SET oncoprotein accumulation regulates transcription through DNA demethylation and histone hypoacetylation. Oncotarget. 2017;8(16):26802–26818. doi:10.18632/oncotarget.15818
9. Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med. 2016;6:56.
10. Lee SH, Yoo C, Im S, Jung JH, Choi HJ, Yoo J. Expression of histone deacetylases in diffuse large B-cell lymphoma and its clinical significance. Int J Med Sci. 2014;11(10):994–1000. doi:10.7150/ijms.8522
11. Weichert W, Roske A, Niesporek S, et al. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008;14(6):1669–1677. doi:10.1158/1078-0432.CCR-07-0990
12. Liu C, Lv D, Li M, et al. Hypermethylation of miRNA-589 promoter leads to upregulation of HDAC5 which promotes malignancy in non-small cell lung cancer. Int J Oncol. 2017;50(6):2079–2090. doi:10.3892/ijo.2017.3967
13. Linares A, Assou S, Lapierre M, et al. Increased expression of the HDAC9 gene is associated with antiestrogen resistance of breast cancers. Mol Oncol. 2019;13(7):1534–1547. doi:10.1002/1878-0261.12505
14. Ler SY, Leung CH, Khin LW, et al. HDAC1 and HDAC2 independently predict mortality in hepatocellular carcinoma by a competing risk regression model in a Southeast Asian population. Oncol Rep. 2015;34(5):2238–2250. doi:10.3892/or.2015.4263
15. Minamiya Y, Ono T, Saito H, et al. Strong expression of HDAC3 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Tumour Biol. 2010;31(5):533–539. doi:10.1007/s13277-010-0066-0
16. Zhang K, Liu Z, Yao Y, et al. Structure-Based Design of a Selective Class I Histone Deacetylase (HDAC) Near-Infrared (NIR) Probe for Epigenetic Regulation Detection in Triple-Negative Breast Cancer (TNBC). J Med Chem. 2021;64(7):4020–4033. doi:10.1021/acs.jmedchem.0c02161
17. Jordaan G, Liao W, Sharma S. E-cadherin gene re-expression in chronic lymphocytic leukemia cells by HDAC inhibitors. BMC Cancer. 2013;13:67.
18. Shankar E, Pandey M, Verma S, et al. Role of class I histone deacetylases in the regulation of maspin expression in prostate cancer. Mol Carcinog. 2020;59(8):955–966. doi:10.1002/mc.23214
19. Xie Y, Tang P, Xing X, et al. In situ exploring Chidamide, a histone deacetylase inhibitor, induces molecular changes of leukemic T-lymphocyte apoptosis using Raman spectroscopy. Spectrochim Acta A Mol Biomol Spectrosc. 2020;241:118669. doi:10.1016/j.saa.2020.118669
20. Liu L, Chen B, Qin S, et al. A novel histone deacetylase inhibitor Chidamide induces apoptosis of human colon cancer cells. Biochem Biophys Res Commun. 2010;392(2):190–195. doi:10.1016/j.bbrc.2010.01.011
21. Wang H, Liu YC, Zhu CY, et al. Chidamide increases the sensitivity of refractory or relapsed acute myeloid leukemia cells to anthracyclines via regulation of the HDAC3 -AKT-P21-CDK2 signaling pathway. J Exp Clin Cancer Res. 2020;39(1):278. doi:10.1186/s13046-020-01792-8
22. Que Y, Zhang XL, Liu ZX, et al. Frequent amplification of HDAC genes and efficacy of HDAC inhibitor chidamide and PD-1 blockade combination in soft tissue sarcoma. J Immunother Cancer. 2021;9(2):98.
23. Tu K, Yu Y, Wang Y, et al. Combination of Chidamide-Mediated Epigenetic Modulation with Immunotherapy: boosting Tumor Immunogenicity and Response to PD-1/PD-L1 Blockade. ACS Appl Mater Interfaces. 2021;13(33):39003–39017. doi:10.1021/acsami.1c08290
24. He Y, Jiang D, Zhang K, et al. Chidamide, a subtype-selective histone deacetylase inhibitor, enhances Bortezomib effects in multiple myeloma therapy. J Cancer. 2021;12(20):6198–6208. doi:10.7150/jca.61602
25. Ding N, You A, Tian W, Gu L, Deng D. Chidamide increases the sensitivity of Non-small Cell Lung Cancer to Crizotinib by decreasing c-MET mRNA methylation. Int J Biol Sci. 2020;16(14):2595–2611. doi:10.7150/ijbs.45886
26. Li X, Yuan X, Wang Z, et al. Chidamide Reverses Fluzoparib Resistance in Triple-Negative Breast Cancer Cells. Front Oncol. 2022;12:819714. doi:10.3389/fonc.2022.819714
27. Liu L, Qiu S, Liu Y, et al. Chidamide and 5-flurouracil show a synergistic antitumor effect on human colon cancer xenografts in nude mice. Neoplasma. 2016;63(2):193–200. doi:10.4149/203_150422N214
28. Zhou Y, Pan DS, Shan S, et al. Non-toxic dose chidamide synergistically enhances platinum- induced DNA damage responses and apoptosis in Non-Small-Cell lung cancer cells. Biomed Pharmacother. 2014;68(4):483–491. doi:10.1016/j.biopha.2014.03.011
29. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–681. doi:10.1124/pr.58.3.10
30. Duarte D, Vale N. Evaluation of synergism in drug combinations and reference models for future orientations in oncology. Curr Res Pharmacol Drug Discov. 2022;3:100110. doi:10.1016/j.crphar.2022.100110
31. Barnard GF, Mori M, Staniunas RJ, et al. Ubiquitin fusion proteins are overexpressed in colon cancer but not in gastric cancer. Biochim Biophys Acta. 1995;1272(3):147–153. doi:10.1016/0925-4439(95)00079-8
32. Mu Q, Luo G, Wei J, et al. Apolipoprotein M promotes growth and inhibits apoptosis of colorectal cancer cells through upregulation of ribosomal protein S27a. Excli j. 2021;20:145–159. doi:10.17179/excli2020-2867
33. Luo J, Zhao H, Chen L, Liu M. Multifaceted functions of RPS27a: an unconventional ribosomal protein. J Cell Physiol. 2022. doi:10.1002/jcp.30941
34. Wang H, Zhao J, Yang J, et al. PICT1 is critical for regulating the Rps27a-Mdm2-p53 pathway by microtubule polymerization inhibitor against cervical cancer. Biochimica et Biophys Acta Mol Cell Res. 2021;1868(10):119084. doi:10.1016/j.bbamcr.2021.119084
35. Cardoso R, Guo F, Heisser T, et al. Colorectal cancer incidence, mortality, and stage distribution in European countries in the colorectal cancer screening era: an international population-based study. Lancet Oncol. 2021;22(7):1002–1013. doi:10.1016/s1470-2045(21)00199-6
36. Li Y, Gan Y, Liu J, et al. Downregulation of MEIS1 mediated by ELFN1-AS1/EZH2/DNMT3a axis promotes tumorigenesis and oxaliplatin resistance in colorectal cancer. Signal Transduct Target Ther. 2022;7(1):87. doi:10.1038/s41392-022-00902-6
37. Shi Y, Dong M, Hong X, et al. Results from a multicenter, open-label, pivotal Phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann Oncol. 2015;26(8):1766–1771. doi:10.1093/annonc/mdv237
38. Mithraprabhu S, Kalff A, Chow A, Khong T, Spencer A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics. 2014;9(11):1511–1520. doi:10.4161/15592294.2014.983367
39. Wang W, Zhao M, Cui L, et al. Characterization of a novel HDAC/RXR/HtrA1 signaling axis as a novel target to overcome cisplatin resistance in human non-small cell lung cancer. Mol Cancer. 2020;19(1):134. doi:10.1186/s12943-020-01256-9
40. Delcuve GP, Khan DH, Davie JR. Targeting class I histone deacetylases in cancer therapy. Expert Opin Ther Targets. 2013;17(1):29–41. doi:10.1517/14728222.2013.729042
41. Xiong W, Yang S, Zhang W, Chen Y, Wang F. MiR-761 inhibits colorectal cancer cell proliferation and invasion through targeting HDAC1. Pharmazie. 2019;74(2):111–114. doi:10.1691/ph.2019.8756
42. Ye P, Xing H, Lou F, et al. Histone deacetylase 2 regulates doxorubicin (Dox) sensitivity of colorectal cancer cells by targeting ABCB1 transcription. Cancer Chemother Pharmacol. 2016;77(3):613–621. doi:10.1007/s00280-016-2979-9
43. Li J, Hu M, Liu N, et al. HDAC3 deteriorates colorectal cancer progression via microRNA-296-3p/TGIF1/TGFβ axis. J Exp Clin Cancer Res. 2020;39(1):248. doi:10.1186/s13046-020-01720-w
44. Zhijun H, Shusheng W, Han M, Jianping L, Li-Sen Q, Dechun L. Pre-clinical characterization of 4SC-202, a novel class I HDAC inhibitor, against colorectal cancer cells. Tumour Biol. 2016;37(8):10257–10267. doi:10.1007/s13277-016-4868-6
45. Lamoine S, Cumenal M, Barriere DA, et al. The Class I HDAC Inhibitor, MS-275, Prevents Oxaliplatin-Induced Chronic Neuropathy and Potentiates Its Antiproliferative Activity in Mice. Int J Mol Sci. 2021;23(1):98.
46. Ridinger J, Koeneke E, Kolbinger FR, et al. Dual role of HDAC10 in lysosomal exocytosis and DNA repair promotes neuroblastoma chemoresistance. Sci Rep. 2018;8(1):10039. doi:10.1038/s41598-018-28265-5
47. Huang X, Bi N, Wang J, et al. Chidamide and Radiotherapy Synergistically Induce Cell Apoptosis and Suppress Tumor Growth and Cancer Stemness by Regulating the MiR-375-EIF4G3 Axis in Lung Squamous Cell Carcinomas. J Oncol. 2021;2021:4936207. doi:10.1155/2021/4936207
48. Qiao Z, Ren S, Li W, et al. Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells. Biochem Biophys Res Commun. 2013;434(1):95–101. doi:10.1016/j.bbrc.2013.03.059
49. Sun XX, DeVine T, Challagundla KB, Dai MS. Interplay between ribosomal protein S27a and MDM2 protein in p53 activation in response to ribosomal stress. J Biol Chem. 2011;286(26):22730–22741. doi:10.1074/jbc.M111.223651
50. Wong JM, Mafune K, Yow H, et al. Ubiquitin-ribosomal protein S27a gene overexpressed in human colorectal carcinoma is an early growth response gene. Cancer Res. 1993;53(8):1916–1920.
51. Wang Q, Cai Y, Fu X, Chen L. High RPS27A Expression Predicts Poor Prognosis in Patients With HPV Type 16 Cervical Cancer. Front Oncol. 2021;11:752974. doi:10.3389/fonc.2021.752974
52. Li H, Zhang H, Huang G, et al. Loss of RPS27a expression regulates the cell cycle, apoptosis, and proliferation via the RPL11-MDM2-p53 pathway in lung adenocarcinoma cells. J Exp Clin Cancer Res. 2022;41(1):33. doi:10.1186/s13046-021-02230-z
53. Rimal R, Desai P, Marquez AB, Sieg K, Marquardt Y, Singh S. 3-D vascularized breast cancer model to study the role of osteoblast in formation of a pre-metastatic niche. Sci Rep. 2021;11(1):21966. doi:10.1038/s41598-021-01513-x
54. Wang H, Feng J, Zhou T, Wei L, Zhou J. P-3F, a microtubule polymerization inhibitor enhances P53 stability through the change in localization of RPS27a. Int J Biochem Cell Biol. 2017;92:53–62. doi:10.1016/j.biocel.2017.09.010
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.