")
Back to Journals » Drug Design, Development and Therapy » Volume 17
Chemistry, Biosynthesis and Pharmacology of Streptonigrin: An Old Molecule with Future Prospects for New Drug Design, Development and Therapy
Authors Nabihah Nasir N, Sekar M , Ravi S, Wong LS , Sisinthy SP, Gan SH , Subramaniyan V , Chidambaram K , Mat Rani NNI, Begum MY , Ramar M , Safi SZ, Selvaraj S , Chinna Maruthu SK , Fuloria S, Fuloria NK, Lum PT, Djearamane S
Received 7 September 2022
Accepted for publication 20 January 2023
Published 8 April 2023 Volume 2023:17 Pages 1065—1078
DOI https://doi.org/10.2147/DDDT.S388490
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Naurah Nabihah Nasir,1 Mahendran Sekar,2,3 Subban Ravi,4 Ling Shing Wong,5 Sreenivas Patro Sisinthy,6 Siew Hua Gan,2 Vetriselvan Subramaniyan,7 Kumarappan Chidambaram,8 Nur Najihah Izzati Mat Rani,6 M Yasmin Begum,9 Mohankumar Ramar,10 Sher Zaman Safi,11 Siddharthan Selvaraj,12 Senthil Kumar Chinna Maruthu,13 Shivkanya Fuloria,14 Neeraj Kumar Fuloria,14 Pei Teng Lum,1 Sinouvassane Djearamane15
1Department of Pharmaceutical Chemistry, Faculty of Pharmacy and Health Sciences, Royal College of Medicine Perak, Universiti Kuala Lumpur, Ipoh, Perak, 30450, Malaysia; 2School of Pharmacy, Monash University Malaysia, Bandar Sunway, Selangor, 47500, Malaysia; 3Center for Transdisciplinary Research, Department of Pharmacology, Saveetha Institute of Medical and Technical Sciences, Saveetha Dental College and Hospital, Saveetha University, Chennai, Tamil Nadu, 600077, India; 4Department of Chemistry, Karpagam Academy of Higher Education, Coimbatore, Tamil Nadu, 641021, India; 5Faculty of Health and Life Sciences, INTI International University, Nilai, 71800, Malaysia; 6Faculty of Pharmacy and Health Sciences, Royal College of Medicine Perak, Universiti Kuala Lumpur, Ipoh, Perak, 30450, Malaysia; 7Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia, Bandar Sunway, Subang Jaya, Selangor, Malaysia; 8Department of Pharmacology, College of Pharmacy, King Khalid University, Abha, 62529, Saudi Arabia; 9Department of Pharmaceutics, College of Pharmacy, King Khalid University, Abha, 61421, Saudi Arabia; 10Department of Surgical Research, Rhode Island Hospital, Alpert Medical School, Brown University, Providence, RI, 02903, USA; 11Faculty of Medicine, Bioscience and Nursing, MAHSA University, Jenjarom, Selangor, 42610, Malaysia; 12Faculty of Dentistry, AIMST University, Bedong, Kedah, 08100, Malaysia; 13Department of Pharmacology, Karpagam College of Pharmacy, Coimbatore, Tamilnadu, 641032, India; 14Faculty of Pharmacy, AIMST University, Bedong, Kedah, 08100, Malaysia; 15Department of Biomedical Science, Faculty of Science, Universiti Tunku Abdul Rahman, Kampar, Perak, 31900, Malaysia
Correspondence: Ling Shing Wong, Faculty of Health and Life Sciences, INTI International University, Nilai, 71800, Malaysia, Tel +6014 – 3034057, Email [email protected] Sinouvassane Djearamane, Department of Biomedical Science, Faculty of Science, Universiti Tunku Abdul Rahman, Kampar, 31900, Perak, Malaysia, Tel +6016 – 4037685, Email [email protected]
Abstract: Streptonigrin is an aminoquinone alkaloid isolated from Streptomyces flocculus and is gaining attention as a drug molecule owing to its potential antitumor and antibiotic effects. It was previously used as an anticancer drug but has been discontinued because of its toxic effects. However, according to the most recent studies, the toxicity of streptonigrin and its structurally modified derivatives has been reduced while maintaining their potential pharmacological action at lower concentrations. To date, many investigations have been conducted on this molecule and its derivatives to determine the most effective molecule with low toxicity to enable new drug discovery. Therefore, the main objective of this study is to provide a comprehensive review and to discuss the prospects for streptonigrin and its derived compounds, which may boost the molecule as a highly interesting target molecule for new drug design, development and therapy. To complete this review, relevant literature was collected from several scientific databases, including Google Scholar, PubMed, Scopus and ScienceDirect. Following a complete screening, the obtained information is summarized in the present review to provide a good reference and accelerate the development and utilization of streptonigrin and its derivatives as pharmaceuticals.
Keywords: streptonigrin, chemistry, biosynthesis, pharmacology, anticancer, antibacterial
Graphical Abstract:
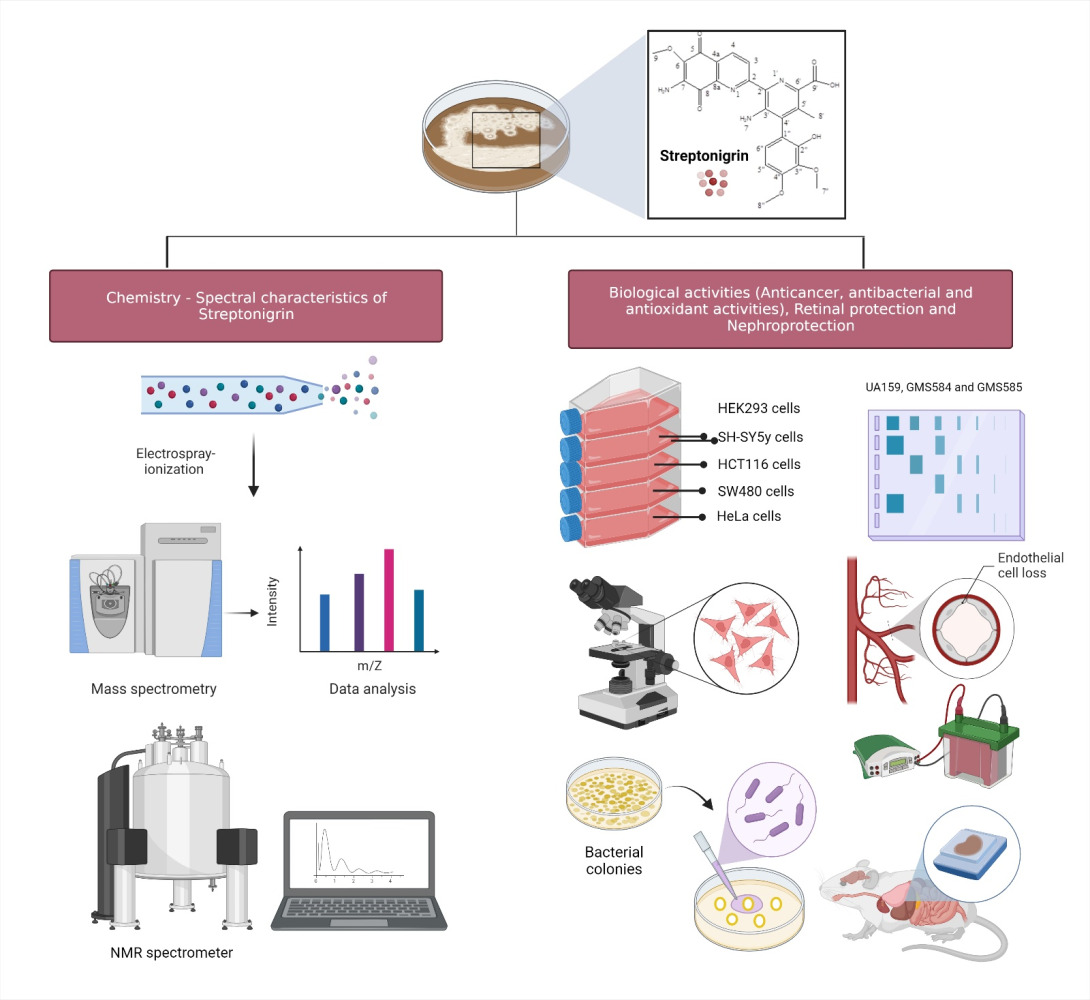
Introduction
Streptonigrin is a metabolite of the bacterium Streptomyces flocculus which was first isolated in 1959 by Rao and Cullen using a combination of spectral and degradation methods.1 Since the initial determination of its structure, streptonigrin has been the subject of extensive synthetic, biosynthetic and biological investigations, which have resulted in continued interest in its confirmed potential biological properties (Figure 1). Subsequently, the structure of the isolated metabolite was further investigated by Rao, Biemann and Woodward in 1963,1 through a series of spectroscopic and chemical degradation studies. The data gathered by previous researchers were further confirmed by a study conducted by Chiu and Lipscomb in 1975 via X-ray crystallographic analysis.2 Soon afterwards, as streptonigrin was demonstrated to possess a broad spectrum of anticancer activity, clinical trials of streptonigrin were conducted in the late 1970s, to measure its ability to treat various medical conditions, and to monitor their safety and possible side effects. However, the studies stopped at phase II clinical trials owing to the unavoidable toxicity demonstrated by streptonigrin.1 In addition, a study carried out Chiu and Lipscomb in 1975 revealed that streptonigrin caused several undesirable side effects, including severe bone marrow depression, nausea, vomiting, diarrhea and alopecia.2 It is plausible that streptonigrin 1) causes chromosomal damage when added to cultures of human leukocytes, 2) inhibits DNA synthesis in tissue culture cells as well as in bacteria, and 3) inhibits partial RNA and protein synthesis.2 However, the investigation into determining the best pathways for the synthesis for streptonigrin did not stop there and was continued by Boger,3 who successfully modified its structure by implementing an inverse electron demand Diels–Alder reaction of electron-deficient heterocyclic azadienes.
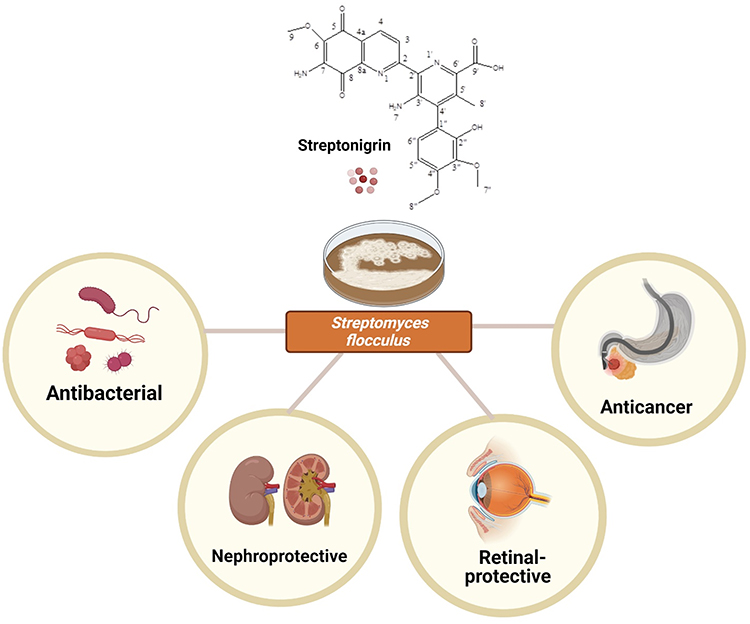 |
Figure 1 Role of streptonigrin in the management of various diseases. Notes: Created with BioRender.com. |
Streptonigrin has been confirmed to have structurally and biosynthetically related antitumor and antibiotic properties. Therefore, several structure–activity investigations involving streptonigrin and a series of truncated analogues, as well as genome scanning studies, have led to a proposed model of biosynthesis and established the cytotoxic mode of action. Dreyton et al4 have developed a structure–activity relationship for streptonigrin that provides unique insights into streptonigrin-induced inactivation of the peptidyl arginine deiminases (PADs). Since then, researchers’ interest in the impact of PADs in facilitating the inhibitory action of streptonigrin has increased, especially regarding its significant anticancer effects against several types of cancer, as well as in retinal-protective5 and nephroprotective agents.6,7
Quinones constitute a structurally diverse class of phenolic compounds with a wide range of pharmacological properties, which are the basis for different applications in the broad field of pharmacy and medicine. In traditional medicine all over the world, plants that are rich in quinones are used for the treatment of a variety of diseases.8–17
Therefore, in this review, we focus on providing an up-to-date overview of the chemistry, biosynthesis and biological properties of streptonigrin, in order to increase the utility of the molecule as an intriguing target molecule for new drug design, development and therapy.
Chemistry of Streptonigrin
Spectral Characteristics of Streptonigrin
Streptonigrin (Figure 2), with a melting point (m.p.) of 275°C, showed a pseudo-molecular ion peak at a mass/charge ratio (m/z) of 529.135 [M+Na]+ ion in positive-mode electrospray-ionization mass spectrometry (ESI-MS) and at m/z 506.1443 in MS (negative ion NH3 chemical ionization). It was analyzed and determined to have the molecular formulae C25H22N4O8. Streptonigrin has a maximum ultraviolet (UV) absorption in methanol at 248 nm (e 38,400) and 375–380 nm (e 17,400). It is a monobasic acid with a pKa of between 6.2 and 6.4 in dioxane:water.
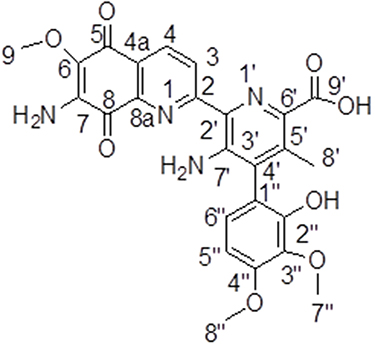 |
Figure 2 Chemical structure of streptonigrin. |
The 1H-nuclear magnetic resonance (1H-NMR) spectrum exhibited four singlets in the upfield region [at δ 2.38 (s, 3H), 3.84 (s, 3H), 3.88 (s, 3H) and 3.97 (s, 3H)]. The four singlets were attributed to H-8ʹ, H-7″, H-8″ and H-9, respectively. It also exhibited four doublets in the downfield aromatic region [at δ 6.68 (d, J=7.5 Hz, 1H), 6.72 (d, J=7.4 Hz, 1H), 8.37 (d, J=8.4 Hz, 1H), 9.01 (d, J=8.38 Hz, 1H)], which were attributed to H-5″, H-6″, H-4 and H-3 respectively. In addition, two broad singlets at δ 6.27 for two protons attributable to the H-10 amino group and 8.18 for one phenolic proton were also observed.
The proton assignments were further supported by a nuclear Overhauser effect spectroscopy (NOESY) experiment. The aromatic proton H-5″ was assigned from an NOE to the adjacent methoxy group H-8″. Hence, the second aromatic proton at δ 6.72 was assigned to H-6”, which was confirmed by an NOE to the methyl group on ring C. Differentiation of the remaining two methoxy signals at δ 3.84 and 3.97 was achieved by correlating ring “A” amino protons, H-7a/H-7b, and the signal at δ 3.92 at low temperature. However, when the spectrum was run at 300 K, the H-7 amino protons were not visible. Nevertheless, when the experiment was repeated at 240 K, where the rate of rotation around the biaryl bond between rings B and C was slow and the amino protons on ring C, H-7′c and H-7′d were sharp, well-separated signals at δ 5.66 and 10.08 ppm, respectively, were seen.18
The 13C-NMR spectrum of streptonigrin contained 25 distinct signals. A heteronuclear single quantum coherence (HSQC) experiment was conducted to assign the methyl carbon C-8’ (δ 17.5), the methoxy carbons C-9 (δ 60.3), C-7″ (δ 60.7) and C-8″ (δ 56.1), and the aromatic carbons C-3 (δ 126.3), C-4 (δ 134.1), C-5″ (δ 105.2) and C-6″ (δ 125.4). Nevertheless, owing to the presence of only four aromatic protons, the experiment yielded only limited information. Therefore, additional experiments on heteronuclear multiple bond correlation (HMBC) were further optimized to detect long-range couplings. The quaternary phenyl ring carbons C-1″ (δ 115.8), C-2″ (δ 149.6), C-3″ (δ 138.4) and C-4″ (δ 154.5) were assigned through HMBC correlations from the aromatic protons H-5″ and H-6″, the two methoxy signals from C-7″ and C-8″, the hydroxy and the methyl protons in the three HMBC experiments. Carbons C-3″ and C-4″ were easily distinguished based on the strong correlations with the methoxy groups substituted on these carbons, while C-1″ and C-2″ both had the expected long-range correlations with H-5″, H-6″, H-8″ and OH. The carbons C-6 (δ 137.5), C-7 (δ 141.6) and C-5″ (δ 138.9) were assigned via HMBC correlations with the methoxy (H-9, δ 3.97), amino (H-7, δ 5.66 and 10.08) and methyl groups (H-8’, δ 2.38) substituted at each of these carbons, respectively.
The three most downfield peaks were assigned as the three carbonyl carbons C-5 (δ 177.0), C-8 (δ 181.0) and C-9’ (δ 165.8). The carbonyl carbon at δ 165.8 was assigned to C-9’ on the basis of cross peaks with the adjacent methyl carbon C-8’. The two carbonyl carbons in ring A possess similar chemical shifts where both signals were correlated to H-3, H-4 as well as the amino protons H-a/H-b. These signals were assigned based on the relative intensity of the correlations; in particular, the strong correlations between C-8 and the adjacent amino protons, H-a/H-b. Assignment of the more downfield carbon resonance as C-8 is consistent with hydrogen bonding to the adjacent amino group.
Furthermore, the carbons C-4a (δ 127.9) and C-8a (δ 145.4) from the quinoline dione portion of streptonigrin were assigned based on the strong correlations observed from the three-bond couplings between C-4a and H-3 as well as C-8a and H-4. Carbon C-2 (δ 161.2) also showed strong coupling to H-3 and H-4, in addition to a very weak correlation with the methyl signal at C-8ʹ. The final four signals were assigned as follows: C-2ʹ at δ 130.5 via cross peaks with H-3, H-4 and the methyl protons H-8’; C-4’ as δ 135.6 with cross peaks to H-5″, H-6″ and H-8’; C-6ʹ as δ 133.7 from correlations with the methyl protons H-8’ and, weakly, the hydroxyl; and finally, C-3’ at δ 147.9 was assigned owing to an extremely weak correlation with methyl protons H-8’.18
In the 15N-NMR spectra (40.5 MHz), streptonigrin showed four nitrogen resonances, from the two amino groups on rings A and C as well as the two pyridyl nitrogens on rings B and C. The 15N resonances were obtained at natural abundance from HSQC and long-range HMBC experiments. The aromatic nitrogens N-1 (δ 287.5 ppm) and N-10 (δ 299.0 ppm) were assigned from HMBC experiments based on the correlations to H-3 and the methyl group on ring C, respectively. The amino nitrogens that were assigned from HSQC experiments were optimized to detect single-bond N–H coupling. When the spectrum was recorded at 300 K, it indicated a resonance assigned to N-10 (δ 60.5 ppm), although no signal was detected for N-7′. The failure to detect the second nitrogen signal was attributed to the weak and significantly exchange-broadened signal from the amino protons H-c and H-d. This amino group is involved in stabilization of a coplanar arrangement of rings A, B and C owing to the formation of an intramolecular hydrogen bond to the ring B pyridyl nitrogen. When the spectrum was recorded at 240 K, where the rate of rotation around the biaryl bond between rings B and C is slow, it allowed the detection of N-10 (δ 65.0 ppm) and N-7′ (δ 75.0 ppm). These chemical shifts are in the range previously seen for nitrogens in analogous systems.18
Biosynthesis of Streptonigrin
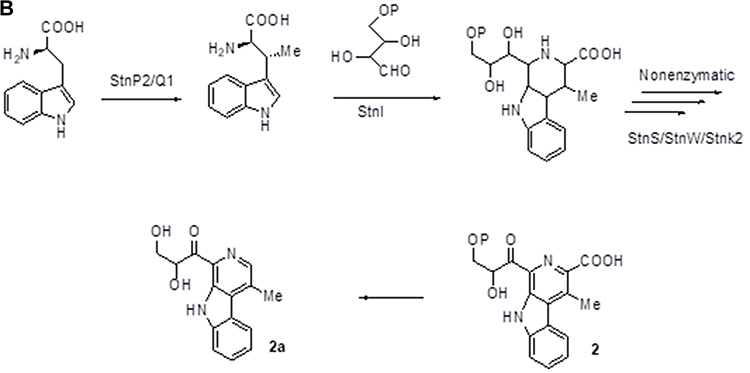
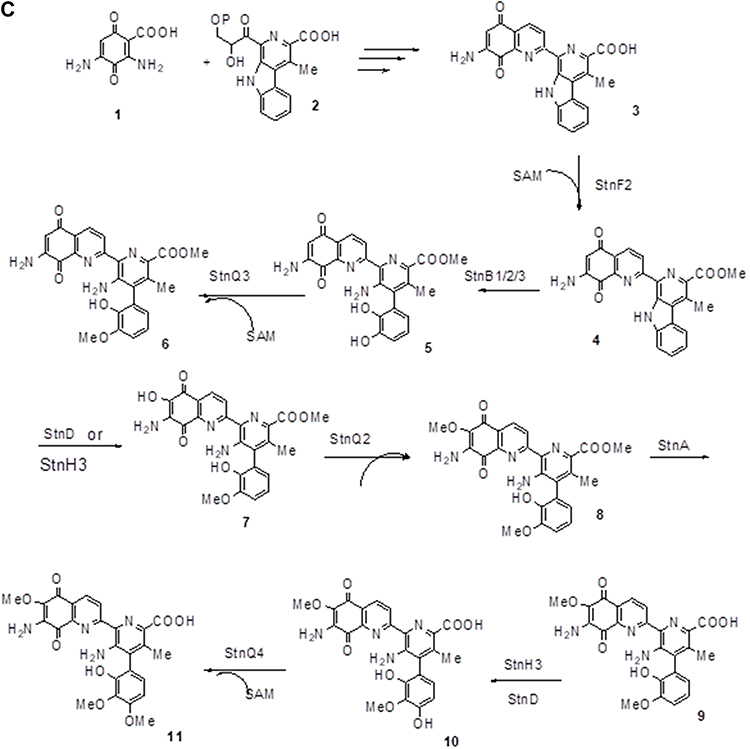
Figure 3A–Csummarizes the current genetic, chemical and biochemical data, along with bioinformatics analysis of the biosynthetic pathway for streptonigrin. Recently, the stn gene cluster responsible for the biosynthesis of streptonigrin has been cloned and has been found to contain 48 genes. Lavendamycin containing a β-carboline moiety has been isolated from the mutant derived from the inactivation of stnB1, encoding a dioxygenase, and confirmed to be an intermediate of the biosynthesis of streptonigrin, based on both genetic and biochemical studies.18
Figure 3 Continued. Figure 3 (A) Biosynthesis of the key intermediate 1 for streptonigrin. (B) Biosynthesis of the intermediate 2 for streptonigrin. (C) Final step in the biosynthesis of streptonigrin.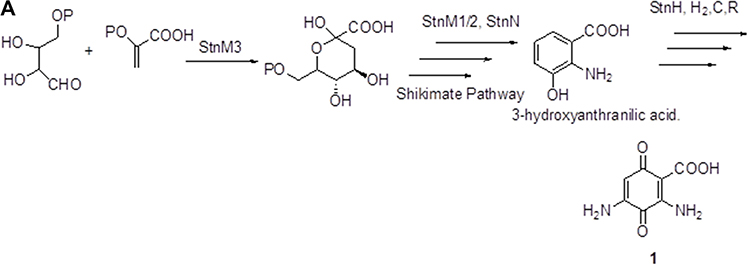
Biological Activities of Streptonigrin
Anticancer Activity
Several studies have confirmed the efficacy of streptonigrin at 0.2 mg/kg against renal cancer, purported to occur through the inhibition of transglutaminase-2.19 Streptonigrin (100 nM) has also been confirmed to be effective against pancreatic cancer at a plating efficiency of 0.25±0.02 in calcium carbonate cells, while at a concentration as low as 0.04 μM, it was highly effective against melanoma cells.20 Ambaye et al21 identified streptonigrin as a small molecule inhibitor of biogenic selenium nanoparticles (SENPs) with higher inhibitory effect against SUMO-specific protease-1 (SENP1) (IC50=0.518±0.100 μM) compared to SENP2 (IC50=6.919±0.676 μM), and SENP6 activity with similar potency to that of SENP2 (IC50=5.205±0.853 μM). The findings indicate that streptonigrin has a higher potency in inhibiting SENP1 relative to other SENPs.21
The ubiquitin-like processing protease (Ulp)/SENP family members are predominantly concentrated in distinct subnuclear regions.22 Wang et al,23 who generated SENP1 knockout mice to delineate the contribution of SENP1, demonstrated that SENP1 contributes to the formation of secondary tumors in the bone and is therefore important in metastatic prostate cancer.23 In fact, inactivation of the SENP1 gene causes severe fetal anemia in mid-gestation, indicating the important physiological role of SENP1 in the hypoxic response through the regulation of HIF1a stability and the fact that SUMOylation can also target a protein for both ubiquitination and degradation.24 Therefore, one of the mechanisms of action of streptonigrin in combating cancer is by inhibiting SENP1.
A study by Park and Chun25 showed that streptonigrin has a cytotoxic effect against the β-catenin-activated type of human cancer cells by targeting β-catenin activity, where streptonigrin acts as an inhibitor of β-catenin/Tcf signaling (Figure 4). Streptonigrin showed an 86% inhibition against β-catenin/Tcf-DNA complex formation at 5 μM.25 Nevertheless, the finding was consistent with the notion that inhibition of β-catenin/Tcf signaling was likely to be ascribed not only to a direct blocking of the complex formation but also to an indirect effect diminishing β-catenin/Tcf-DNA complex formation.
 |
Figure 4 Anticancer activities of streptonigrin-transfected SW480 cells and HEK293 cells. Streptonigrin interferes with the complex formation of β-catenin/Tcf with DNA by downregulating β-catenin/Tcf signaling by blocking the upstream GSK-3β. Abbreviations: Wnt, wingless-int, LRP, lipoprotein receptor-related protein; GSK-3β, glycogen synthase kinase-3 beta; CKIα, casein kinase Iα; TCF/LEF, T-cell factor/lymphoid enhancer factor. Notes: Created with BioRender.com. |
Streptonigrin affects the upstream components of the β-catenin/Tcf pathway via suppression of GSK-3β phosphorylation. The findings indicate that β-catenin cannot function as a transcription activator in the presence of inhibitors.25 Therefore, changes in the subcellular localization of β-catenin may contribute to the downregulation of β-catenin/Tcf signaling in the presence of streptonigrin. The data collected suggested that there are two possible inhibitory mechanisms of streptonigrin: 1) the nuclear localization of β-catenin may be decreased owing to suppression of upstream GSK-3β, and 2) the inhibitor directly blocks the complex formation of β-catenin/Tcf with DNA.
Most previous studies demonstrated that streptonigrin has strong cytotoxic effects at high concentrations (>1 μM). However, a more recent study established that streptonigrin at concentrations as low as 1 nM can cause an increase in heterochromatin without significantly affecting cell proliferation and viability, thus raising the possibility that it could be a useful anticancer agent with low cytotoxic side effects.26 The overall findings of that study suggest that streptonigrin, especially when used at higher concentrations (>10 nM), causes HeLa cells to enter cell death and to become detached from the culture. In contrast, cells treated with low concentrations of streptonigrin (1 nM) remain and proliferate normally.26
Antibacterial Activity
One of the very latest studies demonstrated that the bacterial ribonucleotide reductase enzyme is a promising target for drug design and development for antibiotics. According to the results from the most recent study on molecular docking and dynamics simulations, streptonigrin can bind to Elizabethkingia anophelis and Elizabethkingia meningoseptica ribonucleotide reductase alpha subunits with good affinity.27 These results indicate that streptonigrin may be a potential inhibitor of these ribonucleotide reductases.27
One of the very early studies showed that the bacteriocidal activity of streptonigrin is similar to that of mitomycin, which also contains an aminoquinone fragment.2 Further, a study reported that a modified bacteria foraging algorithm (mbfA) mutant of Bradyrhizobium japonicum defective in iron export is sensitive to short-term exposure to high levels of iron or hydrogen peroxide.17 Following treatment with streptonigrin (200 μg/mL) or an equal volume of dimethyl sulfoxide (DMSO) for 26 hours, the cells were spotted on non-selective plates. The mbfA bfr double mutant was found to be highly susceptible to killing compared to the wild type (WT) or the single mutants, while those treated with an equal volume of DMSO did not show any difference in sensitivity, indicating the compensatory roles of Bfr and MbfA in maintaining the iron content and, in turn, conferring the antimicrobial protective activity of streptonigrin.28
Bhubhanil et al29 conducted a study on Agrobacterium tumefaciens membrane-bound ferritin (MbfA), which is a member of the erythrin (Er)–vacuolar iron transport family. Agrobacterium tumefaciens mbfA mutant strain was identified to have a 1.5-fold higher total iron content than the WT strain. Furthermore, multi-copy expression of mbfA reduced the total iron content by two- and three-fold in WT and mbfA mutant backgrounds, respectively, thus suggesting that MbfA may function as an iron exporter rather than as an iron storage protein. The mbfA mutant showed a 10-fold increased sensitivity to the iron-activated antibiotic streptonigrin, implying that the mutant had increased accumulation of intracellular free iron,29 indicating the effectiveness of streptonigrone as an antibacterial agent. Further, an earlier study conducted by Herlt et al30 demonstrated that streptonigrin (50 μg/mL) inhibited Streptomyces aureofaciens, Streptomyces fragilis, Bacillus subtilis, Escherichia coli and Saccharomyces cerevisiae at the stated concentration. To investigate the role of SloR in Streptococcus mutans oxidative stress tolerance, Crepps et al31 performed disk assays on streptonigrin as the oxidative stressor. Notably, GMS584 demonstrated a significantly larger average zone of inhibition (4.03±0.019 cm) compared with UA159 (3.85±0.014 cm) and GMS585 (3.72±0.015 cm).31
Retinal Protection
Wizeman and Mohan5 investigated the root cause behind citrullination occurring during retinal gliosis, and found that the expression of peptidyl arginine deiminases type 4 (PAD4) increases in the injured mouse retina. In addition, the PAD isozyme localizes to Müller glial processes, concomitant with the upregulation of glial fibrillary acidic protein (GFAP). Most importantly, the inhibition of PAD4-driven citrullination can downregulate GFAP in the injured retina. Therefore, PAD4 is a potential target of glial reactivity. Previous studies4,32 have demonstrated streptonigrin to be a potent and selective PAD4 inhibitor. An ocular explants system has been developed to measure the effects of streptonigrin, to allow for short-term assessments of retinal glial reactivity. In the study by Wizeman and Mohan,5 streptonigrin downregulated GFAP at 25 nM (vehicle=2.78±0.57; streptonigrin=1.56±0.12), indicating that injury-induced citrullination and soluble GFAP protein levels are reduced by PAD4 inhibition. Streptonigrin, however, did not alter GFAP transcript levels in treated eyes, as confirmed by a quantitative reverse transcription polymerase chain reaction (p=0.7431; t-test), suggesting, overall, that PAD4 inhibition reduces the major citrullinated protein species, corresponding with the downregulation of GFAP protein levels.5
Nephroprotection
Various studies have revealed that acute kidney injury (AKI) is inducible by renal ischemia/reperfusion (I/R). In fact, renal PAD4 activity has been demonstrated to be closely correlated with AKI.6,7,33 In a fluorescence-based assay conducted by Ham et al,6 there was a significant (~15-fold) induction of PAD4 activity in the kidneys of mice subjected to 24 hours of renal I/R compared with sham-operated mice, while PAD2 mRNA expression remained unchanged. Owing to the higher PAD4 levels, an increasing level of citrullinated histone H3 immunoreactivity in the nucleus of the renal proximal tubules was also observed after 24 hours of renal I/R, induced by PAD4 activity and expression. It is hypothesized that peptidyl arginine residue is converted to peptidyl citrulline on the histone tail by PAD4. In contrast, several in vivo studies have demonstrated that PAD4-deficient mice are protected against renal I/R-induced kidney injury. A similar study by Rabadi et al7 supported the hypothesis of increased PAD4 activity following renal I/R, which increased histone H3 citrullination but ameliorated in PAD4-deficient mice.7 In addition, the PAD4-deficient mice had significantly reduced renal tubular inflammation (reduced MIP-2 synthesis, neutrophil infiltration and ICAM-1 induction) as well as reduced renal tubular necrosis and apoptosis.6,7 Moreover, renal I/R injury not only resulted in increased renal tubular PAD4 expression and activity, but also generated translocation of renal tubular PAD4 from the nucleus to the cytosol.
Renal tubular cytosolic peptidyl citrullination and hepatic PAD4 expression as well as activity increased after ischemic AKI. The liver injury that occurs after ischemic AKI is attenuated in PAD4-deficient mice, with less hepatocyte vacuolization and hepatic neutrophil infiltration seen.7 Ham et al6 further investigated the efficacy of two distinct classes of PAD4 inhibitors (2-chloroamidine and streptonigrin) in protecting against ischemic AKI.4,34 As expected, plasma creatinine significantly increased in vehicle-treated mice 24 hours after 30 minutes of renal I/R injury. However, PAD4 inhibition with 2-chloroamidine or streptonigrin treatment before renal ischemia protected against the renal injury, as evidenced by the significantly lower plasma creatinine levels compared with vehicle-treated mice that were subjected to renal I/R.6 Overall, the findings revealed protection against renal I/R injury, with significantly reduced plasma creatinine, renal tubular necrosis, inflammation and apoptosis occurring via irreversible inactivation of PAD4 by covalently modifying the catalytically active cysteine sites.6,7 The PAD4 inhibitors may be useful therapeutic agents that can attenuate both the complications and severity of ischemic AKI, as well as liver injury, or may be applied as an adjunct in AKI therapy.
Structurally Related/Modified Molecules of Streptonigrin for New Drug Discovery, Development and Therapy
Although streptonigrin is not readily amenable to chemical modification, relatively few derivatives of the antibiotic have been reported. Streptomyces species collected from Fujian Province, China, produced the metabolite streptonigrone and also possesses antitumor and antimicrobial antibiotic properties. Lavendamycin is another structurally related streptomycete metabolite and is the precursor for streptonigrin during its biosynthesis.35
Isopropylazastreptonigrin and streptonigrin methyl ester have been prepared by chemists from Pfizer. The former has no anticancer activity while the latter is weakly active owing to partial in vivo hydrolysis to streptonigrin. In related compounds where the 7-NH2 group is replaced with either –OH or –OMe, the activity is reduced further.35 Orsellinamide, which is the amide derivative of streptonigrin, obtained using a strain of Streptomyces griseus, also showed significant in vivo activity against refractive mouse mammary tumor.35
In recent years, a considerable number of streptonigrin analogues have been synthesized. These are primarily variations of the 5,8-quinoline quinone structure with different substituents at C-2, C-6 and C-7. Nevertheless, most of the compounds have only been investigated in in vitro studies related to determining the mechanism of action of streptonigrin. During the structural elucidation process of streptonigrin, a dihydrohexamethyl derivative of streptonigrin was synthesized and its structure characterized using NMR spectra.36
In addition, during the biosynthesis of streptonigrin, intermediate compounds 1–8 (Figure 5) formed following lavendamycin formation and hydroxylation, as well as methylation steps from ∆stnA, ∆stnB1, ∆stnQ3, ∆stnQ4 and ∆stnT4 mutants, and feeding experiments, have been reported.18
 |
Figure 5 Structurally related/modified molecules of streptonigrin. |
Changes in the B-ring of streptonigrin have been made and a series of 7-amino-6-methoxy-quinoxzlinequinones were prepared with varied substitutions at position 3 (ie from H, Cl, OCH3, CN, COOCH3 and OH). When the quinones were tested for cytotoxicity against mouse leukemic cells (L-1210) and for antibacterial activity against Bacillus subtilis, the quinones showed significant activity, indicating their potential.37
A series of streptonigrin analogues, in which the quinoline of ring B is replaced by isoquinoline and the substituted pyridine of ring C is replaced by phenyl, nitrophenyl, aminophenyl or benzyl functions, have also been synthesized and evaluated for their antibacterial and root-growth inhibitory activities.37
Dreyton et al4 developed a structure–activity relationship for streptonigrin that has provided unique insights into streptonigrin-induced inactivation of PADs, focusing on the quinoline-5,8-dione core and, in particular, the 7-amino-quinoline-5,8-dione scaffold. The overexpression of PAD activity could result in various human pathologies, such as rheumatoid arthritis,38 colitis and cancer, thus highlighting the importance of designing inhibitors for these enzymes. Previously, chloroacetamidine (Cl-amidine) and fluoroacetamidine (F-amidine) have been successfully developed as potent and bioavailable PAD4 inactivators. These compounds are mechanism-based inactivators that covalently modify a Cys active site in PAD4, which is critical for catalysis.34
In addition to Cl- and F-amidine being potent PAD4 inhibitors, a study by Knuckley et al32 found that Cl-amidine exhibits a strong inhibitory effect against PADs 1 and 3, indicating its utility as a pan-PAD inhibitor. These compounds are pan-PAD inhibitors since Cl-amidine, and to a lesser extent F-amidine, inhibits all the active PAD isozymes with similar potency. Given the increasing number of diseases in which dysregulated PAD activity has been implicated, the development of PAD-selective inhibitors is of paramount importance. Thus, the identification of PAD-selective inhibitors remains important; such compounds will allow us to determine the individual contributions of PAD isozymes to both normal human physiology and disease.
Streptonigrin has been reported to inhibit PAD4 activity in an in cellulo model.32 Relative to Cl-amidine, the high in cellulo potency reflects the fact that streptonigrin 1) is 33-fold more potent than Cl-amidine, and 2) has a greater bioavailability since Cl-amidine is positively charged and is highly hydrophilic, thus limiting cell permeability, and making streptonigrin a potential drug candidate as a selective PDA4 inhibitor. Overall, the findings suggest that PAD4 inhibitors treat an underlying cause of the disease rather than merely treating the symptoms.
Recently, the structure of streptonigramide has been established by spectroscopic analyses. Its biosynthetic pathway has also been proposed by Wang et al.39 Overall, our findings from this review may provide new insights for the further development of novel quinoline-based drug molecules for the treatment of various disorders.40–46
Challenges and Future Prospects for Streptonigrin in New Drug Design, Development and Therapy
In early clinical studies, the potent antitumor antibiotic streptonigrin was found to suppress several tumors, malignancies and viruses. Despite having a great potency, streptonigrin has numerous pharmaceutical and pharmacological difficulties. Streptonigrin is a poorly soluble drug, making oral administration challenging. The clinical utility of streptonigrin has been reduced because of its high toxicity, and it is currently only utilized as an experimental antitumor drug. However, it is envisaged that these problems can be resolved by improving the drug delivery, specifically targeting it to the tumors.47 Targeted delivery is believed to enhance the efficacy of drugs while reducing their side effects. Commonly used drug-targeting strategies include passive drug targeting, where the drug accumulates in the leaky vasculature areas in the tumor site;48 physical targeting, which is based on abnormal temperature49 and/or pH values50 in the pathological sites; or targeting using a specific carrier molecule,51 which has an increased affinity toward the tumor cells.
Targeted drug delivery can be successfully accomplished by attaching the drug molecule to pharmaceutical carriers. Recent developments in nanotechnology have shown that nanoparticles have excellent potential as drug carriers. Drugs can be encapsulated in nanocarriers to increase their solubility and bioavailability, change their biodistribution and make it easier for them to enter the target cell. Using nanoparticles, drugs can be delivered into tumor cells with minimum drug leakage into normal cells. Nanocarriers tend to accumulate in solid tumors as a result of the enhanced permeability and retention (EPR) of macromolecules, enhancing their antitumor activity.52 Conjugation of nanoparticles with ligands of cancer-specific tumor biomarkers is a potent therapeutic approach to treat cancer diseases with high efficacy.53 Some of the widely studied nanocarriers for cancer targeting include liposomes,54 dendrimers,55 polymeric nanoparticles,56 lipid nanoparticles,57 silica nanoparticles58 and carbon nanoparticles59 (Figure 6).
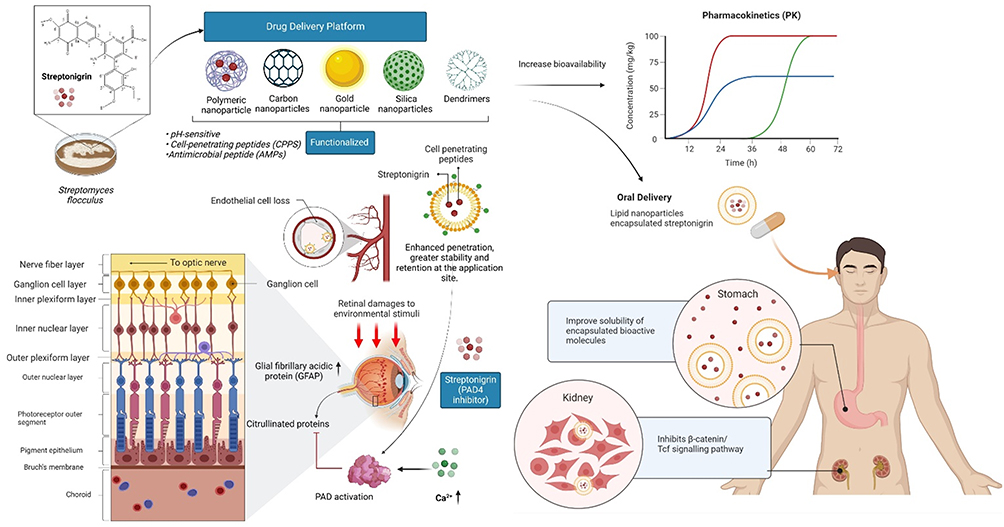 |
Figure 6 Future perspectives for managing retinal damage/injuries utilizing the nanotechnology platform. Efficient intraocular drug/compound delivery is extremely complex. Hence, nanocarriers such as micelles, which have been shown to be ideal drug delivery systems for maintaining compound/drug concentrations in the posterior region of the eyeball, may be conjugated with cell-penetrating peptides to enhance the penetration of streptonigrin. Streptonigrin can inhibit PAD4, and thus would be able to inhibit the citrullination of GFAP, which may contribute to chronic changes in Müller glia. Apart from that, it can also be used for oral delivery to the stomach as well as to the kidney. Notes: Created with BioRender.com. |
Many researchers over the years have also made attempts to reduce the toxicity of streptonigrin by synthesizing its derivatives. The methyl ester of streptonigrin was found to be less toxic than streptonigrin while maintaining its antitumor effectiveness.60 Some of the derivatives retained the antitumor effects while showing changes in antibacterial activities. Demethylstreptonigrin has been reported to elicit weak antibacterial activity.61 Streptonigrin hydroxamic acid and streptonigrin hydrazide also show weak antimicrobial activity compared with streptonigrin. Streptonigrin amide and streptonigrin hydrazide showed cytocidal activity against marine lymphosarcoma L5178Y cells to various extents, although the antibacterial activity against Gram-positive and Gram-negative bacteria was almost lost.62
With the advent of multiple nanoparticulate drug delivery systems and various drug targeting strategies, it is anticipated that streptonigrin could be delivered to targeted sites in the body with minimal toxicity. Moreover, with the use of streptonigrin derivatives, the clinical efficacy of the drug could still be maintained with reduced toxicity.
Conclusions
Streptonigrin has become a candidate of interest owing to its biological properties, with broad anticancer and antibacterial coverage, as well as having retinoprotective and nephroprotective activities. Nevertheless, its unavoidable toxicity is a limitation in designing the most effective formulation with maximum efficacy against targeted diseases while minimizing its toxic effects . By developing 1) synthetic targets possessing significant biological activities or 2) new synthetic methodology especially suited for application in the total synthesis of the structure, it is hoped that the design and preparation of structurally related compounds, yielding the structural characteristics of the naturally occurring compounds responsible for the biological properties, can be achieved. Further studies in these areas could help to produce the desired biological activities of streptonigrin with minimal or no toxicity.
Acknowledgments
All the authors of this manuscript are thankful to their respective departments/universities for the successful completion of the study. The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for financial support through the Research Group (Large) (project number RGP.2/31/43). Figures 1, 4, 6 and Graphical Abstract were created with BioRender.com.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Donohoe TJ, Jones CR, Kornahrens AF, et al. Total synthesis of the antitumor antibiotic (±)-streptonigrin: first-and second-generation routes for de novo pyridine formation using ring-closing metathesis. J Org Chem. 2013;78(24):12338–12350. doi:10.1021/jo402388f
2. Chiu -Y-YH, Lipscomb WN. Molecular and crystal structure of streptonigrin. J Am Chem Soc. 1975;97(9):2525–2530. doi:10.1021/ja00842a033
3. Boger DL. Diels–alder reactions of heterocyclic azadienes: development of a strategy for the total synthesis of streptonigrin, lavendamycin, and synthetic quinoline-5, 8-quinones. In: Strategies and Tactics in Organic Synthesis. Vol. 2. Elsevier; 1989.
4. Dreyton CJ, Anderson ED, Subramanian V, Boger DL, Thompson PR. Insights into the mechanism of streptonigrin-induced protein arginine deiminase inactivation. Bioorg Med Chem. 2014;22(4):1362–1369. doi:10.1016/j.bmc.2013.12.064
5. Wizeman JW, Mohan R. Expression of peptidylarginine deiminase 4 in an alkali injury model of retinal gliosis. Biochem Biophys Res Commun. 2017;487(1):134–139. doi:10.1016/j.bbrc.2017.04.031
6. Ham A, Rabadi M, Kim M, et al. Peptidyl arginine deiminase-4 activation exacerbates kidney ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2014;307(9):F1052–F1062. doi:10.1152/ajprenal.00243.2014
7. Rabadi M, Kim M, D’Agati V, Lee HT. Peptidyl arginine deiminase-4-deficient mice are protected against kidney and liver injury after renal ischemia and reperfusion. Am J Physiol Renal Physiol. 2016;311(2):F437–F449. doi:10.1152/ajprenal.00254.2016
8. Chen DB, Gao HW, Peng C, et al. Quinones as preventive agents in Alzheimer’s diseases: focus on NLRP3 inflammasomes. J Pharm Pharmacol. 2020;72(11):1481–1490. doi:10.1111/jphp.13332
9. Dahlem Junior MA, Nguema Edzang RW, Catto AL, Raimundo J-M. Quinones as an efficient molecular scaffold in the antibacterial/antifungal or antitumoral arsenal. Int J Mol Sci. 2022;23(22):14108. doi:10.3390/ijms232214108
10. Nilufer B, Hatice Y, Amac TF, et al. Synthesis, computational study, and evaluation of in vitro antimicrobial, antibiofilm, and anticancer activities of new sulfanyl aminonaphthoquinone derivatives. Lett Drug Des Discov. 2017;14(6). doi:10.2174/157018081406170606155530
11. Lee HW, Choi H, Nam SJ, Fenical W, Kim H. Potent inhibition of monoamine oxidase B by a piloquinone from marine-derived streptomyces sp. CNQ-027. J Microbiol Biotechnol. 2017;27:785–790. doi:10.4014/jmb.1612.12025
12. Alfadhli A, Mack A, Harper L, Berk S, Ritchie C, Barklis E. Analysis of quinolinequinone reactivity, cytotoxicity, and anti-HIV-1 properties. Bioorg Med Chem. 2016;24(21):5618–5625. doi:10.1016/j.bmc.2016.09.028
13. Patel OPS, Beteck RM, Legoabe LJ. Antimalarial application of quinones: a recent update. Eur J Med Chem. 2021;210:113084. doi:10.1016/j.ejmech.2020.113084
14. Benites J, Valderrama JA, Ramos M, Muccioli GG, Buc Calderon P. Targeting Akt as strategy to kill cancer cells using 3-substituted 5-anilinobenzo[c]isoxazolequinones: a preliminary study. Biomed Pharmacother. 2018;97:778–783. doi:10.1016/j.biopha.2017.10.108
15. Lee HW, Ryu HW, Kang MG, Park D, Oh SR, Kim H. Selective inhibition of monoamine oxidase A by purpurin, an anthraquinone. Bioorg Med Chem Lett. 2017;27(5):1136–1140. doi:10.1016/j.bmcl.2017.01.085
16. Pingaew R, Prachayasittikul V, Worachartcheewan A, et al. Novel 1,4-naphthoquinone-based sulfonamides: synthesis, QSAR, anticancer and antimalarial studies. Eur J Med Chem. 2015;103:446–459. doi:10.1016/j.ejmech.2015.09.001
17. Yildirim H, Bayrak N, Tuyun AF, Kara EM, Celik BO, Gupta GK. 2,3-disubstituted-1,4-naphthoquinones containing an arylamine with trifluoromethyl group: synthesis, biological evaluation, and computational study. RSC Adv. 2017;7:25753–25764. doi:10.1039/C7RA00868F
18. Xu F, Kong D, He X, et al. Characterization of streptonigrin biosynthesis reveals a cryptic carboxyl methylation and an unusual oxidative cleavage of a N–C bond. J Am Chem Soc. 2013;135(5):1739–1748. doi:10.1021/ja3069243
19. Lee S-H, Lee W-K, Kim N, et al. Renal cell carcinoma is abrogated by p53 stabilization through transglutaminase 2 inhibition. Cancers. 2018;10(11):455. doi:10.3390/cancers10110455
20. Sztiller-Sikorska M, Koprowska K, Majchrzak K, Hartman M, Czyz M. Natural compounds’ activity against cancer stem-like or fast-cycling melanoma cells. PLoS One. 2014;9(3):e90783. doi:10.1371/journal.pone.0090783
21. Ambaye N, Chen C-H, Khanna S, Li Y-J, Chen Y. Streptonigrin inhibits SENP1 and reduces the protein level of hypoxia-inducible factor 1α (HIF1α) in cells. Biochemistry. 2018;57(11):1807–1813. doi:10.1021/acs.biochem.7b00947
22. Nayak A, Müller S. SUMO-specific proteases/isopeptidases: sENPs and beyond. Genome Biol. 2014;15(7):1–7. doi:10.1186/s13059-014-0422-2
23. Wang Q, Xia N, Li T, et al. SUMO-specific protease 1 promotes prostate cancer progression and metastasis. Oncogene. 2013;32(19):2493–2498. doi:10.1038/onc.2012.250
24. Cheng J, Kang X, Zhang S, Yeh ET. SUMO-specific protease 1 is essential for stabilization of HIF1α during hypoxia. Cell. 2007;131(3):584–595. doi:10.1016/j.cell.2007.08.045
25. Park S, Chun S. Streptonigrin inhibits β-Catenin/Tcf signaling and shows cytotoxicity in β-catenin-activated cells. Biochimica et Biophysica Acta. 2011;1810(12):1340–1345. doi:10.1016/j.bbagen.2011.06.023
26. Loyola AC, Dao K, Shang R, et al. Streptonigrin at low concentration promotes heterochromatin formation. Sci Rep. 2020;10(1):1–11. doi:10.1038/s41598-020-60469-6
27. Mahfuz AB, Iqbal MN, Opazo FS, Zubair-Bin-Mahfuj A. Characterization of ribonucleotide reductases of emerging pathogens Elizabethkingia anophelis and Elizabethkingia meningoseptica and streptonigrin as their inhibitor: a computational study. J Biomol Struct Dyn. 2021;40:1–13.
28. Sankari S, O’Brian MR. Synthetic lethality of the bfr and mbfA genes reveals a functional relationship between iron storage and iron export in managing stress responses in Bradyrhizobium japonicum. PLoS One. 2016;11(6):e0157250. doi:10.1371/journal.pone.0157250
29. Bhubhanil S, Chamsing J, Sittipo P, Chaoprasid P, Sukchawalit R, Mongkolsuk S. Roles of Agrobacterium tumefaciens membrane-bound ferritin (MbfA) in iron transport and resistance to iron under acidic conditions. Microbiology. 2014;160(5):863–871. doi:10.1099/mic.0.076802-0
30. Herlt A, Rickards R, Wu J-P. The structure of streptonigrone, and a comment on the biosynthesis of the streptonigrin antibiotics. J Antibiot. 1985;38(4):516–518. doi:10.7164/antibiotics.38.516
31. Crepps SC, Fields EE, Galan D, Corbett JP, Von Hasseln ER, Spatafora GA. The SloR metalloregulator is involved in the Streptococcus mutans oxidative stress response. Mol Oral Microbiol. 2016;31(6):526–539. doi:10.1111/omi.12147
32. Knuckley B, Jones JE, Bachovchin DA, et al. A fluopol-ABPP HTS assay to identify PAD inhibitors. Chem Commun. 2010;46(38):7175–7177. doi:10.1039/c0cc02634d
33. Raup-Konsavage WM, Wang Y, Wang WW, Feliers D, Ruan H, Reeves WB. Neutrophil peptidyl arginine deiminase-4 has a pivotal role in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2018;93(2):365–374. doi:10.1016/j.kint.2017.08.014
34. Luo Y, Arita K, Bhatia M, et al. Inhibitors and inactivators of protein arginine deiminase 4: functional and structural characterization. Biochemistry. 2006;45(39):11727–11736. doi:10.1021/bi061180d
35. Anderberg PI, Luck IJ, Harding MM. Assignment of the 13C and 15N NMR spectra of the antitumour antibiotic streptonigrin. Magn Reson Chem. 2002;40(4):313–315. doi:10.1002/mrc.997
36. Kametani T, Ogasawara K, Shio M, Kozuka A. Streptonigrin and related compounds. VI. The NMR spectra of 4-(2, 3, 4-trimethoxyphenyl)-2, 3-dimethylpyridine derivatives. (Studies on the syntheses of heterocyclic compounds. CLXX). Yakugaku zassh. 1967;87(3):260–265. doi:10.1248/yakushi1947.87.3_260
37. Rao KV, Rock CP. Streptonigrin and related compounds. 6. Synthesis and activity of some quinoxaline analogues. J Heterocycl Chem. 1996;33(2):447–458. doi:10.1002/jhet.5570330238
38. Auger I, Charpin C, Balandraud N, Martin M, Roudier J. Autoantibodies to PAD4 and BRAF in rheumatoid arthritis. Autoimmun Rev. 2012;11(11):801–803. doi:10.1016/j.autrev.2012.02.009
39. Wang X, Xu F, Huang T, Deng Z, Lin S. A novel streptonigrin type alkaloid from the Streptomyces flocculus CGMCC 4.1223 mutant Δ stnA/Q2. Nat Prod Res. 2022;36(13):3337–3345. doi:10.1080/14786419.2020.1856840
40. Bayrak N, Ciftci HI, Yildiz M, et al. ‘Structure based design, synthesis, and evaluation of anti-CML activity of the quinolinequinones as LY83583 analogs’. Chem Biol Interact. 2021;345:109555. doi:10.1016/j.cbi.2021.109555
41. Ciftci HI, Bayrak N, Mahmut Y, et al. Design, synthesis and investigation of the mechanism of action underlying anti-leukemic effects of the quinolinequinones as LY83583 analogs. Bioorg Chem. 2021;114:105160. doi:10.1016/j.bioorg.2021.105160
42. Mataraci-Kara E, Bayrak N, Yildiz M, Yildirim H, Ozbek-Celik B, Tuyun AF. Discovery and structure-activity relationships of the quinolinequinones: promising antimicrobial agents and mode of action evaluation. Drug Dev Res. 2022;83:628–636. doi:10.1002/ddr.21893
43. Mataraci-Kara E, Bayrak N, Yildiz M, Yildirim H, TuYuN AF. Active quinolinequinones against methicillin-resistant staphylococcus spp. Chem Biodivers. 2022;19:e202100616. doi:10.1002/cbdv.202100616
44. Yildirim H, Bayrak N, Yildiz M, et al. Highly active small aminated quinolinequinones against drug-resistant Staphylococcus aureus and Candida albicans. Molecules. 2022;27(9):2923. doi:10.3390/molecules27092923
45. Yildiz M, Bayrak N, Yildirim H, et al. Discovery of quinolinequinones with N-phenylpiperazine by conversion of hydroxyquinoline as a new class of antimicrobial agents targeting resistant pathogenic microorganisms. Bioorg Chem. 2022;128:106045. doi:10.1016/j.bioorg.2022.106045
46. Yildirim H, Bayrak N, Yildiz M, et al. Aminated quinolinequinones as privileged scaffolds for antibacterial agents: synthesis, in vitro evaluation, and putative mode of action. ACS Omega. 2022;7(46):41915–41928. doi:10.1021/acsomega.2c03193
47. Attia MF, Anton N, Wallyn J, Omran Z, Vandamme TF. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J Pharm Pharmacol. 2019;71(8):1185–1198. doi:10.1111/jphp.13098
48. Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63(3):131–135. doi:10.1016/j.addr.2010.03.011
49. Osaki F, Kanamori T, Sando S, Sera T, Aoyama Y. A quantum dot conjugated sugar ball and its cellular uptake. On the size effects of endocytosis in the subviral region. J Am Chem Soc. 2004;126(21):6520–6521. doi:10.1021/ja048792a
50. Rowinsky EK, Rizzo J, Ochoa L, et al. A Phase I and pharmacokinetic study of pegylated camptothecin as a 1-hour infusion every 3 weeks in patients with advanced solid malignancies. J Clin Oncol. 2003;21(1):148–157. doi:10.1200/JCO.2003.03.143
51. Yoo HS, Park TG. Folate receptor targeted biodegradable polymeric doxorubicin micelles. J Control Release. 2004;96(2):273–283. doi:10.1016/j.jconrel.2004.02.003
52. Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2014;4(1):81. doi:10.7150/thno.7193
53. Noble GT, Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. Ligand-targeted liposome design: challenges and fundamental considerations. Trends Biotechnol. 2014;32(1):32–45. doi:10.1016/j.tibtech.2013.09.007
54. Liu Y, Ran R, Chen J, et al. Paclitaxel loaded liposomes decorated with a multifunctional tandem peptide for glioma targeting. Biomaterials. 2014;35(17):4835–4847. doi:10.1016/j.biomaterials.2014.02.031
55. Choudhury H, Sisinthy SP, Gorain B, Kesharwani P. History and introduction of dendrimers. In: Dendrimer-Based Nanotherapeutics. Elsevier; 2021.
56. Graf N, Bielenberg DR, Kolishetti N, et al. αVβ3 integrin-targeted PLGA-PEG nanoparticles for enhanced anti-tumor efficacy of a Pt (IV) prodrug. ACS Nano. 2012;6(5):4530–4539. doi:10.1021/nn301148e
57. Lasa-Saracibar B, Estella-Hermoso de Mendoza A, Guada M, Dios-Vieitez C, Blanco-Prieto MJ. Lipid nanoparticles for cancer therapy: state of the art and future prospects. Expert Opin Drug Deliv. 2012;9(10):1245–1261. doi:10.1517/17425247.2012.717928
58. Watermann A, Brieger J. Mesoporous silica nanoparticles as drug delivery vehicles in cancer. Nanomaterials. 2017;7(7):189. doi:10.3390/nano7070189
59. Holmannova D, Borsky P, Svadlakova T, Borska L, Fiala Z. Carbon nanoparticles and their biomedical applications. Appl Sci. 2022;12(15):7865. doi:10.3390/app12157865
60. Humphrey E, Dietrich F. Clinical experience with the methyl ester of streptonigrin (Ncs-45384). Cancer Chemother Rep. 1963;33:21–26.
61. Isshiki K, Sawa T, Miura K, et al. A new antitumor antibiotic: demethylstreptonigrin. J Antibiot. 1986;39(7):1013–1015. doi:10.7164/antibiotics.39.1013
62. Inouye Y, Okada H, Roy SK, et al. Biological properties of streptonigrin derivatives I. antimicrobial and cytocidal activities. J Antibiot. 1985;38(10):1429–1432. doi:10.7164/antibiotics.38.1429
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.