Back to Journals » Drug Design, Development and Therapy » Volume 20
Characterization of the Second-Generation Covalent Fragment Library (CovLib Gen2): Thiol Reactivity Profiling and p53-Y220C Rescue
Authors Schwer M ![]() , Aldea SR
, Aldea SR ![]() , Engelhardt MU
, Engelhardt MU ![]() , Stahlecker J
, Stahlecker J ![]() , Rheinganz J
, Rheinganz J ![]() , Langkamp A
, Langkamp A ![]() , Boeckler FM
, Boeckler FM ![]()
Received 24 January 2026
Accepted for publication 5 May 2026
Published 18 May 2026 Volume 2026:20 598622
DOI https://doi.org/10.2147/DDDT.S598622
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yan Zhu
Martin Schwer,1 Sven R Aldea,1 Marc U Engelhardt,1 Jason Stahlecker,1 Janosch Rheinganz,1 Aaron Langkamp,1 Frank M Boeckler1,2
1Department of Pharmacy and Biochemistry, Laboratory for Molecular Design & Pharmaceutical Biophysics, Eberhard Karls Universität Tübingen, Institute of Pharmaceutical Sciences, Tübingen, 72076, Germany; 2Interfaculty Institute for Biomedical Informatics (IBMI), Eberhard Karls Universität Tübingen, Tübingen, 72076, Germany
Correspondence: Frank M Boeckler, Department of Pharmacy and Biochemistry, Laboratory for Molecular Design & Pharmaceutical Biophysics, Eberhard Karls Universität Tübingen, Institute of Pharmaceutical Sciences, Tübingen, 72076, Germany, Tel +49 7071 29 74567, Email [email protected]
Purpose: Covalent Fragment-Based Drug Discovery (FBDD) has emerged as a powerful strategy for unlocking challenging pharmacological targets and engaging shallow or “cryptic” binding pockets. In this study, we present the design and characterization of the Second Generation Covalent Fragment Library (CovLib Gen2), an expanded collection of 81 structurally diverse electrophiles tailored for Covalent Fragment-Based Drug Discovery (FBDD) using an electrophile-first approach. The library spans five distinct warhead classes, including epoxides, vinyl sulfones, acrylamides, α-cyanoacrylamides, and a core set of SɴAr-reactive heteroarenes.
Methods: We comprehensively profiled the library for physicochemical properties and intrinsic thiol reactivity using high-throughput 5,5’-dithiobis-(2-nitrobenzoic acid) (DTNB) and high-performance liquid chromatography (HPLC)-based glutathione (GSH) reactivity assays. To demonstrate the library’s utility, we performed differential scanning fluorimetry (DSF) screening against the oncogenic, thermally unstable p53-Y220C mutant and subsequent specificity testing with two control mutants.
Results: The library exhibited a broad dynamic range of reactivities with a clear correlation between the assay methods. Additionally, we identified 12 fragments with desirable mild reactivity profiles (t1/2GSH = 1– 10 h). The DSF screen yielded 15 hits, primarily SɴAr-reactive heteroarenes and vinyl sulfones. Notably, the fragment SN054 emerged as the most potent stabilizer, inducing a maximal thermal shift of 4.5 °C. Specificity was confirmed using a cysteine-light variant (T-p53C-Y220C-CL), where SN054 retained significant stabilizing activity.
Conclusion: Our findings validate CovLib Gen2 as a versatile tool for ligand discovery, including electrophilic fragments covering a broad range of reactivity, and provide tractable starting points for the pharmacological rescue of p53-Y220C. An illustration shows molecular structures of fragment size resembling keys labeled ‘CovLib Gen2’ on the left. These keys are designed with chemical structures featurting warheads for covalent bond formation extending from a central point. On the right, there is a large, complex structure resembling a lock with a keyhole emitting light, symbolizing a cryptic lock. The background features a cosmic or abstract landscape, enhancing the theme of unlocking complex molecular structures.Illustration of fragments with covalent warheads as molecular keys labeled ‘CovLib Gen2’ unlocking a cryptic lock protein structure.
Keywords: covalent fragment-based drug discovery, differential scanning fluorimetry, 5, 5’-dithiobis-(2-nitrobenzoic acid), glutathione, tumor suppressor p53, warheads
Introduction
The landscape of drug discovery has been significantly impacted by the resurgence of targeted covalent inhibitors (TCIs). While historical concerns regarding off-target toxicity persisted, modern TCI design has demonstrated that covalent binding offers distinct advantages, including high potency, prolonged duration of action, and the ability to target shallow binding pockets.1–10 Their success is underlined by the FDA approval of several covalent-acting drugs, primarily kinase inhibitors, as well as antiviral drugs like Nirmatrelvir, used for the treatment of COVID-19.8,11–16 Typically, these inhibitors function via a two-step mechanism: initial reversible association followed by the formation of a covalent bond through the reaction of a covalent reactive group (CRG, often referred to as “warhead”) with a nucleophilic residue, most commonly cysteine.17–19 The standard TCI design strategy relies on a structure-based approach, where a warhead is attached to a known non-covalently binding ligand to target a specific nucleophilic amino acid residue.9,19–22 Moreover, covalent fragment-based drug discovery (FBDD) has emerged as a powerful complementary strategy. Often termed the “electrophile-first” approach, covalent FBDD screens small, reactive fragments to identify novel binding sites—including allosteric or transient protein binding sites—without relying on a pre-existing non-covalent ligand binding next to a targetable amino acid residue.23–34
Previously, our group developed a halogen-enriched fragment library (HEFLib) intended to investigate the application of halogen bonding in FBDD.35–39 During characterization, we observed that one specific fragment (compound 4482) stabilized the tumor suppressor p53 not through non-covalent interactions, but by covalently modifying surface cysteines via nucleophilic aromatic substitution (SɴAr).38 This fragment is identical to the compound PK11000, which was previously shown by Bauer et al to covalently bind and stabilize p53-Y220C.40 In principle, halogen bonding is driven by the interaction between a Lewis base and the electron-deficient σ-hole of a halogen atom.41,42 While the strength of this interaction (Vmax) can be enhanced by adding electron-withdrawing groups to the aromatic scaffold, increasing the electrophilicity carries the risk of triggering unintended nucleophilic aromatic substitution (SɴAr) at the arene core, leading to the observed unintended protein arylation.43–45 This reveals not only the necessity for thiol-reactivity profiling of electron-deficient aromatic fragments, but highlights the potential of SNAr-reacting heterocycles as a warhead class additional to the frequently used Michael-type acceptors. Building on these observations, in addition to the previously mentioned advantages of TCIs and covalent FBDD, we aimed to design a diverse Covalent Fragment Library (CovLib).46 Effective covalent library design merges classical FBDD parameters47 (“rule of three”) with warhead characteristics like reactivity, stability, and reaction geometry. To ensure maximum diversity, the library should feature diverse warhead classes spanning a wide dynamic range of reactivities.23,25,48,49 Due to the pandemic and other restrictions, we started our CovLib with a first subset comprising 20 fragments across four warhead classes: α-cyanoacrylates, epoxides, vinyl sulfones, and SɴAr-reactive heteroarenes. The physicochemical profiling of this subset, including computational and experimental solubility assessments, as well as reactivity profiling against cysteine surrogates and the validation through differential scanning fluorimetry (DSF) screening against c-Jun N-terminal kinase 3 (JNK3), ubiquitin-specific protease 7 (USP7), and the tumor suppressor p53 core domain followed by intact protein mass spectrometry, was published previously by our group.46 This investigation revealed first interesting hits, including the highly cysteine-reactive SNAr-type electrophiles SN001, SN006, and SN007. These hits were then examined further for their potential to rescue the cancer mutant p53-Y220C.50,51
The tumor suppressor p53 is a central regulator of cell cycle arrest, apoptosis, DNA-repair, and cellular senescence52–55 Its inactivation, driven by murine double minute 2 homologue (MDM2) and murine double minute 4 homologue (MDM4) upregulation or missense mutations in the core domain, is a frequent driver of tumorigenesis and chemoresistance56–61 These mutations are generally categorized as either DNA-contact alterations (eg R273H) or structural mutations that induce thermal destabilization (eg Y220C).62–64 The Y220C mutant, accounting for approximately 100,000 cancer cases annually, is a prime target for pharmacological rescue.62,63,65–69 The mutation creates a solvent-accessible, hydrophobic crevice that lowers the protein’s melting temperature (Tm) by ~8 °C, causing rapid denaturation.62–64,70 Because this crevice is distant from the DNA-binding interface, it serves as an ideal pocket for stabilizing ligands. This has led to the development of various binders,71,72 including the carbazole PK083 (ΔTm = 0.8 °C at 125 μM PK083),65 the iodophenole PK5176 (ΔTm = 2.6 °C at 250 μM PK5176),71,73 (the aminobenzothiazole MB710 (ΔTm = 2.0 °C at 250 μM MB710),74 and the potent clinical candidate PC14586 (Rezatapopt).75 Notably, the presence of Cys220 in this pocket has also enabled the design of TCIs such as KG13, an acrylamide derivative that restores wild-type stability.76 Based on the screening of the first CovLib subset, our group identified multiple SNAr reactive covalent p53-Y220C cleft binders, among them, the highly reactive tyrosine mimicking pyridine derivative SN001 and the pyrazine derivative SN006/7-8 stabilizing the protein up to 4.5 °C and 5.0 °C, respectively.46,50,51
These promising results, considering the small number of compounds in the initial test set, encouraged us to expand the CovLib to a Second Generation Covlib (CovLib Gen2). Hence, we assembled a library of 81 fragments featuring epoxide (EO), vinyl sulfone (VS), acrylamide (AA), and α-cyanoacrylamide (CA) warheads, as well as electron-deficient heteroarenes capable of SɴAr reactions (SN). Herein, we profile the physicochemical properties of the CovLib Gen2 relevant to drug discovery. We initially evaluated solubility through a combination of computational predictions (LogP/LogS) and experimental turbidimetry. Subsequently, we assessed electrophilic reactivity against cysteine surrogates using a high-throughput 5,5’-dithiobis-(2-nitrobenzoic acid) (DTNB) assay, followed by a glutathione (GSH) stability assay to mimic physiologically relevant conditions. We subsequently screened CovLib Gen2 against p53-Y220C via DSF. Hits were validated by time- and concentration-dependent measurements, alongside specificity checks using p53 and a cysteine-light variant of p53-Y220C (p53-Y220C-CL).
Materials and Methods
Material
The compounds studied were purchased from Aldrich Market Select (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) at purity levels of 90% or higher. Purity was confirmed by high performance liquid chromatography (HPLC) on an Ultimate 3000 HPLC-System with UV-detection (Thermo Fisher Scientific, Dreieich, Germany). Compounds EO004, EO006, EO009, and VS006 were not suitable for HPLC-UV analysis, probably due to a lack of UV absorption. As an alternative, 1H nuclear magnetic resonance (NMR) spectroscopy (Bruker Avance III HD 400 MHz) was performed. For compounds EO007, SN034, and SN066, the purity determined did not meet the criteria of ≥ 90% and were therefore excluded from the following investigations. The detected impurities could be caused by degradation during transport or storage at the vendor. Results of the purity confirmation are summarized in Table S 1.1. The corresponding chromatograms, NMR spectra, and chemical shifts are presented in the Figures S 1.1 – S 1.81.
Structural Filtering Process
The Aldrich Market Select Data Bank (~18 million compounds as of April 2023) was filtered employing a customized python script using the RDKit package. Compounds larger than 22 non-hydrogen atoms and smaller than six non-hydrogen atoms were removed. Further, compounds that show a logP > 2 were excluded. LogP values were calculated by RDKit according to Wildman et al.77
Calculation of Molecular Properties
The SMILES codes of all investigated compounds, including CovLib Generation 1, are listed in Table S 2.1. The molecular properties Heavy Atoms, Molecular Weight, Hydrogen-Bond Donors, Hydrogen-Bond Acceptors, Rotatable Bonds, QPLogS, QPLlogPo/w, and Polar Surface Area of the fragments were calculated using the QikProp module of Schrödinger suite version 2021−1.78 Molecules were protonated and preprocessed using Schrödingers’ LigPrep module with default parameters.78 All calculations were then carried out using the default parameters and the normal processing mode of QikProp. The results are depicted in Table S 2.2.
Turbidimetric Solubility Assay
The turbidimetric solubility assay was performed as previously described46 using phosphate buffered saline (PBS) pH 7.4 at 25 °C in a 200 μL scale. The 100 mM fragment stocks in dimethyl sulfoxide (DMSO) were diluted by a factor of 4/5 with DMSO in a dilution series. Diluted compounds were added to the buffer, resulting in fragment concentrations of 5 mM, 4 mM, 3.2 mM, 2.56 mM, 2.048 mM, 1.638 mM, 1.311 mM, 1.049 mM, 0.839 mM, 0.671 mM, 0.537 mM, and 0.429 mM and 5% DMSO. Due to lower solubility in DMSO, the compounds AA004, AA005, and SN070 were measured at 2.048 mM as the highest concentration. Extinction at 600–800 nm was measured in a Lumox® multiwell, 96-well-plate (SARSTEDT, Nürnbrecht, Germany) using the CLARIOstar Plate Reader (BMG Labtech, Ortenberg, Germany) for 2 h with 300 rpm double orbital shaking for 60s every 2 min. The concentration-dependent extinction spectra for all compounds, measured at the beginning (0 min), an intermediate cycle (57 min), and at the end (117 min), are depicted in the Figures S 3.1 – S 3.78. The highest concentration up to which no scattering is observed is reported as “minimal instant solubility” (MIS) based on the first measurement cycle and as “minimal final solubility” (MFS) based on the last measurement cycle. MIS and MIC values can be found in Tables 1 and 2.
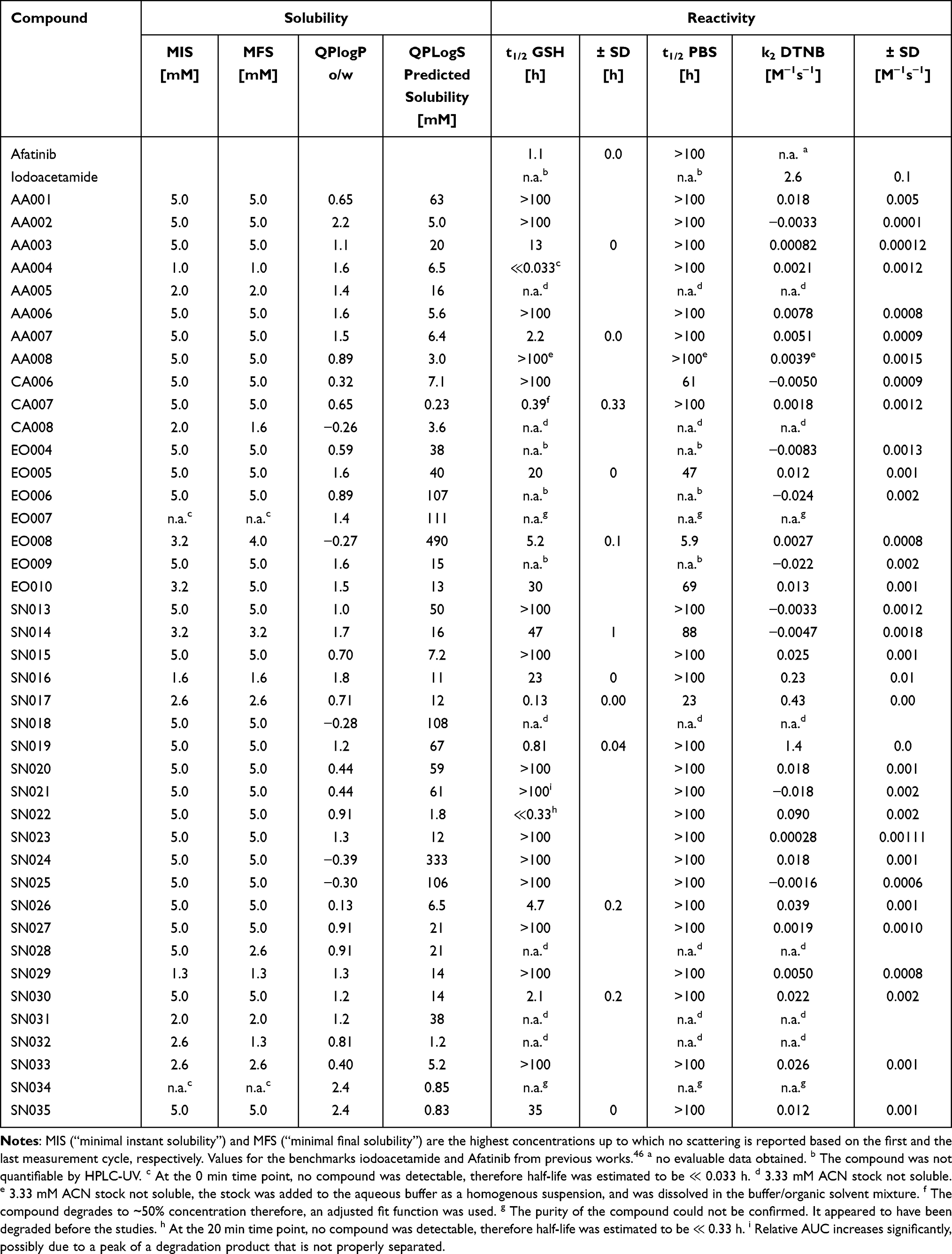 |
Table 1 Results of the Experimental Turbidimetric Solubility Characterization, Computational QLogPo/w and QPLogS Calculations, and the Reactivity Assessment via GSH and DTNB Assay |
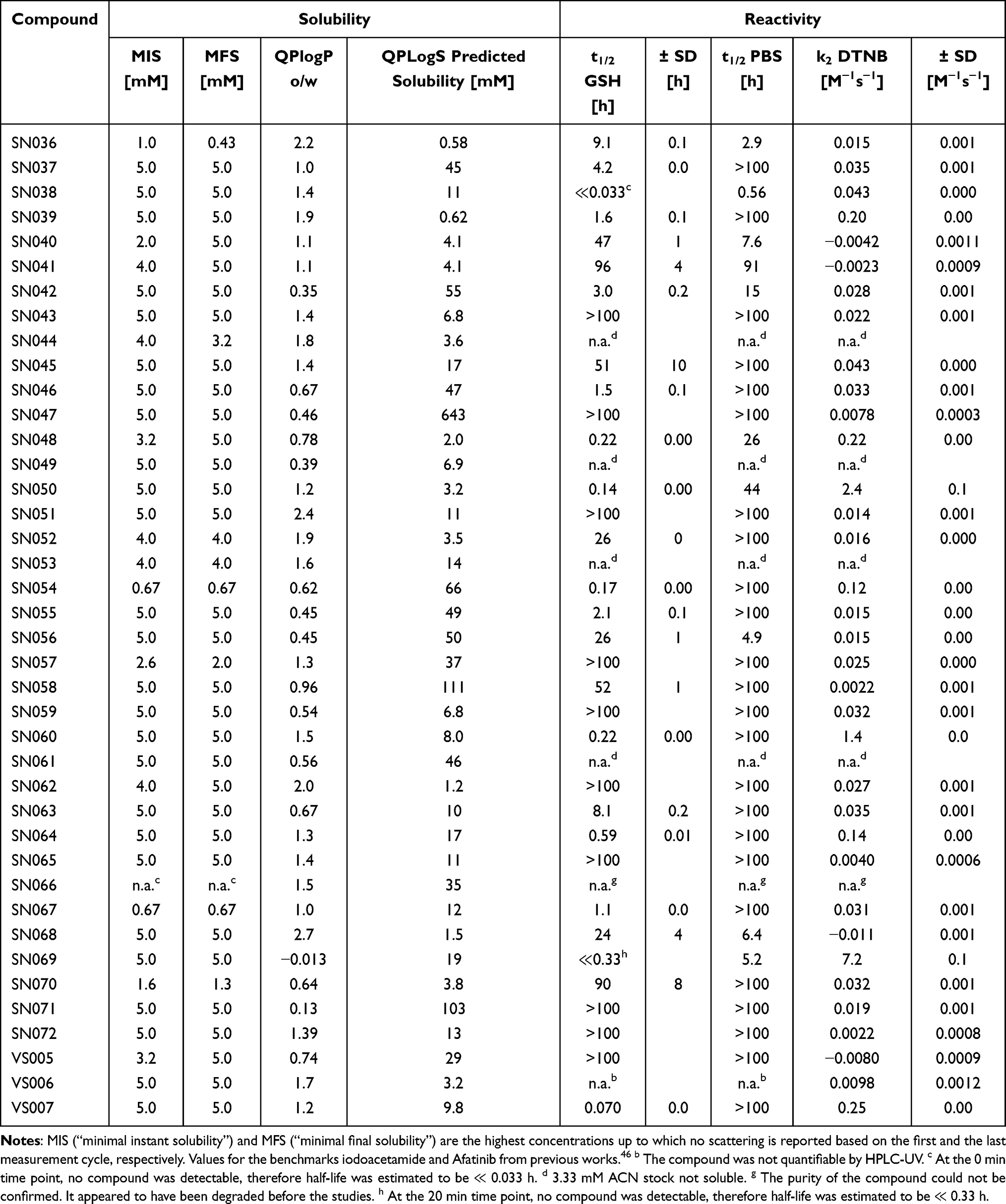 |
Table 2 Results of the Experimental Turbidimetric Solubility Characterization, Computational QLogPo/w and QPLogS Calculations, and the Reactivity Assessment via GSH and DTNB Assay |
DTNB Assay
The thiol-reactivity assay was performed as previously described46 according to the protocol published by Resnick et al27 with some modifications.79 The cysteine surrogate 2-nitro-5-thiobenzoate anion (TNB2-) was prepared in situ by reduction of 5,5′- dithiobis(2-nitrobenzoic acid) (DTNB) in the presence of tris(2-carboxyethyl) phosphine hydrochloride (TCEP). Reaction conditions were 200 μL buffer (20 mM NaPi pH 7.4, 150 mM NaCl), 10% acetonitrile, 100 μM fragment, 25 μM DTNB, and 100 μM TCEP (yielding to 50 μM TNB2-) at 37° C. The reaction was performed in a Lumox® multiwell, 96-well-plate (SARSTEDT, Nürnbrecht, Germany) covered with a lid. TNB2- absorbance at 412 nm was monitored every 5 min for 4 h using the CLARIOstar Plate Reader (BMG Labtech, Ortenberg, Germany). Measurements were performed in triplicate, and a parallel experiment without DTNB was conducted to determine the background absorption of the compounds. A measurement without fragment was performed to calculate the extinction coefficient of TNB2- from the absorption of the first time point. The compound background absorbance was subtracted from each measurement, and the remaining TNB2- and compound concentrations were calculated for each time point. The data were then fitted in OriginPro2020 (OriginLab, Northampton, MA, USA) and the second order rate constant k2 was calculated using the integrated rate equation of second-order kinetics with multiple reactants:
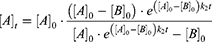
[A]0 and [B]0 are the initial concentration of the fragment and TNB2- respectively, and [A]t is the remaining compound concentration as a function of time. The [A]0 value for the initial compound concentration was set on the calculated compound concentration at 0 min. [B]0 was set by default on the experimentally used concentration of 50 μM. All parameters used for the fitting calculations and the computed results can be found in Table S 4.1 and S 4.2. The k2 values are given as the mean value of the triplicate determination with the respective standard deviation, which was calculated according to the rules of error propagation. The corresponding plots are depicted in the Figures S 4.1 – S 4.10.
Glutathione Assay
The glutathione (GSH) assay was performed as previously described38,46,51 using a method established by Keeley et al24 The following reaction conditions were applied: 250 μM fragment, 100 μM ketoprofen or ibuprofen as an internal standard, 5 mM GSH excess, 10% acetonitrile, and phosphate buffered saline (PBS) pH 7.4 at 37 °C. The samples were analyzed on an Ultimate 3000 HPLC-System (Thermo Fisher Scientific, Dreieich, Germany) with UV-detection over a period of 24 h. Highly reactive fragments (half-life t1/2 < 1 h) were analyzed every 20 min. The reaction of the compounds with GSH was detected by measuring the decreasing area under the curve (AUC) of the compounds relative to the internal standard. OriginPro2020 (OriginLab, Northampton, MA, USA) was used to fit the relative AUC to the integrated rate equation of pseudo-first order kinetics:
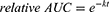
For the fitting of the GSH reaction of CA007, an additional +c term was used, because the CA007 concentration approaches ~0.5 instead of zero, possibly due to the reversible reaction.
The half-life t1/2 was calculated from the pseudo-first order rate constant k according to the following equation:
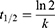
The GSH measurements were performed in duplicates and multiple runs were averaged using error propagation. In addition, measurements were performed in PBS buffer without GSH to test the hydrolytic degradation. Contrary to the calculations of Keeley et al,46 the rate constants kGSH and the corresponding t1/2 were not corrected for the degradation reaction in pure buffer For comparison, the half-lives for degradation in PBS (t1/2 PBS) are also given. The corresponding degradation plots are presented in the Figures S 5.1 – S 5.10.
Molecular Biology
For the conducted experiments, the superstable quadruple mutant (M133L/V203A/N239Y/N268D) of the p53 core domain (94–312) T-p53C was used. T-p53C-Y220C additionally contains the investigated Y220C mutation. The cysteine light control variant (T-p53C-Y220C-CL) further has mutations of the surface-exposed cysteines Cys124, Cys182, Cys229, Cys275, and Cys277 to serine residues. The expression and purification of T-p53C (94–312, M133L/V203A/N239Y/N268D) and its mutants using a pET24a(+)_HLT vector was carried out as previously described.38 The purity of the expressed protein was monitored by sodium dodecyl sulfate - polyacrylamide gel electrophoresis (SDS-PAGE). The protein sequences of all used constructs are displayed in Table S 6.1.
Differential Scanning Fluorimetry (DSF)
Differential Scanning Fluorimetry was used to investigate the impact of the compounds on the melting temperatures of T-p53C and its mutants. DSF measurements were performed as previously described38,46,51 using a Qiagen Rotor-Q Model-5-Plex HRM real-time PCR instrument (Qiagen, Hilden, Germany). SYPRO Orange (Life Technologies Corporation, Eugene, OR, USA) served as a fluorescent dye (5x final concentration). A constant heating rate of 270 °C/h was used, while the temperature was raised from 28 °C to 60–70 °C. The excitation and emission filters were set at 470 nm and 610 nm, respectively. The DSF measurements were performed with 8 μM protein in phosphate buffer (25 mM KPi pH 7.2, 150 mM NaCl, 1 mM TCEP, 5% DMSO). For the primary screening, measurements were carried out after 4 and 24 h with final fragment concentrations of 250 and 1000 µM. For concentration-dependent DSF measurements, the compound concentration was varied by diluting the compounds in pure DMSO before added to the protein solution. All samples were incubated at room temperature while shaking. The Tm of T-p53C and its mutants was obtained by calculating the maxima of the first derivative of the melting curves using OriginPro2020 (OriginLab, Northampton, MA, USA). Finally, the Tm of the protein sample was subtracted from the Tm of the sample containing both protein and compound to calculate ΔTm. All measurements were performed at least in triplicate and multiple runs were averaged according to the rules of error propagation. The results of the primary screening are depicted in the Tables S 7.1 and S 7.2. Results for the and time and concentration-dependent measurements are presented in the Figures S 7.1 and S 7.2, respectively.
Results and Discussion
Selection Process
Our goal was to assemble a library of covalent fragments (CovLib) with a broad range of structural diversity and reactivity. After the initial test set of 20 fragments,46 we aimed for expanding the library. The first part of the CovLib consists of four different warhead types to achieve a wide spectrum of covalent binding modes and reactivity. The selected warheads include Michael acceptors, specifically α-cyano acrylamides/acrylates and vinylsulfones. Furthermore, epoxides were added as compact reactive groups featuring two electrophilic centers. Finally, electron-deficient heteroarenes bearing leaving groups capable of undergoing nucleophilic aromatic substitution (SNAr) were selected. The latter type is less studied in drug design but has the advantages of structural rigidity, broad tunability, and, above all, good commercial availability due to its well-established reactions and synthesis in medicinal chemistry.46 For this expansion, we decided to add acrylamides as a fifth type of warhead. They are well-studied and already present in covalent-reacting FDA-approved drugs.7 Furthermore, they are generally less reactive Michael-type warheads compared to the other two used in this study, and therefore could balance the reactivity spectrum of the library.80,81
We used the same procedure for selecting the fragments as we did for the original library.46 The basis for the selection was the Aldrich Market Select Library (Sigma-Aldrich Chemie GmbH). Compounds larger than 22 non-hydrogen atoms, smaller than six non-hydrogen atoms, and a logP >2 were removed, and structural SMARTs filters were applied.
The final selection was accomplished by manual choice. In addition to steric and structural features, the assumed reactivity of the compounds was a primary selection criterion. While our initial test set comprised fragments with either very high or low reactivity,46 this study specifically targeted compounds exhibiting moderate reactivity. However, the selection process was constrained by commercial availability and cost. Owing to their frequent use as synthetic building blocks, electron-deficient heterocycles are readily available and generally more cost-effective than the other warhead types. Additionally, our group and others have reported the successful application of SNAr-reactive heteroarenes as a viable strategy for covalent FBDD.23–25,30,46,50,51,82,83 Consequently, approximately 75% of the acquired fragments are different electron-deficient heteroarenes with diverse leaving groups and various attached electron-withdrawing and donating groups. The distribution of these heteroaromatic scaffolds and respective substitutions is shown in Figure 1.
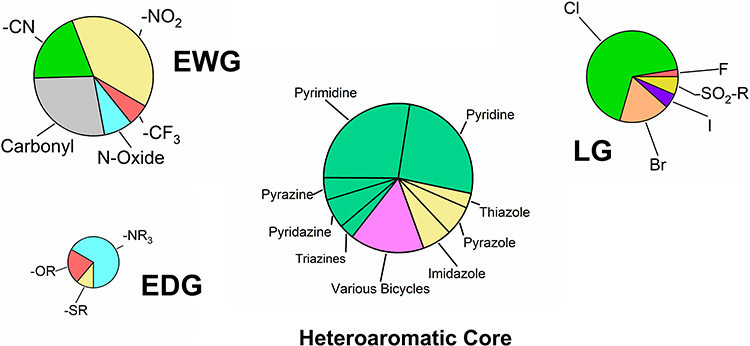 |
Figure 1 Distribution of the heteroaromatic cores, leaving groups (LG), electron-withdrawing groups (EWG), and electron-donating groups (EDG) of the acquired compounds with SNAr-type warhead. The size of the pie charts reflects the approximate number of substituents. Heteroaromatic cores are color-coded by ring type: six-membered (green), five-membered (yellow), and bicyclic (pink). |
The selected library consists predominantly of 42 six-membered rings, particularly pyridines and pyrimidines, due to their well-established reactivity profiles.9,84–89 Additionally, ten five-membered rings such as imidazoles, pyrazoles, and thiazoles were included. These scaffolds typically possess higher electron density and are therefore anticipated to be less reactive than the six-membered analogues.24,90 Finally, ten bicyclic compounds were acquired to investigate interesting compounds that have been sparsely used as warheads to date, but are very common in approved drugs.91
Chlorine was selected as the predominant leaving group, offering a theoretical intermediate reactivity within the halide series.90 In addition to the four halogens (F, Cl, Br, I), alkyl sulfonyl groups were included to represent highly reactive leaving groups with well-studied reactivity profiles.86 It is important to mention that all selected leaving groups also exert an electron-withdrawing effect on the aromatic core. Regarding the substitution pattern, the library turned out to feature a strong preference for electron-withdrawing groups (51 moieties) over electron-donating groups (10 moieties), with a particular emphasis on functional groups with a strong negative mesomeric effect, such as nitro and nitrile moieties. These strong EWGs could enable SNAr reactions at intrinsically more unreactive heteroaromatic scaffolds like five-membered ring systems. In addition to these 60 SNAr-type electrophiles, we purchased eight acrylamides, three α-cyanoacrylamides, seven epoxides, and three vinyl sulfones. The structures of the complete library are depicted in Figure 2.
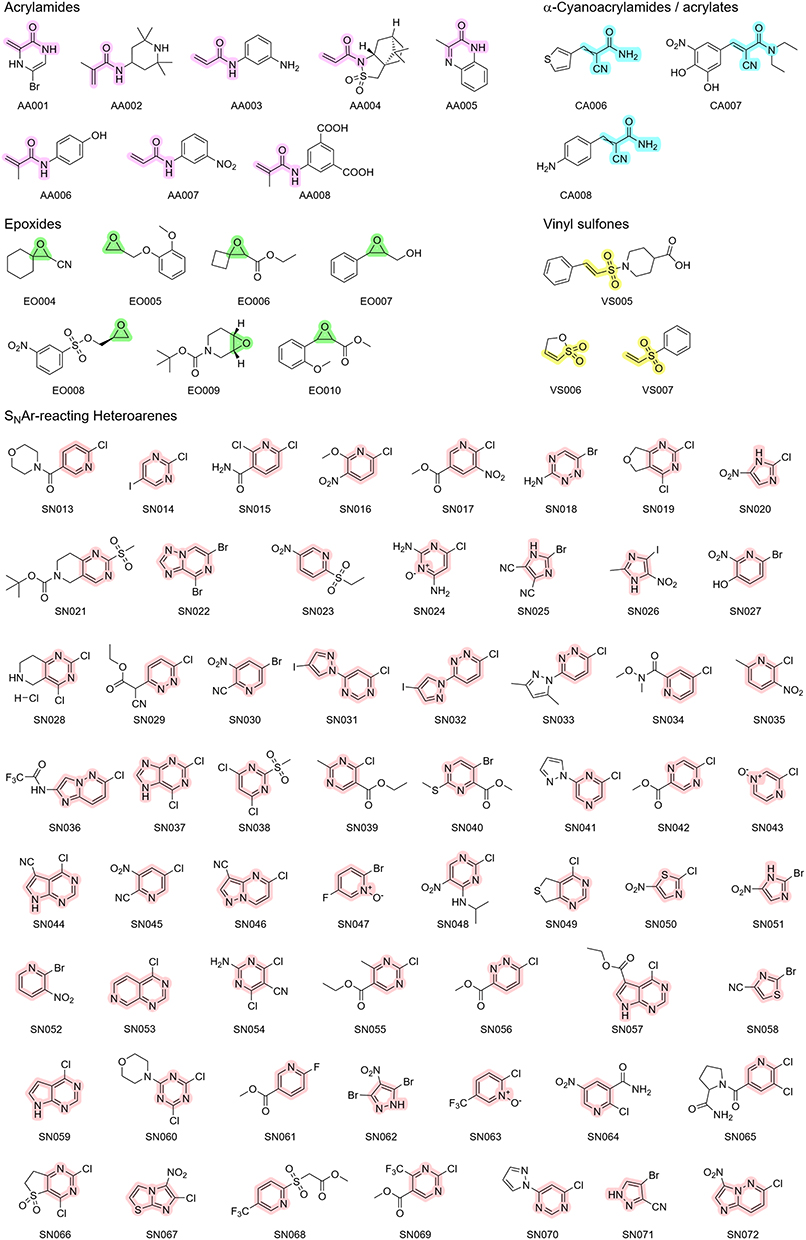 |
Figure 2 Chemical structures of the fragments purchased for the CovLib Gen2 The scaffolds for the respective warhead types are highlighted in purple for acrylamides, cyan for α-cyanoacrylamides, in green for epoxides, in yellow for vinyl sulfones, and in light red for SNAr-type electrophiles. For clarity, these color assignments are used consistently in all following schemes and illustrations. |
Theoretical and Experimental Characterisation of the Library
Distribution of Molecular Parameters for Fragment Libraries
The distribution of molecular parameters, which are often considered crucial for the construction of efficient fragment libraries (rule of three)47 is illustrated in Figure 3. Due to the potential for our substructure filters to bias the library toward molecules containing heavy atoms (S, Cl, Br, I), the count of non-hydrogen atoms (heavy atoms) was used as the primary molecular size cutoff criterion instead of molecular weight. Despite this consideration, only four compounds exceeded the 300 g/mol limit, primarily due to the inclusion of heavy halogen atoms. Only three compounds exceeded the threshold of three hydrogen-bond donors, contrasting with ~60% of the fragments violating the threshold for hydrogen bond acceptors. This elevated count could stem from the intrinsic nature of the selected warhead scaffolds, which are rich in heteroatoms that could act as hydrogen bond acceptors (e g, Michael acceptors and electron-withdrawing groups) The majority of compounds also slightly exceed the polar surface area threshold of 60 Å2, with a mean of 63.8 Å2. High values of these two parameters typically cause higher solubility, which we aimed for. Interestingly, the number of rotatable bonds differs strongly among the warhead classes. The SNAr-type electrophiles have the lowest mean value of 1.10 and thus exhibit high molecular rigidity, which is beneficial for further drug development.92
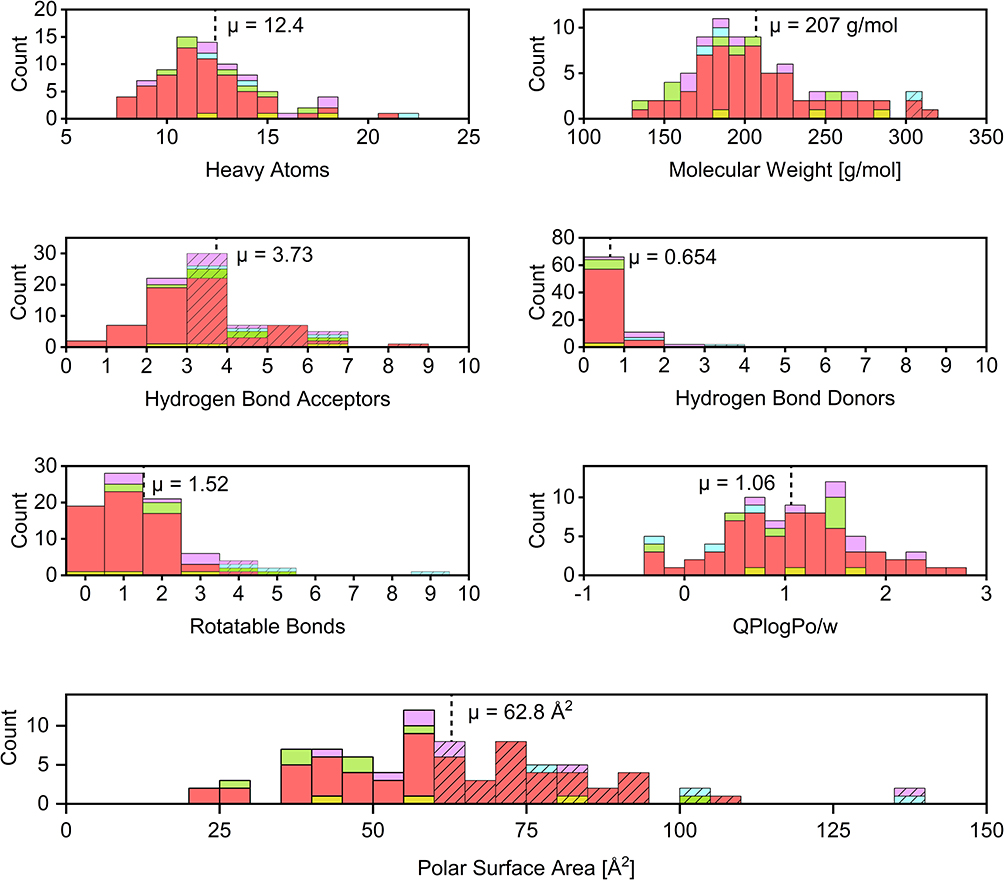 |
Figure 3 Distribution of heavy atoms, molecular weight, number of hydrogen bond acceptors, number of hydrogen bond donors, number of rotatable bonds, QLogPo/w, and polar surface area. Dashed lines indicate mean values. The hatched bars indicate bins that violate the rule of three. The colours of the bars indicate the respective warhead types. Purple for acrylamides, cyan for α-cyanoacrylamides, green for epoxides, yellow for vinyl sulfones, and light red for SNAr-type electrophiles. |
Experimental Solubility Assessment
Sufficient solubility is crucial for experimental compound testing and eventual further drug development. Consequently, we used logP values ≤ 2 calculated by RDKit as initial filtering criterion during library assembly. For more sophisticated theoretical characterisation, we calculated the QPLogPo/w and the QPLogS using the Schrödinger QikProp software. Fragment solubility was also experimentally evaluated in PBS buffer (pH 7.4) containing 5% (v/v) DMSO using a turbidimetric assay. Calculated and measured values are presented in Tables 1 and 2, with QPLogPo/w further illustrated in Figure 3. The three compounds SN036, SN054, and SN067 exhibit MIS or MFS values below 1 mM, used for DSF measurements. QPLogS calculations, however, predicted solubility > 1 mM for SN054 and SN067, but not for SN036. Despite the poor measured values, no precipitation was observed during the DSF experiments for any of these compounds. This discrepancy likely arises because the turbidimetric assay is highly sensitive to intrinsic compound absorption, which can interfere with the optical readout and potentially yield falsely low MIS or MIF values despite actual solubility being sufficient.
Experimental Reactivity Assessment
Thiol reactivity of the library was characterized using GSH and TNB2- as cysteine surrogates. The high-throughput DTNB assay was employed as a primary screen, followed by specific validation via a GSH HPLC-UV assay. To control for non-thiol-mediated degradation, reference measurements were performed in nucleophile-free PBS using HPLC. To ensure comparability, iodoacetamide and Afatinib were evaluated as established benchmarks for the DTNB and GSH assays, respectively.27,46,79,82,85 The determined values are presented in Tables 1 and 2 and illustrated in Figure 4. Corresponding plots and fitting parameters can be found in the Supporting Information.
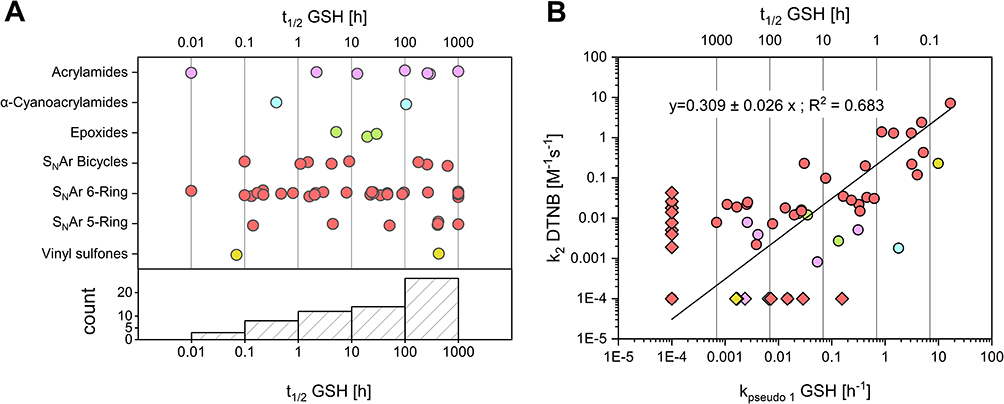 |
Figure 4 Distribution of experimental thiol reactivites of the CovLib. Data points are color-coded by warhead type: acrylamides (purple), α-cyanoacrylamides (cyan), epoxides (green), vinyl sulfones (yellow), SNAr-type electrophiles (light red). (A): Distribution of half-lives across warhead classes measured via GSH assay. The SNAr warheads are subcategorized into 5-membered, 6-membered, and bicyclic ring systems. Values calculated as >1000 h or <0 h were capped at 1000 h. Compounds undetectable at the first (0 min) or second (20 min) time points were assigned half-lives of 0.01 h or 0.1 h, respectively. (B): Correlation between GSH and DTNB reactivity. The line represents a linear regression (R2 = 0.683). Kinetic constants below 0.0001 h−1 (for GSH assay) or M−1s−1 (for DTNB assay), or negative values were set to 0.0001 and are depicted as diamonds. |
Linear regression analysis of GSH kpseudo 1 versus DTNB k2 values revealed a correlation of R2 = 0.683 (Figure 4B). A stronger correlation was observed for compounds with high reaction rates compared to more stable fragments. This correlation is regarded as satisfactory considering the known sensitivity limits of plate-based assays for low-reactivity electrophiles.9 These data align with previous comparisons of GSH and DTNB assays for acrylamide and chloroacetamide warheads described by Resnick et al27 These results underline the potential of the DTNB assay as a primary high-throughput screening method for the estimation of thiol reactivites.
A broad distribution of GSH-reactivities was observed across the entire library and the evaluated warhead classes (Figure 4A), indicating the versatile applicability of CovLib Gen2 for targets with different kinetic requirements. Notably, 12 compounds exhibited mild reactivity (t1/2 = 1–10 h); ten of these exhibit SNAr warheads, underscoring their potential as mild arylating agents. Selected findings and insights derived from the extensive GSH reactivity characterization of these diverse warheads are discussed in the following section.
The evaluation of epoxide-based fragments was complicated by handling difficulties related to their partly liquid physical state and the absence of an intrinsic π-system, which hindered accurate quantification via HPLC-UV. While moderate reactivity was observed for the three analyzable compounds (t1/2 = 5.2–30 h), notable degradation in PBS was also detected. Consequently, it remained unclear whether compound consumption was driven by nucleophilic substitution at the oxirane ring or by competing hydrolytic pathways, such as (sulfonate) ester hydrolysis. The observed reactivity profiles of the Michael acceptors were generally consistent with previously published trends.80,81,93 The influence of electronic effects was illustrated by the comparison of AA003 and AA007, where the presence of an electron-withdrawing nitro group on the phenyl ring was found to enhance reactivity relative to the amino-substituted analogue. Furthermore, a distinct structure-reactivity relationship was observed for acrylamides and vinyl sulfones, wherein substitution at the α- or β-position induced a substantial reduction in thiol-reactivity, resulting in half-lives exceeding 100 h.
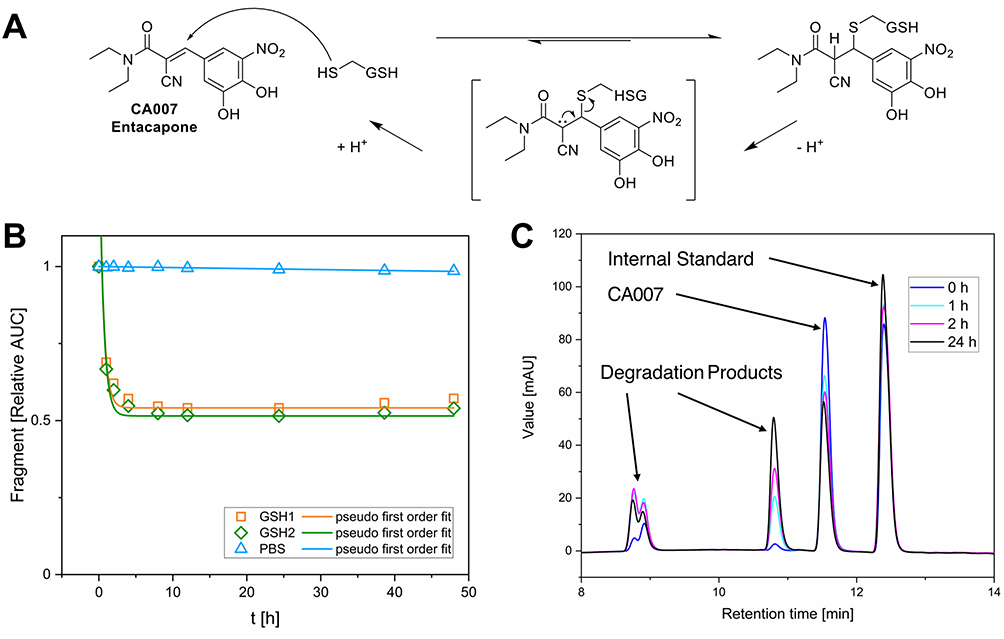
Despite that, a notable observation was made regarding the α-cyano acrylamide CA007. It should be mentioned that CA007 is the FDA-approved reversible catechol-O-methyltransferase (COMT) inhibitor Entacapone, used for the treatment of Parkinson’s disease94 and was serendipitously purchased as part of this library. While α-cyanoacrylamides are known for their potential to undergo reversible cysteine addition - driven by β-elimination facilitated through enhanced acidity of the Cα-proton - the experimental verification of this behaviour remains challenging.2,7 The results obtained from the GSH assay indicate a covalent reversible reaction with GSH. A proposed reaction scheme is shown in Figure 5A. In contrast to other fragments, CA007 was rapidly depleted to ~50% relative AUC and remained at this level over 48 h rather than approaching complete compound consumption (Figure 5B and C). Consequently, we had to adjust the fitting function by adding a constant to the pseudo-first-order rate equation. This adjustment limits the comparability of the calculated t1/2-values. We could not find any evidence in the literature regarding a covalent reversible reaction involved in the COMT inhibitory effects of Entacapone.
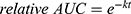
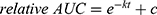
Compounds SN036, SN040, SN056, and SN068 exhibited shorter half-lives in PBS than in PBS containing 5 mM GSH, indicating intrinsic instability in aqueous buffer rather than SNAr with GSH. Their degradation is likely due to hydrolysis of their esters or trifluoroacetamide moieties. Another explanation is the potential precipitation due to poor solubility in PBS/ACN.
Despite the strong electron-withdrawing nature of N-oxides typically enhancing SNAr reactivity,95,96 derivatives SN024, SN043, and SN047 were unreactive (t1/2 > 100 h). Only SN063 showed mild reactivity (t1/2 = 8.1 h), suggesting that N-oxide activation could be a suitable strategy for generating heterocyclic scaffolds with balanced reactivities. Reactivity within five-membered rings varied significantly. While generally less susceptible to SNAr due to higher electron density at the ring carbon atoms, some compounds, like SN026 (t1/2 = 4.7 h), were found unexpectedly reactive. Notably, the thiazole SN050 degraded rapidly (t1/2 = 0.14h), while the corresponding imidazoles (SN020, SN051) remained inert (t1/2 > 100 h), highlighting the higher SNAr reactivity of thiazoles.83 Finally, bicyclic compounds displayed relatively unpredictable stability profiles, ranging from complete depletion within 20 minutes (SN022) to high stability of t1/2 > 100 h (SN057).
Biophysical Evaluation of the CovLib Gen2 Against p53 and Its Mutants Using DSF
We evaluated the influence of the CovLib Gen2 compounds on the melting temperature (ΔTm) of the p53 mutant T-p53C-Y220C. In the primary screen, we tested all fragments except SN031 and SN032, which were found insoluble under the used conditions. In contrast, the turbidimetric data indicated low but sufficient solubility for SN031 and SN032. Compounds were tested at 250 and 1000 µM with incubation times of 4 and 24 h at RT. A compound was classified as a hit if it induced a thermal shift greater than 1 °C under at least one of the tested conditions. Thermal shifts, for all tested compounds, are reported in the SI. Chemical structures and thermal shifts for the 15 hit compounds are presented in Figure 6. The hits show a strong tendency to SNAr-reacting warheads and vinyl sulfones. This is consistent with previous observations.46 The efficacy of these scaffolds is likely due to the nature of the Y220C mutation: the loss of the aromatic tyrosine residue creates a cavity that can be stabilized by the binding of (hetero)aromatic moieties. Furthermore, we observed a preference for compounds with higher thiol reactivity. Notably, all fragment hits exhibited glutathione half-lives of less than 5 h, which is an indicator of covalent fragment binding.
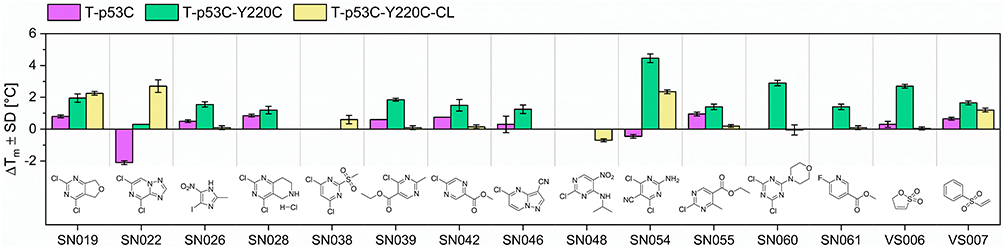 |
Figure 6 Results of the DSF measurements (1000 µM compound concentration and 24 h incubation time) for the initial fragment hits and their chemical structures. Purple bar: p53 core domain (T-p53C). Green bar: p53 core domain with Y220C mutation. Yellow bar: “cysteine-light” p53 core domain with Y220C mutation and surface-exposed cysteins mutated to serine residues (T-p53C-Y220C-CL). |
We investigated the time-dependent stabilization kinetics of all hit compounds using DSF measurements conducted over 24 hours. The time points where thermal stabilization first exceeds 1 °C are presented in Table 3. To compare the kinetic stabilization effect on T-p53C-Y220C with the compounds’ intrinsic thiol reactivity, we plotted the time-dependent GSH depletion alongside the DSF results. Representative plots are shown in Figure 7; the complete data set is provided in the Supporting Information.
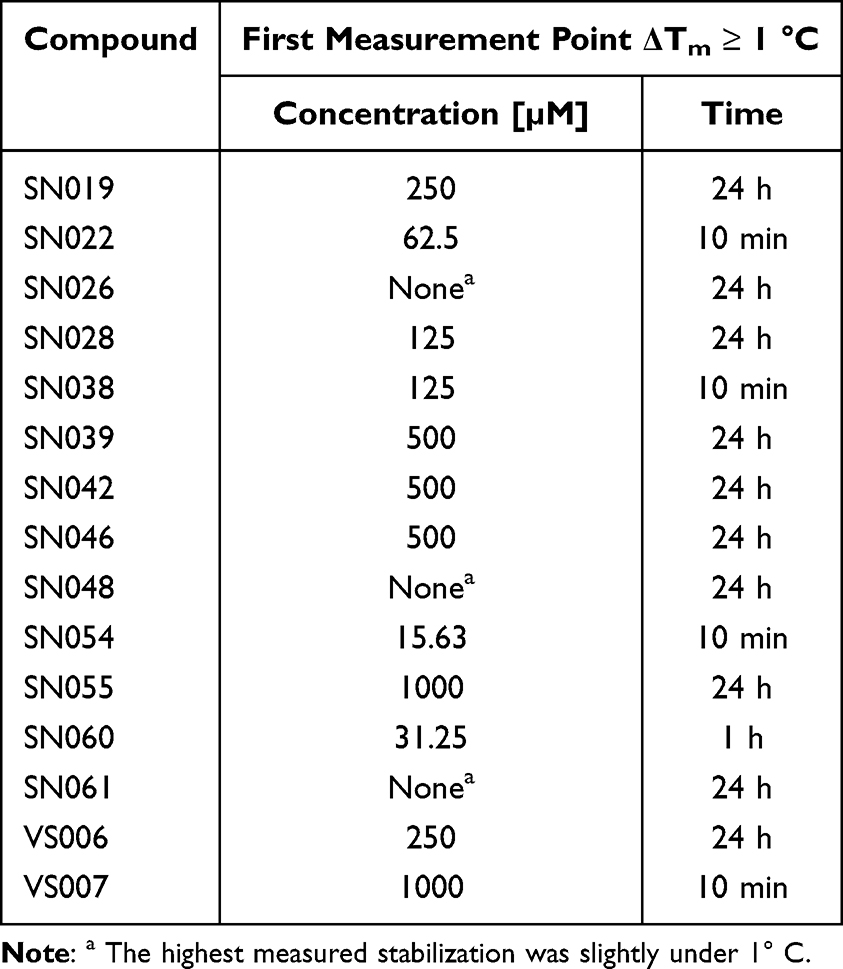 |
Table 3 Results of the Time- and Concentration Dependent DSF Measurements. Concentration-Dependent Experiments Were Carried Out After 24 h Incubation Time. Time-Dependent Measurements Were Carried Out with a Fragment Concentration of 1000 µM Except SN028 and SN038, with 250 µM and 500 µM, Respectively, to Obtain Evaluable Melting Curves |
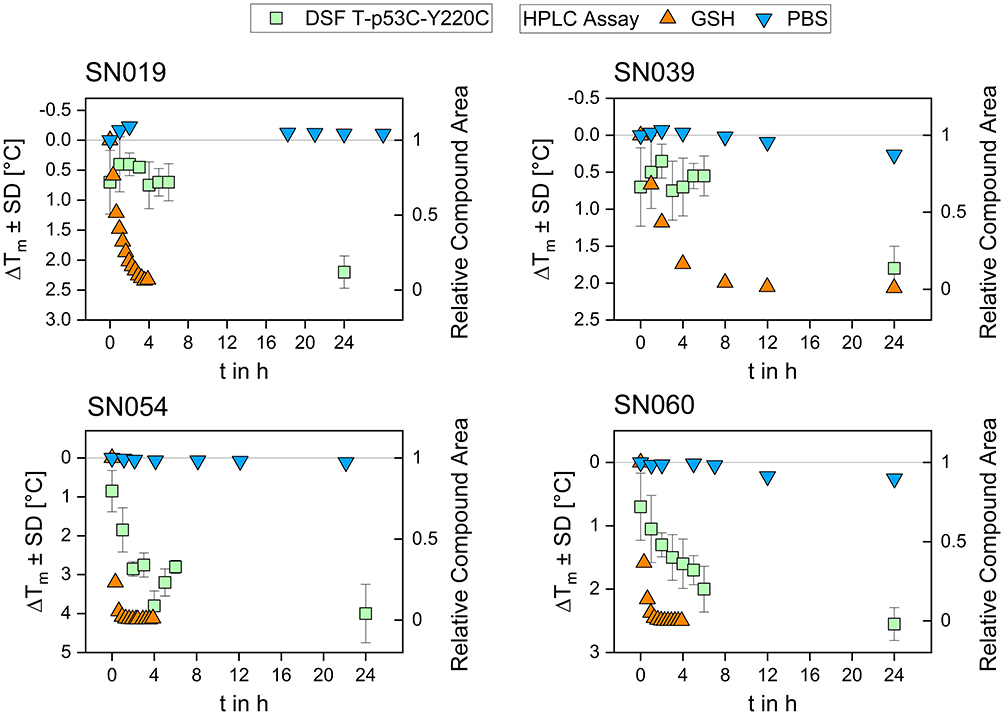 |
Figure 7 Results for the time-dependent DSF measurement (1000 µM compound concentration) and GSH reactivity assessment for the compounds SN019, SN039, SN054, and SN060. Thermal shifts obtained in the DSF assay are plotted on the left y-axis, while the relative compound area determined in the GSH assay is plotted on the right y-axis. |
Generally, the reaction rates observed with the protein were lower than those with GSH, a difference likely attributable to the experimental temperatures (37 °C for GSH vs. RT for protein incubation). The diagrams reveal a clear correlation: compounds exhibiting rapid GSH consumption (eg SN054 and SN060) showed faster kinetic stabilization of T-p53C-Y220C, whereas fragments with slower GSH reactivity required longer incubation times to achieve maximum thermal stabilization. These results highlight the potential of our standardized GSH assay as a predictive indicator for covalent protein binding kinetics, enabling the early-stage prioritisation of mildly reactive fragments.
To get first insights into reaction selectivity of our hits with T-p53C-Y220C, we performed concentration dependent DSF measurements. The concentrations where thermal stabilization exceeds 1 °C are presented in Table 3. Melting curves and plots of representive fragments are illustrated in Figure 8, the complete data set is provided in the Supporting Information.
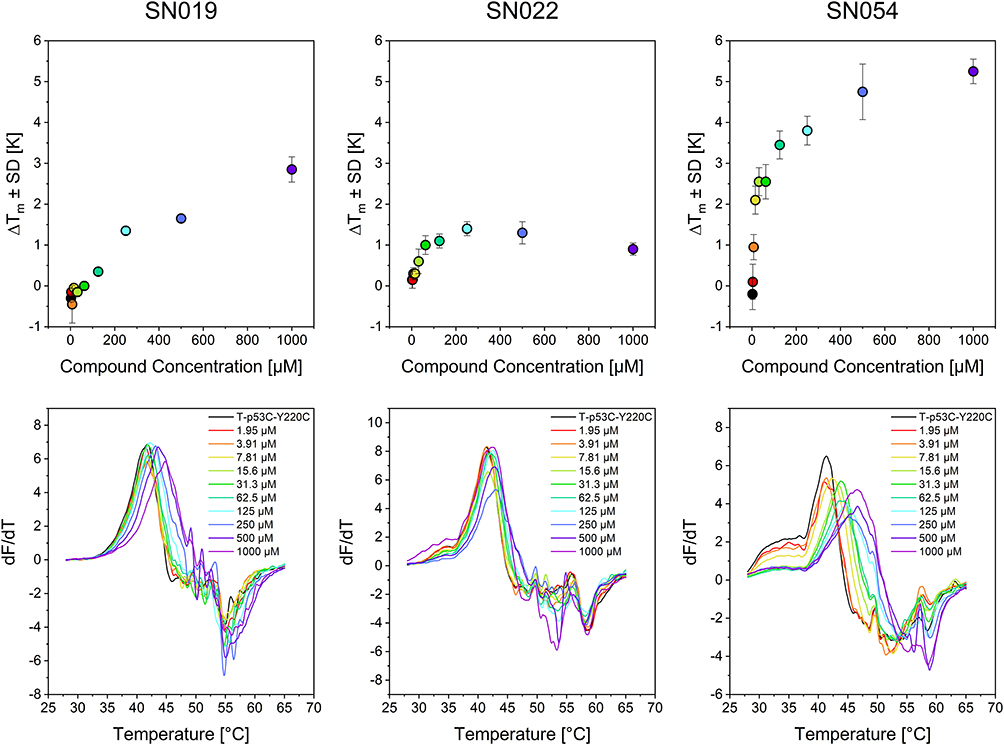 |
Figure 8 Results of the concentration-dependent DSF measurements after a 24 h incubation time for compounds SN019, SN022, and SN054. The upper diagrams show the thermal shift depending on the respective compound concentration; the lower diagrams show the first derivative (dF/dT) of the melting curves. The colors of the dots and lines indicate the concentrations listed in the lower plots of the melting curves. |
Significant stabilization (ΔTm ≥ 1 °C) at concentrations < 100 µM was observed for only three compounds (SN022, SN054, SN060). At the highest concentrations, however, certain highly reactive compounds (SN022, SN028, SN038, SN060) caused protein destabilization. These effects are linked to multiple arylations at high compound excess, some of which may have destabilizing effects on the protein.50
To validate the specificity of fragment binding to Cys220, we performed DSF control experiments using the pseudo-wild type T-p53C and the cysteine-light variant T-p53C-Y220C-CL. In the cysteine-light construct, solvent-accessible cysteine residues are mutated to serine to minimize interactions with competing residues. We hypothesized that a selective Cys220 binder would induce significant thermal shifts in T-p53C-Y220C and T-p53C-Y220C-CL, while showing minimal stabilization of T-p53C, which lacks the target cysteine. DSF results for the control mutants T-p53C and T-p53C-Y220C-CL are presented in Figure 6. The 4,6-dichloro- pyrimidine derivative SN054 induced the highest thermal stabilization of T-p53C-Y220C (ΔTm = 4.45 °C). A substantial shift was retained in the cysteine-light mutant (ΔTm = 2.25 °C), whereas the T-p53C exhibited slight thermal destabilization. These results suggest that while SN054 achieves significant stabilization through specific binding to Cys220, the superior shift observed in T-p53C-Y220C likely results from the additive effect of non-specific arylation of other solvent-exposed cysteines. This hypothesis is supported by the high intrinsic reactivity of the fragment (t1/2GSH = 0.17 h). This thermal stabilization of T-p53C-Y220C by SN054 is remarkable regarding the small size of the fragment and comparable to other known strong stabilizing fragments.50,51 Compound SN019 induced comparable stabilization (ΔTm ~2 °C) in both T-p53C-Y220C and its cysteine-light variant. A minor thermal shift was also observed for T-p53C (ΔTm = 0.8 °C). Structurally, SN019 features a pyrimidine core with two chlorine atoms as well, but with the chlorine leaving groups in 2- and 4-position. SN019 exhibits a milder reactivity profile (t1/2 = 0.81 h) compared to SN054.
Compound SN022 displayed a distinct stabilization profile. The cysteine-light mutant (T-p53C-Y220C-CL) exhibited a strong positive shift (ΔTm = 2.7 °C), whereas T-p53C underwent significant destabilization (ΔTm = −2.1 °C). Interestingly, the standard mutant T-p53C-Y220C showed a negligible shift (ΔTm = 0.3 °C). These data suggest a competition between two binding modes: binding to Cys220 stabilizes the protein (as seen in the CL variant), but this effect is likely masked in the T-p53C-Y220C construct by the simultaneous destabilization caused by off-target binding to other solvent-exposed cysteines.
The vinyl sulfone VS007 induced significant thermal stabilization of the cysteine-light mutant (ΔTm = 1.2 °C). However, the compound exhibited excessive glutathione reactivity (t1/2 = 0.07 h).
Conclusion
Following our initial test set, we assembled a second-generation covalent fragment library (CovLib Gen2) of 81 compounds. SNAr-type warheads form the core due to their favorable properties and can alternatively serve as HEFLibs if they lack suitable covalent reactivity. The structural diversity is further expanded by Michael-type electrophiles (acrylamides, α-cyano acrylamides, vinylsulfones) and small epoxides. Although our library is modest in size compared to industrial screening sets, it represents a carefully curated collection tailored to our available academic resources.
To assess intrinsic thiol reactivity, we employed a high-throughput plate-based DTNB assay validated by a subsequent HPLC-UV GSH assay. A linear correlation (R2 = 0.683) was found between the two methods, with agreement being stronger for highly reactive compounds. Overall, CovLib Gen2 displayed a broad distribution of reactivities across all warhead classes, confirming its versatility for targets with diverse kinetic requirements. Notably, 12 fragments exhibited desirable mild reactivity profiles (t1/2GSH = 1–10 h). We also observed an interesting kinetic profile for the α-cyanoacrylamide CA007, the FDA-approved drug Entacapone. This profile indicates a reversible covalent mechanism in the surrogate assay.
DSF screening against T-p53C-Y220C identified 15 hits, mostly SNAr-reactive heteroarenes and vinyl sulfones. SN054 provided the highest stabilization (ΔTm of up to 4.45 °C). All hits exhibited rapid kinetics (GSH half-lives < 5 h). Time-dependent DSF confirmed that GSH reactivity correlates clearly with protein stabilization kinetics. Selectivity was further probed using T-p53C and the cysteine-light variant T-p53C-Y220C-CL. SN054 retained substantial stabilization (ΔTm = 2.25 °C) in the CL variant but caused slight destabilization in T-p53C.
In summary, screening the second-generation CovLib yielded mildly reactive covalent fragments and identified initial hits against T-p53C-Y220C. These small, structurally diverse scaffolds expand the current landscape of p53-Y220C stabilizers. While their specific effects require further investigation, they serve as excellent starting points for fragment-based optimization.
Abbreviations
AUC, area under the curve; COMT, catechol-O-methyltransferase; CovLib, covalent fragment library; CRG, covalent reactive group; DMSO, dimethyl sulfoxide; DSF, differential scanning fluorimetry; DTNB, 5,5’-dithiobis-(2-nitrobenzoic acid); FBDD, fragment-based drug discovery; GSH, glutathion; HEFLib, halogen-enriched fragment library; HPLC, high performance liquid chromatography; JNK3, c-Jun N-terminal kinase 3; MDM2, murine double minute 2 homologue;MDM4, murine double minute homologue; MIF, minimal instant solubility; MIF, minimal final solubility; NMR, nuclear magnetic resonance; PBS, phosphate buffered saline; SNAr, nucleophilic aromatic substitution; t1/2, half-life; TCEP, tris(2-carboxyethyl) phosphine hydrochloride; TMS, tetramethylsilane; T-p53C, quadruple thermostable mutant of the p53 core-domain, 94–312; TCI, targeted covalent inhibitor; TNB2-, 2-nitro-5-thiobenzoate anion; USP7, ubiquitin-specific protease 7.
Acknowledgments
The authors acknowledge support from the state of Baden–Württemberg through bwHPC and the German Research Foundation (DFG) through Grant No. INST 40/575-1 FUGG (JUSTUS 2 cluster). In addition, support is acknowledged from the High Performance and Cloud Computing Group at the Zentrum für Datenverarbeitung of the University of Tübingen and the German Research Foundation (DFG) through Grant No. INST 37/935-1 FUGG (BinAC cluster).
Author Contributions
F.M.B. envisioned the research. M.S., J.S., J.R., and F.M.B. conceptualized the experiments and designed the study. M.S. performed the DTNB Assay, and Glutathione Assay. A.L. performed the Turbidimetric Solubility Assay.S.A. prepared the proteins by heterologous expression and performed the DSF studies. M.E. performed all computational investigation including the customized fragment filtering process. M.S. and F.M.B. selected the final compounds. M.S. and S.A. conducted data analysis and reprocessing of the experimental data. M.S. and F.M.B. wrote the manuscript. S.A., M.E., J.S., J.R., and A.L. critically reviewed the manuscript. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10(4):307–20. doi:10.1038/nrd3410
2. Gehringer M, Laufer SA. Emerging and re-emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J Med Chem. 2019;62(12):5673–5724. doi:10.1021/acs.jmedchem.8b01153
3. Baillie TA. Approaches to mitigate the risk of serious adverse reactions in covalent drug design. Expert Opin Drug Discov. 2021;16(3):275–287. doi:10.1080/17460441.2021.1832079
4. Barf T, Kaptein A. Irreversible protein kinase inhibitors: balancing the benefits and risks. J Med Chem. 2012;55(14):6243–6262. doi:10.1021/jm3003203
5. Spradlin JN, Zhang E, Nomura DK. Reimagining druggability using chemoproteomic platforms. Acc Chem Res. 2021;54(7):1801–1813. doi:10.1021/acs.accounts.1c00065
6. Dalton SE, Campos S. Covalent small molecules as enabling platforms for drug discovery. ChemBioChem. 2020;21(8):1080–1100. doi:10.1002/cbic.201900674
7. Hillebrand L, Gehringer M. Never gonna give you up – current developments in covalent protein kinase inhibitors. CHIMIA. 2022;76:435. doi:10.2533/chimia.2022.435
8. Boike L, Henning NJ, Nomura DK. Advances in covalent drug discovery. Nat Rev Drug Discov. 2022;21(12):881–898. doi:10.1038/s41573-022-00542-z
9. Hillebrand L, Liang XJ, Serafim RAM, Gehringer M. Emerging and re-emerging warheads for targeted covalent inhibitors: an update. J Med Chem. 2024;67(10):7668–7758. doi:10.1021/acs.jmedchem.3c01825
10. Schaefer D, Cheng X. Recent advances in covalent drug discovery. Pharmaceuticals. 2023;16(5):663. doi:10.3390/ph16050663
11. Joyce RP, Hu VW, Wang J. The history, mechanism, and perspectives of nirmatrelvir (PF-07321332): an orally bioavailable main protease inhibitor used in combination with ritonavir to reduce COVID-19-related hospitalizations. Med Chem Res. 2022;31(10):1637–1646. doi:10.1007/s00044-022-02951-6
12. Ito S, Otsuki S, Ohsawa H, et al. Discovery of futibatinib: the first covalent fgfr kinase inhibitor in clinical use. ACS Med Chem Lett. 2023;14(4):396–404. doi:10.1021/acsmedchemlett.3c00006
13. Lanman BA, Allen JR, Allen JG, et al. Discovery of a Covalent Inhibitor of KRAS G12C (AMG 510) for the treatment of solid tumors. J Med Chem. 2020;63(1):52–65. doi:10.1021/acs.jmedchem.9b01180
14. Fell JB, Fischer JP, Baer BR, et al. Identification of the clinical development candidate mrtx849, a covalent krasg12c inhibitor for the treatment of cancer. J Med Chem. 2020;63(13):6679–6693. doi:10.1021/acs.jmedchem.9b02052
15. Metcalf B, Chuang C, Dufu K, et al. Discovery of GBT440, an orally bioavailable r-state stabilizer of sickle cell hemoglobin. ACS Med Chem Lett. 2017;8(3):321–326. doi:10.1021/acsmedchemlett.6b00491
16. Dayalan Naidu S, Dinkova-Kostova AT. Omaveloxolone (Skyclarys(TM)) for patients with Friedreich’s ataxia. Trends Pharmacol Sci. 2023;44(6):394–395. doi:10.1016/j.tips.2023.03.005
17. Heppner DE, Ogboo BC, Urul DA, et al. Demystifying functional parameters for irreversible enzyme inhibitors. J Med Chem. 2024;67(17):14693–14696. doi:10.1021/acs.jmedchem.4c01721
18. Hoyt KW, Urul DA, Ogboo BC, et al. Pitfalls and considerations in determining the potency and mutant selectivity of covalent epidermal growth factor receptor inhibitors. J Med Chem. 2024;67(1):2–16. doi:10.1021/acs.jmedchem.3c01502
19. Lonsdale R, Ward RA. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018;47(11):3816–3830. doi:10.1039/C7CS00220C
20. Serafim RAM, Haarer L, Pedreira JGB, Gehringer M. Covalent chemical probes for protein kinases. Curr Res Chem Biol. 2023;3:100040. doi:10.1016/j.crchbi.2022.100040
21. Baillie TA. Targeted covalent inhibitors for drug design. Angew. Chem. Int. Ed. 2016;55(43):13408–13421. doi:10.1002/anie.201601091
22. De Cesco S, Kurian J, Dufresne C, Mittermaier AK, Moitessier N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017;138:96–114. doi:10.1016/j.ejmech.2017.06.019
23. Ábrányi-Balogh P, Keeley A, Ferenczy GG, et al. Next-generation heterocyclic electrophiles as small-molecule covalent mura inhibitors. Pharmaceuticals. 2022;15(12):1484. doi:10.3390/ph15121484
24. Keeley A, Ábrányi-Balogh P, Keserű GM. Design and characterization of a heterocyclic electrophilic fragment library for the discovery of cysteine-targeted covalent inhibitors. MedChemComm. 2019;10(2):263–267. doi:10.1039/C8MD00327K
25. Keeley A, Petri L, Ábrányi-Balogh P, Keserű GM. Covalent fragment libraries in drug discovery. Drug Discovery Today. 2020;25(6):983–996. doi:10.1016/j.drudis.2020.03.016
26. Orgován Z, Péczka N, Petri L, et al. Covalent fragment mapping of KRasG12C revealed novel chemotypes with in vivo potency. Eur. J. Med. Chem. 2023;250:115212. doi:10.1016/j.ejmech.2023.115212
27. Resnick E, Bradley A, Gan J, et al. Rapid covalent-probe discovery by electrophile-fragment screening. J Am Chem Soc. 2019;141(22):8951–8968. doi:10.1021/jacs.9b02822
28. Rothweiler EM, Brennan PE, Huber KVM. Covalent fragment-based ligand screening approaches for identification of novel ubiquitin proteasome system modulators. Biol. Chem. 2022;403(4):391–402. doi:10.1515/hsz-2021-0396
29. JD SD, Chessari G, Cleasby A, et al. X-ray screening of an electrophilic fragment library and application toward the development of a novel erk 1/2 covalent inhibitor. J Med Chem. 2022;65:12319–12333. doi:10.1021/acs.jmedchem.2c01044
30. Bum-Erdene K, Ghozayel MK, Xu D, Meroueh S. Covalent Fragment Screening Identifies Rgl2 RalGEF Cysteine for Targeted Covalent Inhibition of Ral GTPase Activation. ChemMedChem. 2022;17. doi:10.1002/cmdc.202100750.
31. Kathman SG, Xu Z, Statsyuk AV. A fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. J Med Chem. 2014;57(11):4969–4974. doi:10.1021/jm500345q
32. Nonomiya J, Li KS, Babin BM, Mulvihill MM. Covalent library screening by targeted mass spectrometry for rapid binding site identification. Anal. Chem. 2023;95:3779–3788. doi:10.1021/acs.analchem.2c04967
33. Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J. Electrophilic Fragment-Based Design of Reversible Covalent Kinase Inhibitors. J Am Chem Soc. 2013;135(14):5298–5301. doi:10.1021/ja401221b
34. Joerger AC, Bauer MR, Wilcken R, et al. Exploiting transient protein states for the design of small-molecule stabilizers of mutant p53. Structure. 2015;23(12):2246–2255. doi:10.1016/j.str.2015.10.016
35. Dammann M, Kramer M, Zimmermann MO, Boeckler FM. Quadruple target evaluation of diversity-optimized halogen-enriched fragments (heflibs) reveals substantial ligand efficiency for ap2-associated protein kinase 1 (aak1). original research. Front Chem. 2022;9. doi:10.3389/fchem.2021.815567.
36. Dammann M, Stahlecker J, Zimmermann MO, et al. Screening of a Halogen-Enriched Fragment Library Leads to Unconventional Binding Modes. J Med Chem. 2022;65(21):14539–14552. doi:10.1021/acs.jmedchem.2c00951
37. Heidrich J, Sperl LE, Boeckler FM. Embracing the diversity of halogen bonding motifs in fragment-based drug discovery-construction of a diversity-optimized halogen-enriched fragment library. Front Chem. 2019;7:9. doi:10.3389/fchem.2019.00009
38. Stahlecker J, Klett T, Schwer M, et al. Revisiting a challenging p53 binding site: a diversity-optimized HEFLib reveals diverse binding modes in T-p53C-Y220C.. RSC Med. Chem. 2022;13:1575–1586. doi:10.1039/D2MD00246A
39. Zimmermann MO, Lange A, Wilcken R, et al. Halogen-enriched fragment libraries as chemical probes for harnessing halogen bonding in fragment-based lead discovery. Future Med Chem. 2014;6(6):617–639. doi:10.4155/fmc.14.20
40. Bauer MR, Joerger AC, Fersht AR. 2-Sulfonylpyrimidines: mild alkylating agents with anticancer activity toward p53-compromised cells. Proc Natl Acad Sci U S A. 2016;113(36):E5271–80. doi:10.1073/pnas.1610421113
41. Cavallo G, Metrangolo P, Milani R, et al. The Halogen Bond. Chem Rev. 2016;116(4):2478–2601. doi:10.1021/acs.chemrev.5b00484
42. Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem. 2013;56(4):1363–1388. doi:10.1021/jm3012068
43. Lange A, Heidrich J, Zimmermann MO, Exner TE, Boeckler FM. Scaffold effects on halogen bonding strength. J Chem Inf Model. 2019;59(2):885–894. doi:10.1021/acs.jcim.8b00621
44. Riley KE, Murray JS, Fanfrlík J, et al. Halogen bond tunability I: the effects of aromatic fluorine substitution on the strengths of halogen-bonding interactions involving chlorine, bromine, and iodine. Journal of Molecular Modeling. 2011;17:3309–3318. doi:10.1007/s00894-011-1015-6
45. Heidrich J, Exner TE, Boeckler FM. Predicting the magnitude of σ-holes using vmaxpred, a fast and efficient tool supporting the application of halogen bonds in drug discovery. J Chem Inf Model. 2019;59(2):636–643. doi:10.1021/acs.jcim.8b00622
46. Klett T, Schwer M, Ernst LN, et al. Evaluation of a Covalent Library of Diverse Warheads (CovLib) Binding to JNK3, USP7, or p53. Drug Des Devel Ther. 2024;Volume 18:2653–2679. doi:10.2147/DDDT.S466829
47. Congreve M, Carr R, Murray C, Jhoti H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov Today. 2003;8(19):876–877. doi:10.1016/s1359-6446(03)02831-9
48. Ábrányi-Balogh P, Petri L, Imre T, et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. Chem. 2018;160:94–107. doi:10.1016/j.ejmech.2018.10.010
49. Keserű GM, Erlanson DA, Ferenczy GG, Hann MM, Murray CW, Pickett SD. Design principles for fragment libraries: maximizing the value of learnings from pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J Med Chem. 2016;59(18):8189–8206. doi:10.1021/acs.jmedchem.6b00197
50. Klett T, Stahlecker J, Jaag S, et al. Covalent fragments acting as tyrosine mimics for mutant p53-y220c rescue by nucleophilic aromatic substitution. ACS Pharmacol. Transl. Sci. 2024;7(12):3984–3999. doi:10.1021/acsptsci.4c00414
51. Klett T, Stahlecker J, Schwer M, et al. SNAr reactive pyrazine derivatives as p53-y220c cleft binders with diverse binding modes. Drug Des Devel Ther. 2025;19:4727–4753. doi:10.2147/DDDT.S513792
52. Joerger AC, Fersht AR. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu Rev Biochem. 2016;85:375–404. doi:10.1146/annurev-biochem-060815-014710
53. Kim MP, Lozano G. Mutant p53 partners in crime. Cell Death Differ. 2018;25(1):161–168. doi:10.1038/cdd.2017.185
54. Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13(6):1027–1036. doi:10.1038/sj.cdd.4401910
55. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi:10.1038/35042675
56. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6(12):909–923. doi:10.1038/nrc2012
57. Shangary S, Wang S. Small-molecule inhibitors of the mdm2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009;49:223–241. doi:10.1146/annurev.pharmtox.48.113006.094723
58. He C, Li L, Guan X, Xiong L, Miao X. Mutant p53 gain of function and chemoresistance: the role of mutant p53 in response to clinical chemotherapy. Chemotherapy. 2016;62(1):43–53. doi:10.1159/000446361
59. Knappskog S, Lønning PE. P53 and its molecular basis to chemoresistance in breast cancer. Expert Opin. Ther. Targets. 2012;16(sup1):S23–S30. doi:10.1517/14728222.2011.640322
60. Blagih J, Buck MD, Vousden KH. p53, cancer and the immune response. J Cell Sci. 2020;133(5):jcs237453. doi:10.1242/jcs.237453
61. Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15(1):2–8. doi:10.1038/ncb2641
62. Joerger AC, Fersht AR. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene. 2007;26(15):2226–2242. doi:10.1038/sj.onc.1210291
63. Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–582. doi:10.1146/annurev.biochem.77.060806.091238
64. Bullock AN, Henckel J, Fersht AR. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene. 2000;19(10):1245–1256. doi:10.1038/sj.onc.1203434
65. Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, Fersht AR. Targeted rescue of a destabilized mutant of p53 by anin silico screened drug. Proc Natl Acad Sci. 2008;105(30):10360–10365. doi:10.1073/pnas.0805326105
66. Basse N, Kaar JL, Settanni G, Joerger AC, Rutherford TJ, Fersht AR. Toward the rational design of p53-stabilizing drugs: probing the surface of the oncogenic Y220C mutant. Chem Biol. 2010;17(1):46–56. doi:10.1016/j.chembiol.2009.12.011
67. Bauer M, Kraemer A, Settanni G, et al. Targeting cavity-creating p53 cancer mutations with small-molecule stabilizers: the y220x paradigm. ACS Chem. Biol. 2020;15:657–668. doi:10.1021/acschembio.9b00748
68. Duffy MJ, Tang M, Rajaram S, O’Grady S, Crown J. Targeting mutant p53 for cancer treatment: moving closer to clinical use? Cancers. 2022;14(18):4499. doi:10.3390/cancers14184499
69. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18(2):89–102. doi:10.1038/nrc.2017.109
70. Joerger AC, Fersht AR. The tumor suppressor p53: from structures to drug discovery. Cold Spring Harb Perspect Biol. 2010;2(6):a000919. doi:10.1101/cshperspect.a000919
71. Wilcken R, Liu X, Zimmermann MO, et al. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J Am Chem Soc. 2012;134(15):6810–6818. doi:10.1021/ja301056a
72. Stephenson Clarke JR, Douglas LR, Duriez PJ, et al. Discovery of nanomolar-affinity pharmacological chaperones stabilizing the oncogenic p53 mutant Y220C. ACS Pharmacol Transl Sci. 2022;5(11):1169–1180. doi:10.1021/acsptsci.2c00164
73. Wilcken R, Zimmermann MO, Bauer MR, et al. Experimental and theoretical evaluation of the ethynyl moiety as a halogen bioisostere. ACS Chem. Biol. 2015;10(12):2725–2732. doi:10.1021/acschembio.5b00515
74. Baud M, Bauer M, Verduci L, et al. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 2018;152:101–114. doi:10.1016/j.ejmech.2018.04.035
75. Vu BT, Dominique R, Fahr BJ, et al. Discovery of Rezatapopt (PC14586), a First-in-Class, Small-Molecule Reactivator of p53 Y220C Mutant in Development. ACS Med Chem Lett. 2025;16(1):34–39. doi:10.1021/acsmedchemlett.4c00379
76. Guiley KZ, Shokat KM. A small molecule reacts with the p53 somatic mutant y220c to rescue wild-type thermal stability. Cancer Discov. 2023;13(1):56–69. doi:10.1158/2159-8290.Cd-22-0381
77. Wildman SA, Crippen GM. Prediction of physicochemical parameters by atomic contributions. J Chem Inf Comput Sci. 1999;39(5):868–873. doi:10.1021/ci990307l
78. Release S. New. York: Schrödinger. Inc; 2021.
79. Proj M, Knez D, Sosič I, Gobec S. Redox active or thiol reactive? Optimization of rapid screens to identify less evident nuisance compounds. Drug Discovery Today. 2022;27(6):1733–1742. doi:10.1016/j.drudis.2022.03.008
80. Birkholz A, Kopecky DJ, Volak LP, et al. Systematic study of the glutathione reactivity of n-phenylacrylamides: 2. effects of acrylamide substitution. J Med Chem. 2020;63(20):11602–11614. doi:10.1021/acs.jmedchem.0c00749
81. Cee VJ, Volak LP, Chen Y, et al. Systematic study of the glutathione (gsh) reactivity of n-arylacrylamides: 1. effects of aryl substitution. J Med Chem. 2015;58(23):9171–9178. doi:10.1021/acs.jmedchem.5b01018
82. Grabrijan K, Hrast M, Proj M, et al. Covalent inhibitors of bacterial peptidoglycan biosynthesis enzyme MurA with chloroacetamide warhead. Eur J Med Chem. 2022;243:114752. doi:10.1016/j.ejmech.2022.114752
83. Keeley A, Ábrányi-Balogh P, Hrast M, et al. Heterocyclic electrophiles as new MurA inhibitors. Arch Pharm. 2018;351(12):e1800184. doi:10.1002/ardp.201800184
84. Baud M. 2-sulfonylpyrimidines: reactivity adjustable agents for cysteine arylation. 2023.
85. Gerstenecker S, Haarer L, Schröder M, et al. Discovery of a potent and highly isoform-selective inhibitor of the neglected ribosomal protein s6 kinase beta 2 (S6K2). Cancers. 2021;13(20). 10.3390/cancers13205133.
86. Pichon MM, Drelinkiewicz D, Lozano D, et al. Structure–reactivity studies of 2-sulfonylpyrimidines allow selective protein arylation. Bioconjugate Chem. 2023;34(9):1679–1687. doi:10.1021/acs.bioconjchem.3c00322
87. Malona J, Chuaqui C, Seletsky BM, et al. Discovery of CC-99677, a selective targeted covalent MAPKAPK2 (MK2) inhibitor for autoimmune disorders. Transl Res. 2022;249:49–73. doi:10.1016/j.trsl.2022.06.005
88. Gaur R, Mensah KA, Stricker J, et al. CC-99677, a novel, oral, selective covalent MK2 inhibitor, sustainably reduces pro-inflammatory cytokine production. Arthritis Res Ther. 2022;24(1):199. doi:10.1186/s13075-022-02850-6
89. Hall A, Abendroth J, Bolejack MJ, et al. Discovery and characterization of a novel series of chloropyrimidines as covalent inhibitors of the kinase MSK1. ACS Med Chem Lett. 2022;13(7):1099–1108. doi:10.1021/acsmedchemlett.2c00134
90. Mayr H. Modern nucleophilic aromatic substitution. by francois terrier. Angew. Chem. Int. Ed. 2014;53(28):7119. doi:10.1002/anie.201404183
91. Taylor RD, MacCoss M, Lawson ADG. Rings in Drugs. J Med Chem. 2014;57(14):5845–5859. doi:10.1021/jm4017625
92. Lawson ADG, MacCoss M, Heer JP. Importance of rigidity in designing small molecule drugs to tackle protein–protein interactions (ppis) through stabilization of desired conformers. J Med Chem. 2018;61(10):4283–4289. doi:10.1021/acs.jmedchem.7b01120
93. Jackson PA, Widen JC, Harki DA, Brummond KM. Covalent modifiers: a chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-michael addition reactions. J Med Chem. 2017;60(3):839–885. doi:10.1021/acs.jmedchem.6b00788
94. Holm KJ, Spencer CM. Entacapone. Drugs. 1999;58(1):159–177. doi:10.2165/00003495-199958010-00017
95. Londregan AT, Jennings S, Wei L. Mild addition of nucleophiles to pyridine- n -oxides. Org Lett. 2011;13(7):1840–1843. doi:10.1021/ol200352g
96. Kobus M, Friedrich T, Zorn E, Burmeister N, Maison W. Medicinal chemistry of drugs with n-oxide functionalities. J Med Chem. 2024;67(7):5168–5184. doi:10.1021/acs.jmedchem.4c00254
 © 2026 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.
© 2026 The Author(s). This work is published by Dove Medical Press Limited, and licensed under a
Creative Commons Attribution License.
The full terms of the License are available at http://creativecommons.org/licenses/by/4.0/.
The license permits unrestricted use, distribution, and reproduction in any medium, provided the
original author and source are credited.