")
Back to Journals » Infection and Drug Resistance » Volume 11
Characterization of hepatitis C virus resistance to grazoprevir reveals complex patterns of mutations following on-treatment breakthrough that are not observed at relapse
Authors Bonsall D, Black S , Howe AYM, Chase R, Ingravallo P, Pak I , Brown A , Smith DA , Bowden R , Barnes E
Received 9 November 2017
Accepted for publication 31 January 2018
Published 8 August 2018 Volume 2018:11 Pages 1119—1135
DOI https://doi.org/10.2147/IDR.S156581
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Joachim Wink
David Bonsall,1 Stuart Black,2 Anita YM Howe,2 Robert Chase,2 Paul Ingravallo,2 Irene Pak,2 Anthony Brown,1 David A Smith,1 Rory Bowden,1 Eleanor Barnes1
On behalf of the STOP HCV Consortium
1Translational Gastroenterology Unit, University of Oxford, Oxford, UK; 2Department of Infectious Diseases, Merck & Co., Inc., Kenilworth, NJ, USA
Purpose: A detailed analysis of hepatitis C virus (HCV) resistance-associated substitutions (RASs) is required to understand why people fail direct-acting antiviral therapies. This study was conducted to assess RASs in an analysis of 2 trials evaluating the second-generation NS3/4A protease inhibitor grazoprevir (GZR) in combination with peginterferon/ribavirin.
Patients and methods: From a total of 113 participants with HCV genotype 1 infection, RASs were evaluated in 25 patients who relapsed and 6 patients with on-treatment virologic breakthrough using consensus Sanger and clonal sequence analysis of NS3/NS4a genes, with in vitro testing of replicon mutants. Next-generation sequencing (NGS) was used in a subset of participants to assess minority variants and the evolution of the whole viral genome.
Results: Baseline RASs did not predict treatment failure. Relapse was most commonly associated with RASs at D168, although additional RASs (Y56, R155 and A156) were also detected, particularly in participants with on-treatment breakthrough. Treatment-emergent RASs usually reverted to wild-type (WT), suggesting these mutations were associated with a negative fitness cost (confirmed using in vitro assays). NGS was the most sensitive assay for the detection of minor variants. Significant viral sequence divergence (up to 5.9% codons) was observed across whole genomes in association with the acquisition and reversion of RASs.
Conclusion: Relapse with GZR and peginterferon/ribavirin is commonly associated with single RASs in NS3 that generally revert to WT, while breakthrough follows more complex patterns of viral resistance. NGS suggests that large diverse pools of viral quasispecies that emerge with RASs facilitate rapid viral evolution.
Keywords: next-generation sequencing, NS3/4A protease inhibitor, resistance-associated substitution, virologic failure
Introduction
Standard-of-care therapy for people with hepatitis C virus (HCV) genotype 1 (GT1) infection involves the use of direct-acting antiviral agents (DAAs).1,2 These agents disrupt viral replication and cause rapid reductions in HCV RNA titer by targeting virally encoded proteins such as the nonstructural NS3/4A protease that is responsible for cleavage of the viral polyprotein3 and may also counteract the innate immune response.4 Specific amino acid substitutions in the NS3/4A protein are known to confer resistance to protease inhibitors and are thought to have limited impact on viral replication. Consequently, populations of resistance-associated substitutions (RASs) may be selected under conditions of NS3/4A inhibition, preventing viral eradication and causing virologic failure.5–8 Emerging data show that RASs, particularly those emerging after failed treatment with NS5A inhibitors, may endure long after treatment is stopped, thus restricting options for re-treatment of previous virologic failures.9,10 Additionally, there is growing concern that RASs may become fixed and transmitted between hosts, compromising the long-term efficacy of entire classes of DAAs with overlapping resistance profiles. The error-prone replication of HCV, coupled with high viral turnover from infected hepatocytes, leads to a high frequency of mutations11 and divergence of quasispecies with minor genomic differences within hosts, so that deep sequencing may be required to understand the contribution of minor viral variants on treatment outcomes.
Grazoprevir (GZR) (MK-5172) (Merck & Co., Inc., Kenilworth, NJ, USA) is a second-generation macrocyclic NS3/4A protease inhibitor that is active against multiple HCV GTs and variants that are resistant to first-generation protease inhibitors.8 Recently, GZR has become first-line treatment in the UK in combination with the NS5A inhibitor elbasvir, having been endorsed by National Institute for Health and Care Excellence (NICE) and commissioned by National Health Service (NHS) England. Several Phase II and III clinical trials have shown high efficacy and favorable tolerability of GZR in combination with elbasvir in diverse populations with HCV GT1 infection.12–18 Earlier studies of treatment-naive participants with HCV GT1 infection examined the safety/efficacy profile of GZR in combination with both peginterferon (PEG-IFN) alfa-2b and ribavirin (RBV),19 or when combined with RBV alone.20 In this report, we describe a pooled analysis of baseline and treatment-emergent RASs detected in treatment-naive individuals with HCV GT1 infection who received GZR as part of triple therapy with PEG-IFN plus RBV or dual therapy with RBV alone. These 2 studies provided an optimal setting for the evaluation of NS3 RASs associated with GZR without interference by other DAAs.
The aims of these studies were to characterize resistance to GZR by sequencing both aggregate viral populations and individual quasispecies before and after treatment failure. Several groups are now working toward upgrading clinical sequencing assays from traditional Sanger-based approaches to next-generation methods, with advantages in throughput, cost and their ability to detect minor variants through the sequencing of whole genomes. One such next-generation sequencing (NGS) method, veSEQ, combines the Illumina RNAseq protocol with virus-targeted probe-based enrichment, which we recently showed to be unbiased toward viral GTs and considerably more sensitive than similar capture-free Illumina-based methods.21
Our objectives were to compare veSEQ with consensus and clonal sequencing methods and explore the capabilities and limitations of each in detecting RASs. We also explored changing patterns of within-host viral diversity and divergence following treatment failure.
Patients and methods
This analysis was based on pooled data from 2 multicenter trials in treatment-naive participants with HCV GT1 infection evaluating the efficacy and safety of GZR in combination with PEG-IFN alfa-2b and RBV (NCT01710501; protocol PN038)19 or in combination with RBV alone (NCT01716156; protocol PN039).20 Both studies were conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, and in compliance with institutional review board (see Box S1 for list of institutional review boards) and regulatory agency requirements. Written informed consent was obtained from each participant prior to entering the trial.
Trial design and participants
The study design and efficacy and safety results for both studies have been reported previously.19,20 PN038 was a randomized, dose-ranging, multicenter trial conducted in Canada, New Zealand, Sweden and the UK. Participants were randomized to receive oral GZR 25, 50 or 100 mg once daily (QD), in combination with PEG-IFN alfa-2b (1.5 μg/kg/week) and RBV (weight-based dosing of 800–1400 mg/day) for 12 weeks. Participants with quantifiable HCV RNA at treatment week (TW) 4 received an additional 12 weeks of PEG-IFN/RBV.
PN039 was a randomized, open-label, multicenter trial conducted in New Zealand, Israel, Australia and the USA. All participants received oral GZR 100 mg QD in combination with RBV (weight-based dosing of 800–1400 mg/day) for a variable treatment duration according to on-treatment virologic response (arm 1) or to a fixed treatment duration of 24 weeks (arm 2). In arm 1, participants with undetectable HCV RNA at TW4 stopped treatment at TW12, whereas those with detectable but unquantifiable HCV RNA at TW4 (<25 IU/mL) were treated for 24 weeks. Participants with detectable HCV RNA at TW4 (≥25 IU/mL confirmed within 2 weeks) were discontinued due to futility.
Both studies enrolled treatment-naive adults with chronic, compensated HCV GT1 infection (with at least 50% required to have HCV GT1a infection) and all participants had HCV RNA levels ≥10,000 IU/mL at enrollment. Participants with signs or symptoms of advanced liver disease or cirrhosis, co-infection with HIV or hepatitis B virus or evidence or history of chronic hepatitis not caused by HCV infection were excluded. Plasma concentrations of GZR were monitored throughout the treatment period.
Definitions of response and virologic failure
The primary end point of both studies was sustained virologic response (SVR), defined as HCV RNA <25 IU/mL 12 weeks after the end of all study therapy. Virologic failure was categorized as: 1) breakthrough while on treatment (HCV RNA ≥25 IU/mL after being <25 IU/mL); 2) end-of-treatment failure (detectable HCV RNA at the end-of-treatment visit) or 3) relapse (HCV RNA ≥25 IU/mL following end of all study therapy, after becoming undetectable at end of treatment). All cases of virologic failure were confirmed by repeat virologic testing within 2 weeks of the initial failure. The number of participants included in the resistance analyses was based on the full analysis set populations of each study (including all randomized participants who received ≥1 dose of study treatment).
Consensus Sanger and TOPO clonal sequencing of NS3/NS4a RASs
Blood samples for resistance assays were collected from all participants at the baseline visit, at the time of virologic failure or at the end-of-treatment visit and at follow-up visits at weeks 4, 12 and 24. Plasma HCV RNA levels were measured using the Roche COBAS® Taqman® HCV Test v2.0, which has a lower limit of quantitation of 25 IU/mL. To assess the presence of polymorphisms at baseline or time of virologic failure, the HCV NS3/4A gene was amplified using the reverse transcriptase-polymerase chain reaction (PCR) followed by consensus Sanger and selective clonal sequencing. Due to the sensitivity of the consensus sequencing assay, resistance analysis was performed only on samples from participants with HCV viral loads >1000 IU/mL. The limit of variant detection using consensus Sanger sequencing was >25% of viral quasispecies. For clonal sequencing, PCR amplification was performed only on the NS3 protease region (amino acids 1–181) and the resultant amplicons cloned into a TOPO TA vector (Thermo Fisher Scientific, Waltham, MA, USA). Approximately 40 clones were sequenced at each time point and resultant amino acid sequences were compared with wild-type (WT) HCV GT1a (H77) or GT1b (Con1) reference sequences.
Definition of RASs
While all amino acid positions within NS3/4A were examined, single NS3-protease amino-acid substitutions, including NS3 V36A/G/L/M/I, T54A/C/G/S, V55A/I, Y56H, Q80K/R, V107I, S122A/G/R, I132V, R155X, A156S/T/V/F/G, V158I, D168X, I170F/T/V (GT1a virus), V170A/T (GT1b virus) and M175L, were considered clinically relevant because these mutations are commonly identified after treatment failures with protease inhibitors.2,22
NGS using veSEQ
NGS was performed at baseline and at 1–3 sequential post-treatment time points in 4 participants with treatment failure. A total of 5 μL RNA extracted from 1 mL of plasma (maximum 10 ng total RNA) was used to construct NEBNext® Ultra™ Directional RNA sequencing libraries (Illumina®; New England Biolabs, Ipswich, MA, USA) with previously published modifications to the manufacturer’s protocol.21,23 A 500-ng aliquot of the pooled library was enriched using the xGen® Lockdown® protocol from Integrated DNA Technologies (IDT) (Rapid Protocol for DNA Probe Hybridization and Target Capture Using an Illumina TruSeq® Library [v1.0], IDT) with equimolar-pooled 120-nt DNA oligonucleotide probes (IDT) designed according to a bespoke, recently published algorithm.23 The enriched library was sequenced on a single run of the Illumina MiSeq using v2 chemistry. An in-house bioinformatic pipeline demultiplexed and trimmed the 150 base paired-end reads (quantification and annotation of short reads in R [QuasR], Cutadapt), before subtracting host-derived sequences (BWA) and selecting reads of viral origin (blastn) for de novo assembly (Vicuna), read mapping (MOSAIK), genome annotation (VFAT) and interpretation of mutations (genewise2, Vphaser, Vprofile).24,25
Assessment of RASs in vitro
Half maximal effective concentrations (EC50) of GZR were determined for WT and mutated replicons carrying the principal RASs reported for boceprevir, telaprevir, simeprevir and paritaprevir as well as GZR. Stable replicon assays were conducted in gt1a (H77) or gt1b (Con1) Huh 7.5-cell lines, with EC50 titers calculated using quantitative PCR (Taqman) assay as the end point measure.8 Transient replicon assays were only conducted in gt1a (H77) Huh 7.5-cell lines using a luciferase based assay as the end point measure.26
Results
Summary of clinical outcomes
A total of 87 participants were enrolled in PN038 and 26 were enrolled in PN039, and all 113 participants received at least 1 dose of study drug. All participants had viral load suppressed to undetectable levels at one or more time points during treatment (ie, there were no nonresponders). SVR was achieved by 75 participants (66.4%), and 38 (33.6%) failed to attain SVR. Viral loads, treatment durations and drug levels in participants who failed therapy are detailed in Figure S1. Of the 38 participants who failed to attain SVR, 7 discontinued treatment due to administrative reasons or because of an adverse event and 31 experienced virologic failure. Among the 31 participants with virologic failure, 25 relapsed, 6 had on-treatment virologic failure, 18 were treated with GZR doses of 25–50 mg/day and 13 received GZR 100 mg/day (Table 1).
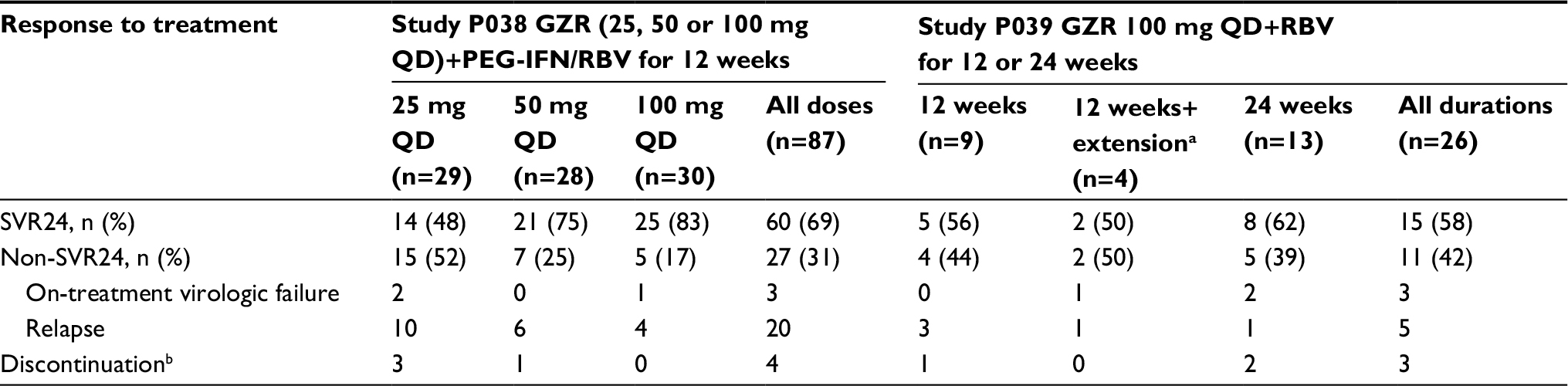 | Table 1 Sustained virologic response rates at follow-up week 24 (SVR24) and reasons for virologic failure with GZR in the full analysis set populations of studies P038 and P039 Notes: aFour participants in the 12-week arm had quantifiable hepatitis C virus RNA at treatment week 4 and received an additional 12 weeks of treatment. bDiscontinuation was due to adverse events or administrative reasons. Abbreviations: GZR, grazoprevir; PEG-IFN/RBV, peginterferon/ribavirin; QD, once daily; RBV, ribavirin; SVR24, sustained virologic response at 24 weeks after end of treatment. |
Of the 113 participants, 83 (73.5%) had HCV GT1a infection and 29 (25.7%) had HCV GT1b infection (1 participant had GT1-other infection). Subgenotype was not associated with treatment outcome: virologic failure was observed in 30/83 (36.1%) participants with GT1a infection and 8/29 (27.6%) participants with GT1b infection.
Undetectable drug levels, suggestive of nonadherence, were observed in 2 participants who relapsed (38-G and 38-N) and 1 participant with virologic breakthrough (38-W) (Figure S1).
Baseline RASs detected by consensus Sanger sequencing and effect on treatment outcome
For all 113 participants, baseline resistance was characterized by consensus Sanger sequencing of NS3 gene segments (aa 1–181), spanning all polymorphic sites known to be associated with resistance to any class of protease inhibitor. Baseline RASs were detected in 38/87 (43.7%) participants enrolled in PN038 and in 5/26 (19.2%) participants in PN039 (Table 2). The most common baseline RASs (occurring in >5% of participants) were T54S, V55A/I, Q80K and I170V (GT1a-specific) in PN038 and T54S, V55A/I and S122G in PN039. The Q80K polymorphism was detected at baseline in 24/71 (33.8%) participants from PN038 with HCV GT1a infection, consistent with the expected frequency and GT association of this polymorphism. Only 12 participants with GT1a infection were enrolled in PN039, and none of these possessed the Q80K mutations at baseline. In addition, R155K and V36M/L RASs were detected at baseline in 1 and 3 participants, respectively, all but 1 of whom achieved SVR. Only WT or common variants not previously associated with resistance were observed at baseline at positions Y56, V107, I132, A156, V158, D168 and M175.
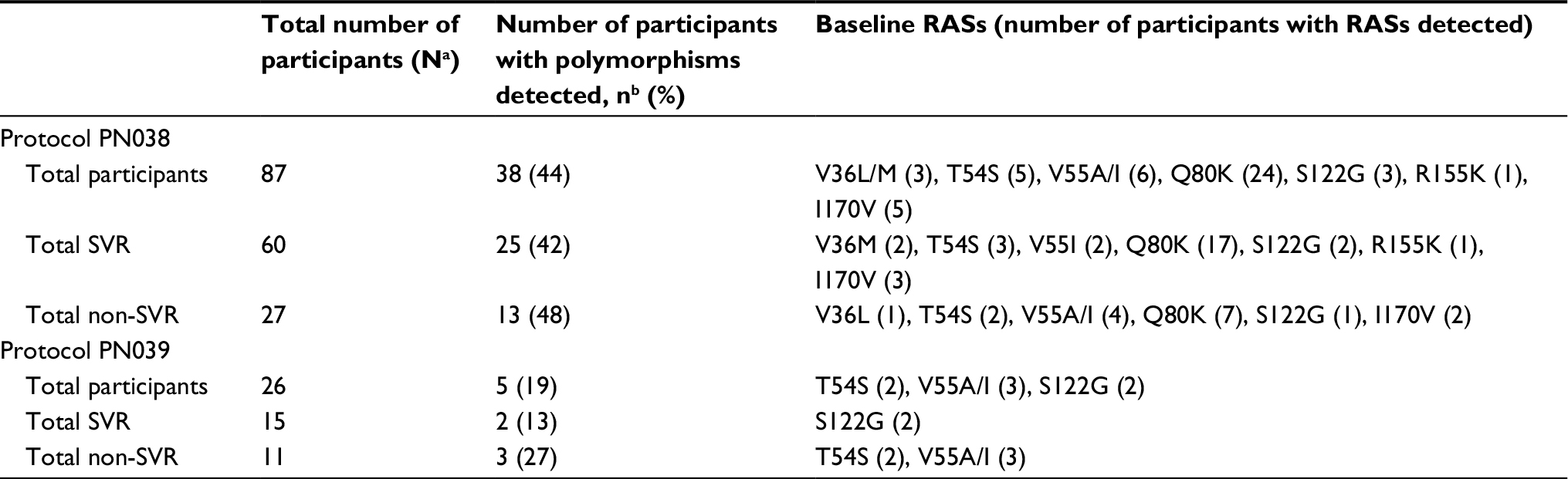 | Table 2 Impact of baseline RASs on treatment outcomes Notes: aN, number of participants who received study therapy; bn, number of participants who had the specified RAS(s) detected at baseline. The following RAS(s) within NS3 were selected for reporting: −36A/G/L/M/I, −54A/C/G/S, −55A/I, −56H, −80K/R, −107I, −122A/G/R, 132V, 155X, 156S/T/V/F/G, 158I, 168X, 170A/F/T/V and 175L. Abbreviations: RAS, resistance-associated substitution; SVR, sustained virologic response. |
No single RAS or combination of RASs at baseline predicted treatment outcome with statistical significance in either study, including the Q80K mutation which was detected in 17/60 (28.3%) participants who achieved SVR compared with 7/27 (25.9%) who failed. In PN038, SVR was achieved by 25/38 (65.8%) participants with ≥1 baseline RASs and by 35/49 (71.4%) participants without baseline RASs (p=0.57, chi-squared). Thirteen participants with baseline RASs failed to achieve SVR: 11 relapsed and 2 discontinued treatment for reasons unrelated to virologic response. Similarly, in PN039, SVR was achieved by 2/5 participants (40.0%) with baseline RASs and 13/21 participants (61.9%) without RASs (p=0.38, chi-squared). Of the 3 participants with baseline RASs who failed to achieve SVR, 2 discontinued due to nonvirologic reasons and 1 relapsed.
Effects of RASs on sensitivity to GZR in vitro
Baseline RASs were assessed for their effects on susceptibility to GZR in vitro (Table 3). Of the common baseline RASs (T54S, V55A/I, Q80K, I170V and S122G), only the I170V mutation had a notable impact, increasing GZR EC50 by 2.6-fold in the GT1a replicon. Two of the 5 participants with GT1a infection carrying this mutation achieved SVR. A modest 3.3-fold increase in GZR EC50 was observed in a replicon carrying the R155K mutation compared with WT, and the 1 participant who carried this mutation achieved SVR.
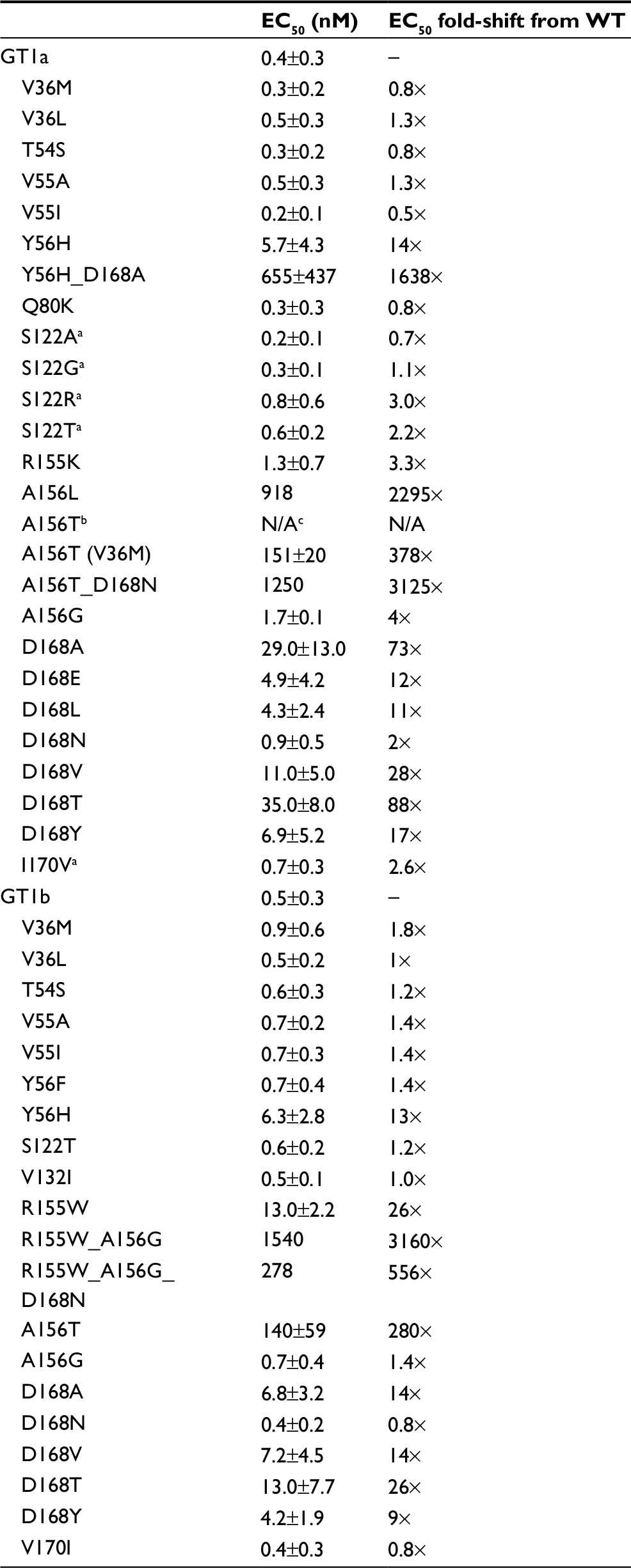 | Table 3 Activities of GZR in HCV replicon variants Notes: All data were obtained from stable replicons from database 07-10-2014. Replicons that harbor this substitution did not survive G418 selection. aTransient Infectious Virus System; gt1a H77=0.27 nM. bReplicon unfit. cN/A, not available. Replicons that harbor this substitution did not survive G418 selection. Abbreviations: EC50, half maximal effective concentration; GT, genotype; GZR, grazoprevir; HCV, hepatitis C virus; WT, wild-type. |
RASs detected by consensus Sanger sequencing emerging after treatment failure
In participants who failed to achieve SVR, further sequencing was performed on samples taken either at the time of breakthrough or following relapse at follow-up week (FW) 4, 12 or 24. Virology data were unavailable for 1 participant with insufficient viral RNA and a second for whom sequencing failed.
A total of 25 participants relapsed (PN038, n=20; PN039, n=5), 16 of whom had received GZR 25 or 50 mg QD, which is below the clinically utilized dose of 100 mg QD (Table 4). In total, 16 participants developed novel resistance mutations post-treatment that were not present at baseline (treatment-emergent), 13 of whom developed a D168A/E/N/T/V/Y polymorphism. Mutation A156T was the only treatment-emergent RAS in participant 38-T and was also found in combination with I170V in participant 38-O, who had GT1a infection. A treatment-emergent S122T mutation was detected in 1 participant who had WT virus at baseline (participant 39-D). Among participants receiving GZR at the current recommended dose of 100 mg, 2 had treatment-emergent D168A RASs (1 participant receiving GZR+RBV and the other receiving GZR+PEG-IFN/RBV) and 1 participant receiving GZR+RBV had a treatment-emergent S122T RAS (Table 4).
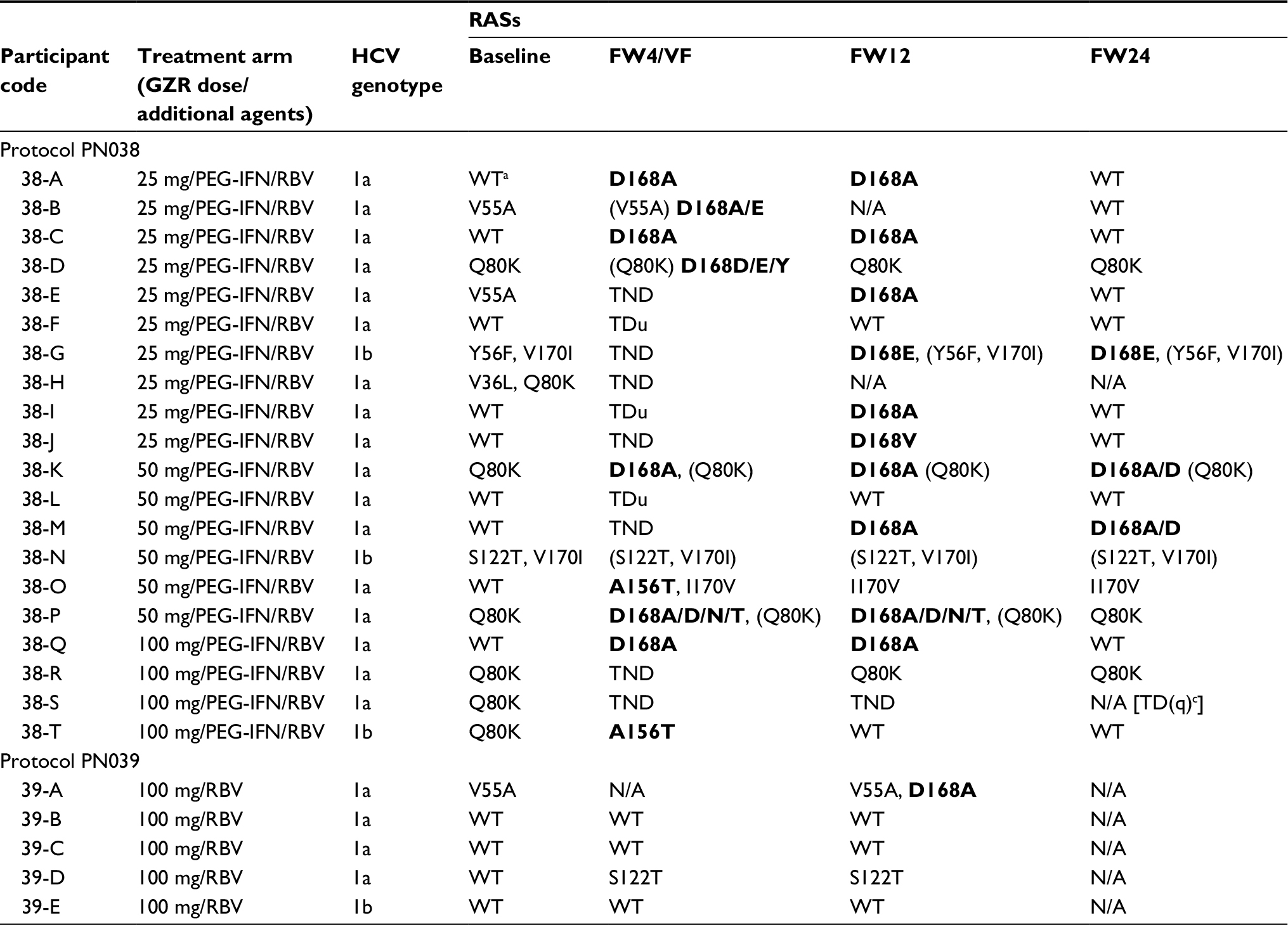 | Table 4 Participants with relapse (consensus Sanger sequence) Notes: Only the RASs V36A/G/L/M/I, T54A/C/G/S, V55A/I, Y56H, Q80K/R, V107I, 122A/G/R, I132V, R155X, A156S/T/V/F/G, V158I, D168X, I/V170A/F/T/V and M175L are indicated. The RASs to GZR are noted in bold. In participants 38-S and 38-H, no data are available due to sequencing failure. aWT, wild-type; GZR RASs not detected by consensus Sanger sequencing. Abbreviations: FW, follow-up week; GZR, grazoprevir; HCV, hepatitis C virus; N/A, not available; PEG-IFN/RBV, peginterferon/ribavirin; RAS, resistance-associated substitution; RBV, ribavirin; TD(q), target detected (quantifiable) (HCV RNA >25 IU/mL); TDu, target detected (unquantifiable) (HCV RNA <25 IU/mL); TND, target not detected; VF, virologic failure; WT, wild-type. |
Six participants had on-treatment virologic breakthrough, including 2 who had viremia detected at the end of treatment (38-U and 38-V) (Table 5). At baseline, all participants with breakthrough had WT virus except participant 38-W, who possessed a Y56F polymorphism. This polymorphism is present in 15% of all GT1b infections, and in vitro did not confer resistance to GZR (Table 3). All but 1 participant with breakthrough (38-V) had treatment-emergent RASs at the time of breakthrough. Participant 38-U developed an A156L substitution at the time of failure that involved 2 nucleotide substitutions from a baseline GCC(A) codon to CTC(L) codon at FW4, which then reverted to the original GCC(A) codon within 63 days. Participant 38-V had breakthrough viremia at the end of treatment, with only WT virus and no RASs detected. GZR concentrations in participant 38-V were persistently below the 25-mg PEG-IFN/RBV Ctrough threshold, suggesting that low drug levels rather than drug resistance contributed to virologic failure.
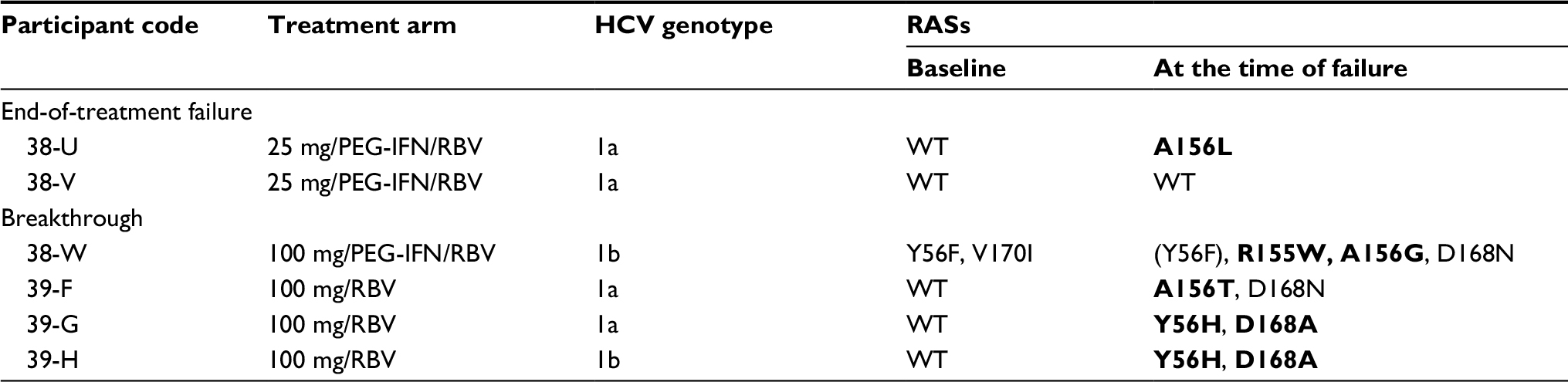 | Table 5 Participants with end-of-treatment failure and breakthrough (consensus Sanger sequence) Note: RASs with grazoprevir resistance are shown in bold. Abbreviations: HCV, hepatitis C virus; PEG-IFN/RBV, peginterferon/ribavirin; RAS, resistance-associated substitution; RBV, ribavirin; WT, wild-type. |
In-depth analysis of viral quasispecies: TOPO TA cloning versus NGS
To further explore the mechanisms of viral evolution that may contribute to virologic failure, we used clonal sequencing and veSEQ (Illumina) of selected samples to characterize the virus quasispecies that existed at low frequency. In particular, we sought to identify patterns of acquisition and reversion of principle RASs that were associated with treatment failure by examining these and other mutations, both in NS3 and along the entire genome. In total, 40 TOPO-cloned NS3 gene fragments from 20 participants with relapse and 5 with breakthrough (including 2 end-of-treatment failures) were sequenced at baseline and at 1–4 time points after treatment failure (Figure 1). In addition, whole genome sequences were obtained at 2–3 time points from 4 of these participants by veSEQ (2 with relapse and 2 with breakthrough) (Figures 2–4). Sequence coverage of the HCV genome correlated with viral load, and ranged from 718 to 51,714 reads mapped, which equated to average depths of 10–741 reads per nucleotide site after removal of PCR-duplicated read pairs.
 | Figure 1 Longitudinal sampling of clonal NS3 sequences after treatment failure. Bars show percentages of resistance-associated substitutions at sites with established in vitro and in vivo evidence for GZR resistance. Clonal sequences were obtained from 40 bacterial colonies transformed with NS3 sequences prior to treatment (day 1; D1), at VF at subsequent clinic visits where VF was confirmed, and at fixed follow-up visits (FW4, 12 and 24). The most complicated patterns of resistance were observed following early treatment breakthrough, characterized by multiple mutations that variably persisted beyond 24-week post-treatment failure. Abbreviations: D, day; FW, follow-up week; GZR, grazoprevir; VF, virologic failure; VF (conf), virologic failure confirmed; WT, wild-type. |
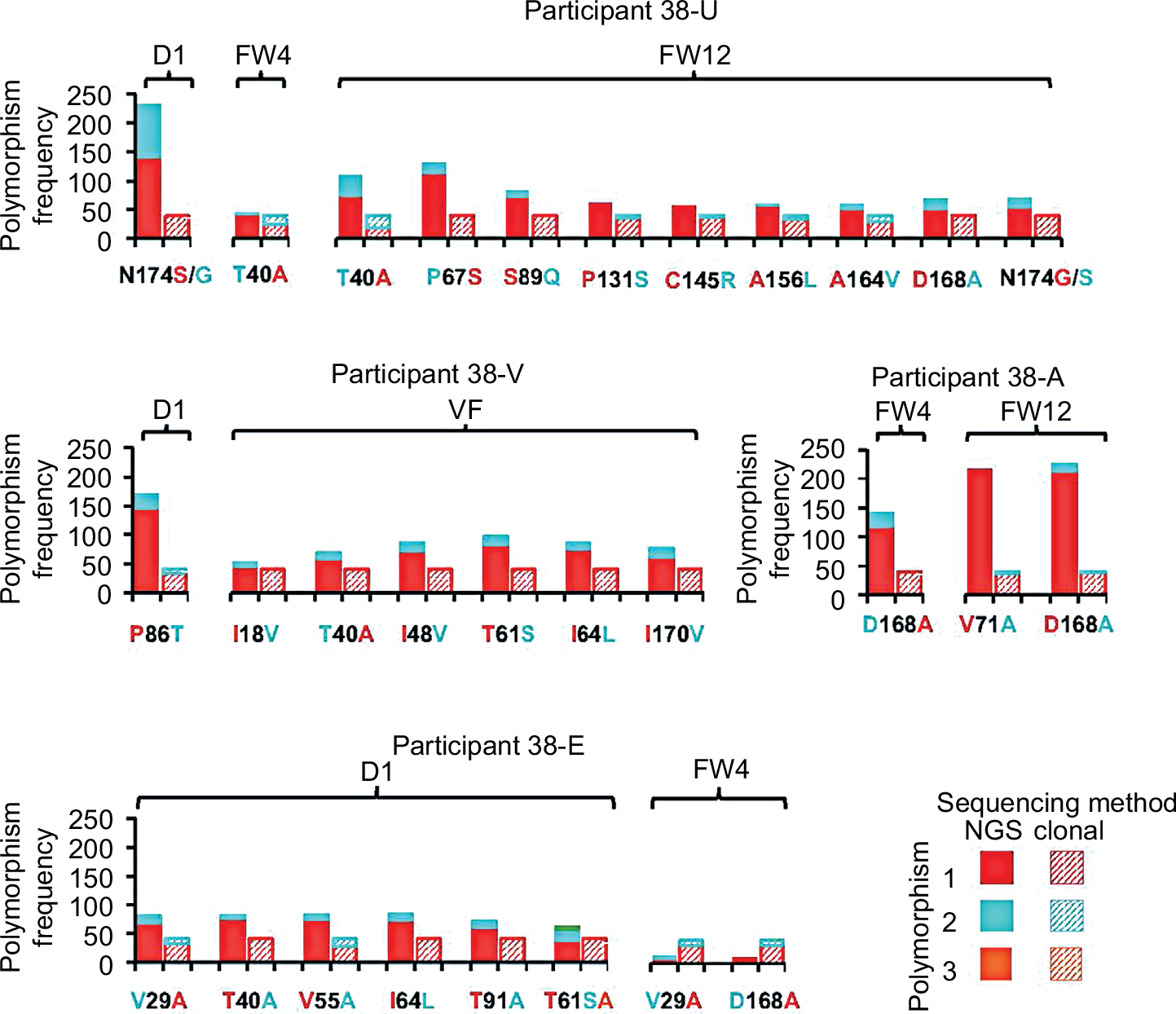 | Figure 2 Detection of low-frequency NS3 polymorphisms by clonal sequencing NGS. Within-host variation at all polymorphic sites within the NS3 gene (amino acids 1–180) are shown for NGS versus clonal sequencing. For each participant time point, 40 TOPO-cloned NS3 gene fragments were sequenced, with the proportion of each amino acid residue shown as hatched bars alongside respective codons sequenced by NGS after bioinformatic removal of PCR duplicates. Abbreviations: D, day; FW, follow-up week; NGS, next-generation sequencing; PCR, polymerase chain reaction; VF, virologic failure. |
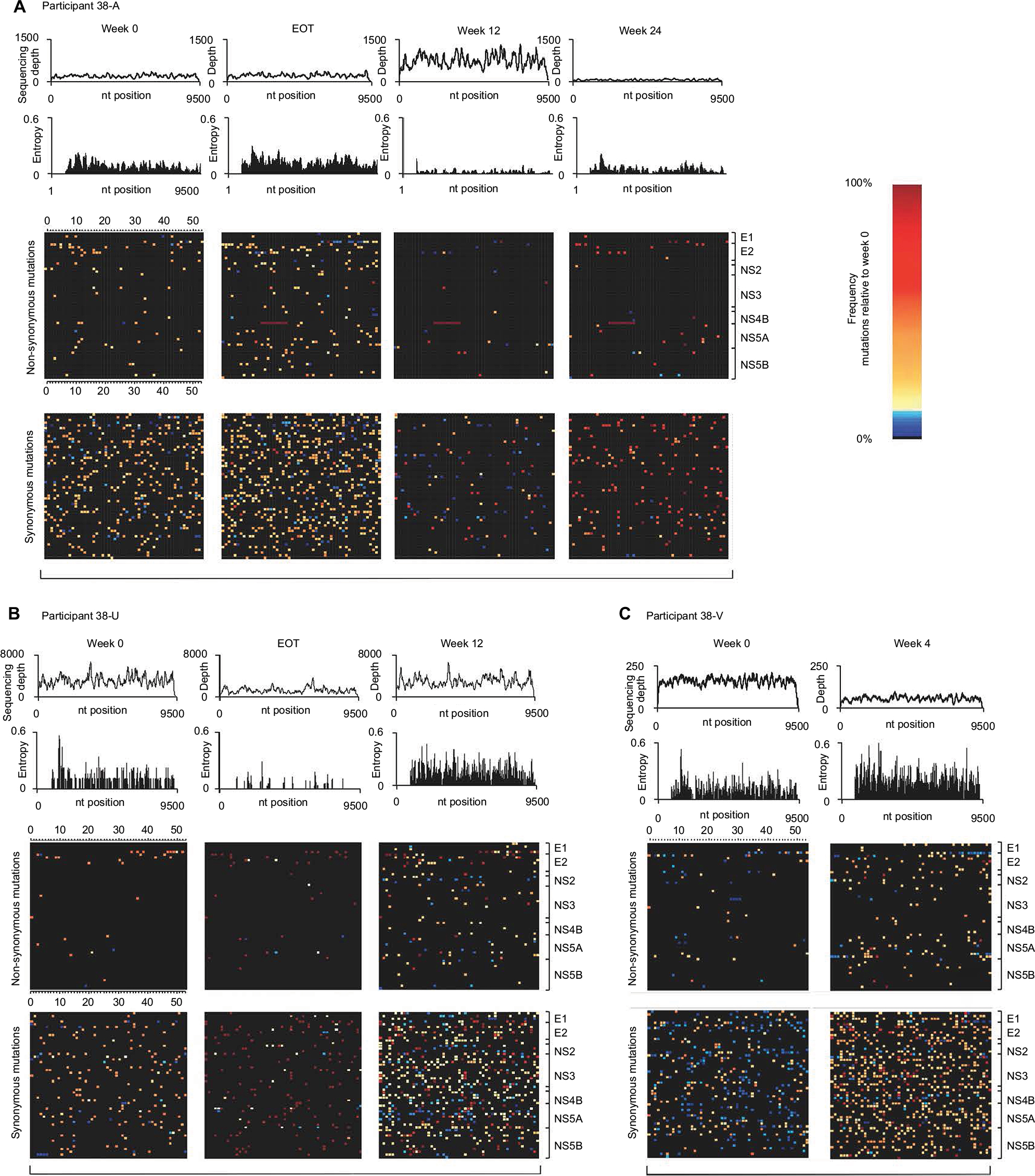 | Figure 3 Impact of treatment failure on within-host viral diversity. The depth of coverage across whole genomes sequenced at baseline and subsequent time points after treatment failure are given by the top histograms for 3 participants (A–C). Viral diversity is depicted by Shannon Entropy for nonstructural and envelope genes, with a breakdown of the nonsynonymous and synonymous changes shown by each heat map. Within each heat map, the percentage diversity at each codon position with respect to the consensus week 0 sequence is shown with minor variations (<10% change from baseline) colored light blue and higher percentages colored yellow through orange. Complete codon changes (all sequence data showing 100% change from baseline codons) are colored dark red. Abbreviations: EOT, end of treatment; nt, nucleotide. |
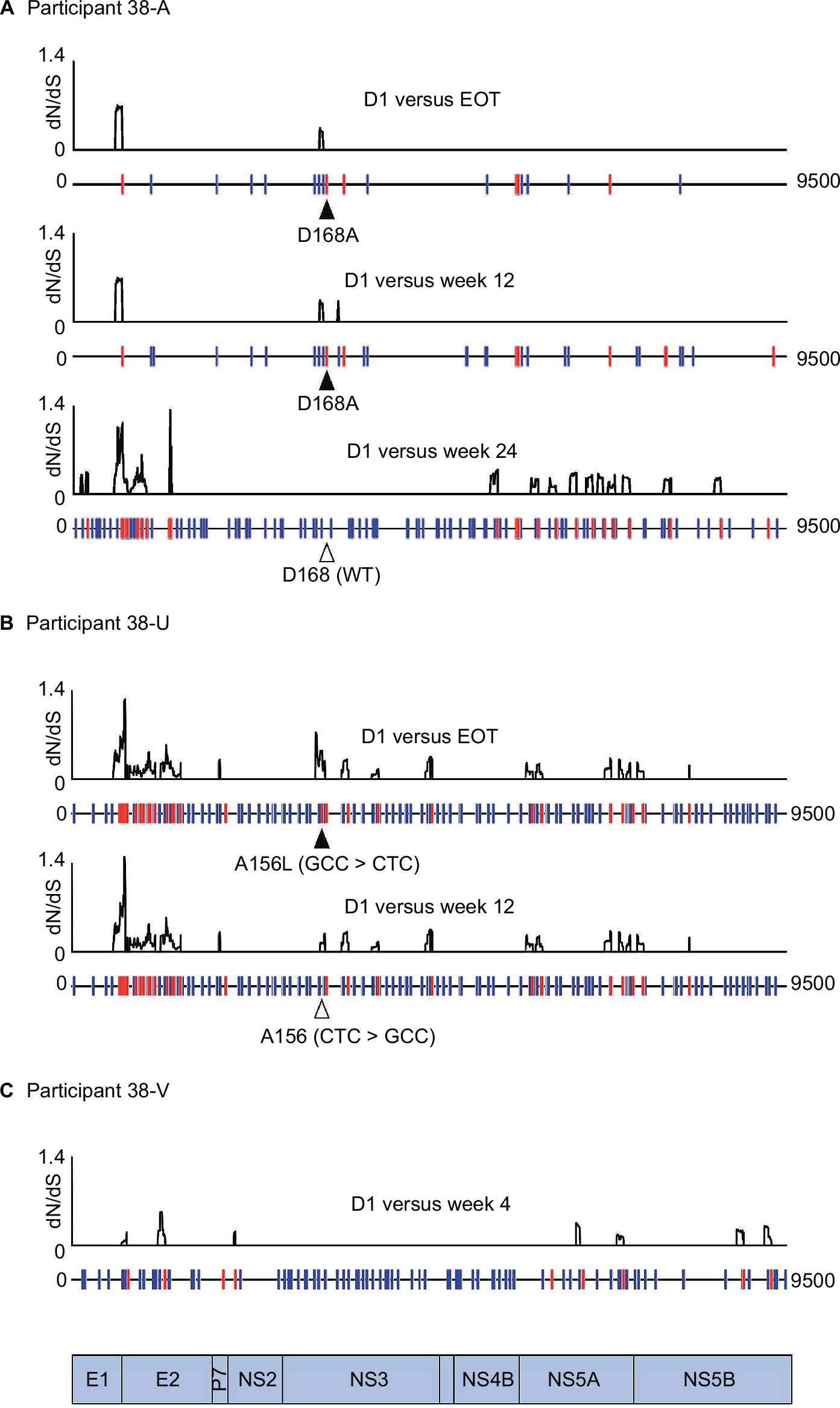 | Figure 4 Divergence of viral genome consensus sequences after treatment failure. Baseline sequences were compared with subsequent time points for 3 participants. The top plots in each panel show dN/dS ratios in 102 nucleotide, codon-aligned pairs of consensus sequences obtained by next-generation sequencing. Synonymous changes and nonsynonymous changes are shown below in blue and red, respectively. Abbreviations: D, day; EOT, end of treatment, WT, wild-type. |
In general, we observed near-perfect concordance between the principal RAS detected by consensus Sanger sequencing and the major variants detected by clonal sequencing and NGS (Figure 1). In all but 1 participant, NGS and clonal sequencing concurred on the major variant; the exception was a dominant V29A mutation detected by clonal sequencing in participant 38-E at FW12, which was the sequence with the lowest depth of coverage by NGS (Figure 2). We also observed good concordance between minor polymorphisms detected by clonal sequencing and NGS, although the added sensitivity of NGS generally uncovered more minor variants. Of the 29 amino acid sites in NS3 with minor polymorphisms detected by either method, 25 were detected by NGS and 13 by clonal sequencing, with concordance at 10 sites. A total of 14 minor variant mutations were detected exclusively by NGS, whereas 4 minor variants were detected exclusively by clonal sequencing. An important caveat to comparisons of sensitivity between NGS and clonal sequencing is that NGS analysis includes a step for removing PCR duplicates that share identical start and end positions, such that total numbers of unique sequences more closely approximate quantities of single-molecule templates. Conversely, TOPO clonal data cannot be de-duplicated, and a greater proportion of “clones” are, therefore, expected to be PCR duplicates.
The most complex combinations of resistance (multiple NS3/4A RASs) were detected in participants with on-treatment breakthrough viremia. No treatment-emergent mutations were detected by either consensus Sanger sequencing or clonal sequencing in 6/25 participants who relapsed (38-F, 38-L, 38-R, 39-B, 39-C and 39-D). A further 16/25 relapsers had a single treatment-emergent RAS detected at positions D168E/A/D/Y/N/T/V (n=13), A156T (n=2) or S122T (n=1). Conversely, D168A/N mutations were detected in 4 participants with breakthrough viremia, always in combination with A156L/G, Y56F/H or R155K polymorphisms (Table 5). In participant 38-W, poor treatment compliance likely contributed to sequential acquisition of multiple RASs: after starting treatment, GZR drug levels became undetectable at 2 sequential visits prior to the detection of breakthrough virus at treatment day 79, which carried novel A156G and R155W mutations (Figure S1, participant 38-W). At subsequent time points, the major variant also included a D168N mutation.
The persistence of RASs over time
Varying patterns of persistence and reversion were observed among RASs detected after relapse or breakthrough. For example, the D168 RAS endured as the major variant for at least 200–260 days in participants 39-G and 39-F. Conversely, in participant 38-N, the process of reversion was already underway 7 weeks (134 days) after treatment ended when the virus was first sampled, with D168H detected in only 5% of clones. In general, D168 variants seemed to be more stable than A156T and Y56H mutations, which were either lost entirely or decreased in frequency with sequential time points. In keeping with this observation, the replicon carrying the A156T mutation was the least fit of all variants assessed in vitro, with noninhibited titers below the quantifiable limits of the drug-resistance assay.
Viral evolution across the entire genome following GZR therapy
To investigate how treatment failure could affect viral evolution at both the nucleotide and amino acid levels, whole genome deep sequences were assessed longitudinally in 3 of the 4 participants sequenced by veSEQ (insufficient post-treatment reads were obtained to assemble a whole genome for participant 38-E). High levels of within-participant diversity were observed at baseline in all 3 participants, with low-level nucleotide polymorphisms detected in 418, 240 and 429 codons spanning the coding sequence for participants 38-A, 38-U and 38-V, respectively. The majority (83%–86%) of these nucleotide polymorphisms resulted in synonymous codon changes, and the majority of nonsynonymous changes were located in the hypervariable regions of the E2 gene (Figure 3A–C).
It was not possible to establish linkage between minor mutations separated by more than the insert size of sequencing libraries (250 nt on average). However, for participants 38-A, 38-U and 38-V, who became viremic at the end of their treatment, consensus whole-virus genomes sequenced at the first viremic time point had diverged from baseline with substitutions at 0.9%–5.9% of a possible 2835 codons across the entire HCV open reading frame (Figure 4A–C). This divergence is likely to reflect comparable levels of diversity among the baseline quasispecies prior to drug-mediated selection. The majority of the mutations detected between each time point were again synonymous with the highest levels of nonsynonymous change observed in the hypervariable regions of E2 and few were detected elsewhere, excepting selection of A156L and D168A variants in the case of participants 38-U and 38-A, respectively. Interestingly, the furthest genetic distance between 2 consecutive time points observed in participant 38-A was between FW12 and FW24, where 1.7% of codons mutated in association with reversion of a D168A mutation back to the WT aspartic acid residue.
In participant 38-U, we were unable to detect the intermediate mutations that are likely to have accompanied the double-nucleotide changes separating A156 (CTC) at baseline from A156L (GCC) at FW4, which subsequently reverted back to A156 (CTC) at FW12. Similarly, for participant 38-V, we were unable to detect RASs at either baseline or FW4, suggesting that the total diversity of viral quasispecies is likely much greater than that sampled by any sequencing method used in this study.
Discussion
GZR is an HCV NS3A protease inhibitor with in vitro and in vivo activity that is less susceptible to resistance than the first-generation protease inhibitors. However, in vitro studies also suggest that GZR has reduced potency against certain HCV variants possessing Y56H, A156T/V or D168A/V/Y NS3A mutations.8 Using multiple methods of sequencing, this study confirmed that these mutations are important in treatment failure with GZR. Clonal sequencing showed contrasting patterns of acquisition and reversion of RASs, with more complex combinations of mutations detected in participants with on-treatment breakthrough compared with those who relapsed. veSEQ also showed surprising levels of divergence and diversification of virus in cases of virologic failure, with up to 1 in 20 codons undergoing change at the consensus level. We postulate that the selective pressures exerted by modern treatments, coupled with the ability of HCV to mutate, may have a significant role in shaping the future HCV pandemic.
Clinically, GZR is utilized with elbasvir in a fixed-dose combination tablet. However, in the PN038 and PN039 studies, GZR was administered in combination with PEG-IFN and/or RBV without a second DAA, providing a valuable opportunity to evaluate RASs that relate solely to GZR therapy. In both this study and published studies that have evaluated GZR plus elbasvir in treatment-naive participants with HCV GT1 infection, we show that baseline NS3 RASs have no effect on treatment outcome.14,17,18 The majority of variants detected at baseline in the present analysis did not significantly modulate resistance to GZR in vitro and were found among both participants achieving SVR and those with virologic failure.
Baseline V55A variants were not found in any participant achieving SVR, but were detected prior to treatment in 3 participants who subsequently relapsed. When assessed in vitro, V55A did not significantly alter resistance to GZR above the intra-assay variance (1.3- to 1.4-fold decrease in EC50). Furthermore, only participant 38-E, who carried the V55A variant in 98% of baseline clones, retained this mutation after treatment failure. Participants 38-E and 39-F lost baseline V55A mutations but gained post-treatment D168N and A156T RASs, respectively, suggesting that V55A does not contribute to GZR resistance. A low-frequency variant characterized by an S122T mutation was present at baseline in participant 39-D and was subsequently selected as the major variant following relapse. S122T increased GZR EC50 by 2.2-fold in the GT1a replicon in this study. However, a previous in vitro study indicated that the S122T mutation was associated with a 26-fold change in simeprevir EC50 in 1 out of 5 variants assessed, suggesting that resistance mediated by S122T may depend on the genetic context of other linked mutations. Further study of this particular mutation is warranted, given its prevalence at baseline in GT1b and 4 isolates.
In the present studies, among the 25 participants with relapse, 16 were receiving doses of GZR of 25–50 mg/day and 9 were receiving a dose of 100 mg/day, which is the dose used clinically in the fixed-dose combination tablet with elbasvir.18,27 D168 mutations were most commonly associated with relapse (present in 13/25 participants) with D168A/E/V being the most commonly detected variants. In vitro, the D168A/E/V variants result in a 73-, 12- and 28-fold shift in GZR EC50, respectively. Two participants had the A156T variant at failure.
Of the 25 participants with relapse, 16 had detectable HCV RNA at FW4, thus relapsing relatively soon after withdrawal of therapeutic pressure, whereas 9 continued to have undetectable HCV RNA at FW4 and did not show re-emergence of detectable virus until FW12. The reasons for the delayed time to relapse in these 9 participants are unclear, but may be related to variable replicative fitness of viruses carrying the same RASs or alternatively may reflect persisting immune stimulation from PEG-IFN therapy maintaining pressure on viral replication after dosing is complete. Consistent with a short-term, post-treatment immunologic “boosting” by PEG-IFN, it is notable that none of the 5 relapsers receiving GZR plus RBV, but 10/20 relapsers receiving GZR plus PEG-IFN/RBV, had undetectable HCV RNA for ≥4 weeks after completion of treatment. Finally, it is also noteworthy that among the 12 participants with new D168 mutations emerging post-treatment, 9 had only WT virus detectable at FW24 and an additional 2 had A156T treatment-emergent RASs; both were WT by FW12/24, indicating a fast reversion to WT which is likely attributable to poor replicative ability of the mutant virus. We cannot exclude complete reversion of RAS mutations prior to sampling in the 4 participants who were sampled at relapse or FW4, in whom no RAS was detected.
Six participants in the present studies had virologic breakthrough or end-of-treatment failure, characterized by re-emergence of the virus while on treatment after previously having undetectable HCV RNA. The 4 who failed earliest on treatment had complex dual- or triple-linked mutations consisting of Y56H, A156T and D168A/N by clonal sequencing. Mutations at sites 56, 155, 156 and 168 were observed in breakthrough participants compared with relapse participants who had RASs at fewer sites (122, 168 and 156). Interestingly, the only participant to exhibit on-treatment breakthrough with a single mutation (participant 38-U) possessed the A156L polymorphism, which produced the highest level of resistance of all RASs when assessed in vitro. Virus from this participant underwent selection of an A156L variant that involved 2 nucleotide changes (GCC>CTC), with no singly mutated intermediate detected either before selection or after reversion. This case indicates 2 points: the high levels of resistance required to facilitate on-treatment breakthrough viremia and the high levels of diversity that must be present below the limit of detection by conventional sequencing and NGS.
The combination of R155W and A156G, observed at breakthrough in participant 38-W, also produced exceptionally high levels of resistance in vitro. This participant went on to acquire a D168N mutation at a subsequent time point, adding further support to the theory that detection of viremia while treatment is ongoing indicates a highly resistant viral phenotype. Precisely which factors distinguish between the complexity and types of mutations observed at relapse compared with on treatment (and end-of-treatment failure) are unknown. However, clonal sequencing confirmed that mutations at sites 156, 155 and 56 were cis-linked, suggesting sequential acquisition (a pattern probably reciprocated by the sequential manner in which they reverted). We speculate that mutations at sites 156, 155 and 56, in particular, led to levels of resistance high enough to support enough virus replication in spite of treatment to allow additional mutations to emerge with further gains to viral fitness and the eventual detection of viremia on treatment. It is also possible that low drug levels may result in optimal selective conditions to on-treatment failure and sequential selection of multiple mutations. Consistent with this, participants 38-V and 38-W had low or undetectable plasma concentrations of GZR prior to failure. These findings highlight the clinical importance of adherence to therapy and cessation of therapy in the event of on-treatment failure.
Consensus Sanger sequencing and clonal sequencing is likely to be superseded by next-generation methods, which have a higher throughput and are capable of characterizing multiple viral genes simultaneously at high depth. We have previously shown whole-genome sequencing to be superior to standard clinical tests for reporting GTs and RASs from multiple genes simultaneously in the same assay.28 In this study, we show how the complexity of different major and minor RASs observed within 1 gene (NS3B) stratifies with clinical outcome in individuals receiving single DAA therapy. The ability to sequence whole genomes at high depth is likely to prove useful in future studies that assess patterns of resistance emerging at breakthrough failure and relapse following combination DAA therapy.
In the 3 participants sampled longitudinally by veSEQ, drug-mediated selection had a profound impact on viral evolution across the entire genome, which was characterized by mostly synonymous mutations that were associated with both acquisition and reversion of RASs. This predominance of synonymous mutations hints at a highly diverse pool of minor variants, maintained under strong negative selection under drug-free conditions, which is consistent with previous studies.29 In this study, selected RAS variants were genetically distinct from the dominant variants present when treatment started and are therefore likely to have descended from archived variants. In turn, this suggests that pre-established viral diversity may be an important predictor of subsequent treatment failure, which will be the subject of future work.
Conclusion
These data provide further insight into the clinical profile of GZR when used in combination with PEG-IFN and RBV. The fixed-dose combination of elbasvir/GZR is currently approved for the treatment of HCV GT1 and 4 infection in the USA, Europe and other countries.27,30 Studies to date suggest that this combination is highly effective, with a favorable tolerability profile in a broad spectrum of people with HCV GT1 infection.12–18 There are no plans to develop GZR as a single-entity tablet for use in combination with PEG-IFN and RBV. These data support the established resistance profile of GZR, indicating a high barrier to resistance and clinical activity across many common RASs. Clearly, suboptimal treatment and subsequent failure has the potential to impact within-person HCV evolution. Precisely how this will shape the phylogeny of the HCV epidemic, baseline susceptibility to treatment and barriers to resistance remains to be seen.
Acknowledgments
Medical writing and editorial assistance were provided by Tim Ibbotson, PhD, of ApotheCom (Yardley, PA, USA). This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. The STOP-HCV Consortium is funded by a grant from the Medical Research Council (MR/K01532X/1). The design, study conduct and financial support for this research were provided by Merck & Co., Inc.
Anita YM Howe is now at the University of British Columbia, Vancouver, British Columbia, Canada.
Author contributions
DB, AYMH and EB conceived, designed or planned the study. DB, AYMH, RC, AB, RB and EB acquired the data. DB, SB, AYMH, IP, DAS and EB analyzed the data. DB, AYMH, PI, IP, DS and EB interpreted the results. DB, AYMH and EB drafted the manuscript. All authors approved the final manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
Paul Ingravallo is an employee of Merck & Co., Inc. Stuart Black, Anita YM Howe, Robert Chase and Irene Pak are former employees of Merck & Co., Inc.
The authors report no other conflicts of interest in this work.
References
AASLD/IDSA HCV Guidance Panel. Hepatitis C guidance: AASLD-IDSA recommendations for testing, managing, and treating adults with hepatitis C virus. Available from: http://www.hcvguidelines.org (version September 21, 2017). Accessed December 14, 2017. | ||
European Association for the Study of the Liver. EASL Recommendations on Treatment of Hepatitis C 2016. Available from: http://www.easl.eu/medias/cpg/HCV2016/Summary.pdf (version September, 2016). Accessed December 14, 2017. | ||
Kolykhalov AA, Mihalik K, Feinstone SM, Rice CM. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J Virol. 2000;74(4):2046–2051. | ||
Li K, Foy E, Ferreon JC, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA. 2005;102(8):2992–2997. | ||
Barnard RJO, Howe JA, Ogert RA, et al. Analysis of boceprevir resistance associated amino acid variants (RAVs) in two phase 3 boceprevir clinical studies. Virology. 2013;444(1–2):329–336. | ||
Kieffer TL, De Meyer S, Bartels DJ, et al. Hepatitis C viral evolution in genotype 1 treatment-naïve and treatment-experienced patients receiving telaprevir-based therapy in clinical trials. PLoS One. 2012;7(4):e34372. | ||
Lenz O, Verbinnen T, Lin T-I, et al. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob Agents Chemother. 2010;54(5):1878–1887. | ||
Summa V, Ludmerer SW, McCauley JA, et al. MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012;56(8):4161–4167. | ||
McPhee F, Hernandez D, Zhou N, et al. Pooled analysis of HCV genotype 1 resistance-associated substitutions in NS5A, NS3 and NS5B pre- and post-treatment with 12 weeks of daclatasvir, asunaprevir and beclabuvir. Antivir Ther. 2017;23(1):53–66. | ||
Wang C, Sun JH, O’Boyle DR, et al. Persistence of resistant variants in hepatitis C virus-infected patients treated with the NS5A replication complex inhibitor daclatasvir. Antimicrob Agents Chemother. 2013;57(5):2054–2065. | ||
Neumann AU, Lam NP, Dahari H, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-α therapy. Science. 1998;282(5326):103–107. | ||
Kwo P, Gane EJ, Peng C-Y, et al. Effectiveness of elbasvir and grazoprevir combination, with or without ribavirin, for treatment-experienced patients with chronic hepatitis C infection. Gastroenterology. 2017;152(1):164–175.e4. | ||
Forns X, Gordon SC, Zuckerman E, et al. Grazoprevir and elbasvir plus ribavirin for chronic HCV genotype-1 infection after failure of combination therapy containing a direct-acting antiviral agent. J Hepatol. 2015;63(3):564–572. | ||
Lawitz E, Gane E, Pearlman B, et al. Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY: a randomised, open-label phase 2 trial. Lancet. 2015;385(9973):1075–1086. | ||
Rockstroh JK, Nelson M, Katlama C, et al. Efficacy and safety of grazoprevir (MK-5172) and elbasvir (MK-8742) in patients with hepatitis C virus and HIV co-infection (C-EDGE CO-INFECTION): a non-randomised, open-label trial. Lancet HIV. 2015;2(8):e319–e327. | ||
Roth D, Nelson DR, Bruchfeld A, et al. Grazoprevir plus elbasvir in treatment-naive and treatment-experienced patients with hepatitis C virus genotype 1 infection and stage 4-5 chronic kidney disease (the C-SURFER study): a combination phase 3 study. Lancet. 2015;386(10003):1537–1545. | ||
Sulkowski M, Hezode C, Gerstoft J, et al. Efficacy and safety of 8 weeks versus 12 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin in patients with hepatitis C virus genotype 1 mono-infection and HIV/hepatitis C virus co-infection (C-WORTHY): a randomised, open-label phase 2 trial. Lancet. 2015;385(9973):1087–1097. | ||
Zeuzem S, Ghalib R, Reddy KR, et al. Grazoprevir–elbasvir combination therapy for treatment-naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection: a randomized trial. Ann Intern Med. 2015;163(1):1–13. | ||
Lagging M, Brown A, Mantry PS, et al. Grazoprevir plus peginterferon and ribavirin in treatment-naive patients with hepatitis C virus genotype 1 infection: a randomized trial. J Viral Hepat. 2016;23(2):80–88. | ||
Gane E, Ben Ari Z, Mollison L, et al. Efficacy and safety of grazoprevir+ribavirin for 12 or 24 weeks in treatment-naïve patients with hepatitis C virus genotype 1 infection. J Viral Hepat. 2016;23(10):789–797. | ||
Bonsall D, Ansari MA, Ip C, et al. ve-SEQ: robust, unbiased enrichment for streamlined detection and whole-genome sequencing of HCV and other highly diverse pathogens. F1000Res. 2015;4:1062. | ||
Lontok E, Harrington P, Howe A, et al. Hepatitis C virus drug resistance-associated substitutions: state of the art summary. Hepatology. 2015;62(5):1623–1632. | ||
Batty EM, Wong THN, Trebes A, et al. A modified RNA-Seq approach for whole genome sequencing of RNA viruses from faecal and blood samples. PLoS One. 2013;8(6):e66129. | ||
Henn MR, Boutwell CL, Charlebois P, et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012;8(3): e1002529. | ||
Yang X, Charlebois P, Gnerre S, et al. De novo assembly of highly diverse viral populations. BMC Genomics. 2012;13:475. | ||
Lahser FC, Bystol K, Curry S, et al. The combination of grazoprevir, a hepatitis C virus (HCV) NS3/4A protease inhibitor, and elbasvir, an HCV NS5A inhibitor, demonstrates a high genetic barrier to resistance in HCV genotype 1a replicons. Antimicrob Agents Chemother. 2016;60(5):2954–2964. | ||
Zepatier (elbasvir and grazoprevir) [package insert]. Kenilworth, NJ: Merck & Co., Inc.; 2017. | ||
Thomson E, Ip CLC, Badhan A, et al. Comparison of next generation sequencing technologies for the comprehensive assessment of full-length hepatitis C viral genomes. J Clin Microbiol. 2016;54(10):2470–2484. | ||
Raghwani J, Rose R, Sheridan I, et al. Exceptional heterogeneity in viral evolutionary dynamics characterises chronic hepatitis C virus infection. PLoS Pathog. 2016;12(9):e1005894. | ||
European Medicines Agency. EPAR summary for the public: Zepatier (elbasvir/grazoprevir) [published July 22, 2016]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004126/WC500211235.pdf. Accessed October 11, 2017. |
Supplementary materials
 | Box S1 Independent Ethics Committees for studies PN038 and PN039 |
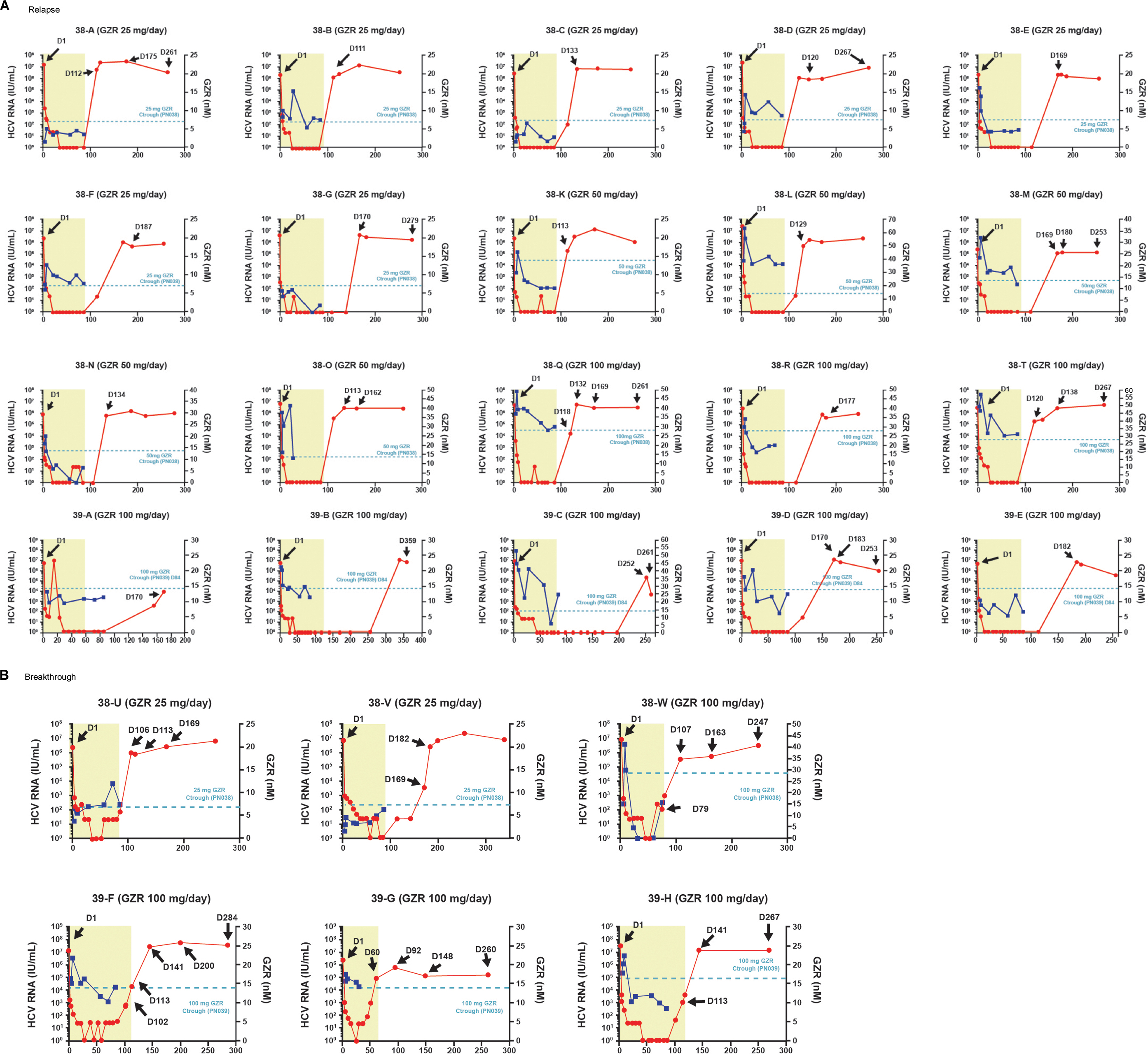 | Figure S1 Viral load, treatment duration and drug levels. Note: Longitudinal data were collected at fixed time points for all 26 participants with clonal sequence analysis; viral load data are shown for participants who relapsed (A) or had virologic breakthrough (B). Abbreviations: GZR, grazoprevir; HCV, hepatitis C virus. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.