")
Back to Journals » OncoTargets and Therapy » Volume 7
Characterization of a fusion protein of RGD4C and the ß-lactamase variant for antibody-directed enzyme prodrug therapy
Authors Zhou X, Wang H, Shi P, Meng A
Received 17 December 2013
Accepted for publication 30 January 2014
Published 10 April 2014 Volume 2014:7 Pages 535—541
DOI https://doi.org/10.2147/OTT.S59346
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Xiaoliang Zhou, Hao Wang, Peiji Shi, Ai-min Meng
Institute of Radiation Medicine, Tianjin Key Laboratory of Molecular Nuclear Medicine, Tianjin, People's Republic of China
Abstract: Antibody-directed enzyme prodrug therapy (ADEPT) delivers chemotherapeutic agents in high concentration to tumor tissue, while minimizing systemic drug exposure. ADEPT has been reported to be an attractive approach for improving the efficacy of cancer therapy. A previously reported β-lactamase was found to contain four cluster of differentiation (CD)4+ T cell epitopes; however, single amino acid changes in the enzyme resulted in significantly reduced proliferative responses, while retaining stability and activity of the enzyme. The β-lactamase variant with reduced immunogenicity is an attractive alternative for constructing the ADEPT fusion protein. In this study, we fused the peptide, RGD4C, known to target integrin αvβ3, to the β-lactamase variant for use in ADEPT. Biological function studies revealed that RGD4C- β-lactamase had a high hydrolytic effect on nitrocefin and cephalosporin–melphalan, and high plasma stability was observed. In addition, fusion of the RGD4C moiety to β-lactamase had little effect on immunogenicity compared with β-lactamase in the proliferation of peripheral blood mononuclear cells. The ability of this fusion protein to both target the central region of αvβ3 and induce toxicity in the non-small-cell lung cancer cell NCI-H460 makes it a promising therapeutic approach in the treatment of cancer.
Keywords: ADEPT, immunogenicity, integrinαvβ3
Introduction
ADEPT (antibody-directed enzyme prodrug therapy) is a novel therapeutic approach that targets a tumor enzyme to convert a relatively nontoxic prodrug into an active cytotoxic agent. This method has a number of advantages, including a reduction in systemic toxicity; however, to date, it has not realized its full potential. A critical component of ADEPT is the choice of the monoclonal antibody used to target the enzyme in the tumor mass. Numerous fusion proteins have been designed, via molecular engineering, to combine the specific cell-binding characteristics of antibodies or growth factors with the cytotoxic or growth-modulatory effects of toxins, cytokines, or proapoptotic proteins.1 Of these strategies, antibodies have shown the greatest potential as delivery vehicles for various classes of therapeutic agents. However, a major limitation of antibody-based constructs is their inadequate uptake and poor distribution in tumors, due in part to the relatively large molecular mass of antibody molecules. One approach to remedy this limitation is to use considerably smaller, genetically engineered antibody fragments. For example, single-chain Fv fusion constructs can penetrate tumor tissue more easily and have improved pharmacokinetics and lower immunogenicity than do intact murine antibodies. To further improve the penetration of targeted molecules, we and others have used small peptides for tumor vasculature-specific delivery, to enhance tumor uptake of the enzyme.
RGD4C (ACDCRGDCFCG) is a high-affinity ligand of αvβ3 integrin, which is highly expressed in tumor cells and angiogenic vessels, and is upregulated in tumor cells and tumor angiogenic vessels compared with normal tissues. Thus, the integrin αvβ3 can be used as a target for drug delivery.2 The RGD4C peptide has four cysteine residues close to each other, and it is a challenge to have all four cysteine forms arranged in the correct disulfide bonds for high integrin-binding affinity. In the peptide, RGD4C, two disulfide bonds are formed between C2-C10 and C4-C8 for optimized αvβ3 or αvβ5, and its affinity is 20-fold greater than that of similar peptides with a single disulfide bond and is 200-fold greater than that of linear peptides.3
In a previous study,4 we constructed and expressed a targeted enzyme as a fusion protein, which consisted of RGD4C as the targeting motif and a β-lactamase variant with mutations at K21A and S324A. The β-lactamase variant had two T-cell epitopes removed to reduce the immunogenicity of the wild-type enzyme. The results revealed that the fusion protein, RGD4C-β-lactamase (RGD4C-βL), retained the targeted effect of RGD4C when used in a targeted enzyme prodrug system. The engineered variant, RGD4C-βL, can be expressed in a soluble form at an acceptable yield in Escherichia coli. In this study, the antibody binding specificity and enzymatic activity of the fusion protein was analyzed and compared with that of the wild-type RGD4C and β-lactamase, respectively. We also investigated the activating capacity of the fusion protein in the presence of a cephalosporin–melphalan prodrug (C-Mel) in vitro.
Materials and methods
Materials
Arctic Express™(DE3) RP (GenScript USA Inc., Piscataway, NJ, USA), and plasmid pCold II were maintained in the laboratory; primers and sequence encoding RGD4C-βL were synthesized by GenScript USA Inc. Ni-NTA resin was obtained from Shanghai ShineGene Molecular Biotechnology Co, Ltd, Shanghai, People’s Republic of China. Nitrocefin was from Oxoid™ (Thermo Fisher Scientific Inc., Waltham, MA, USA). Human plasma and peripheral blood buffy coat samples were from the Tianjin Blood Center (Tianjin, People’s Republic of China). C57 mice were obtained from the Experimental Animal Center of the Academy of Military Medical Sciences (Beijing, People’s Republic of China). NCI-H460 cells were from the Cell Resource Center, IBMS, CAMS/PUMC (Beijing, People’s Republic of China) and were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM)/high glucose containing 100 units/mL penicillin, and 100 μg/mL streptomycin, supplemented with 10% fetal bovine serum. C-Mel was synthesized according to the literature.5 RGD4C-βL was directly labeled with fluorescein isothiocyanate (FITC) as previously described.6 Endotoxin detection and removal kits, cell counting kits (CCK-8 kits), and X-vivo medium were from Shanghai Beyotime Institute of Biotechnology (Shanghai, People’s Republic of China) and used according to the manufacturer’s instructions. All materials were handled in accordance with the guidelines of the Ethics Committee of Chinese Academy of Medical Sciences.
Production and characterization of RGD4C-βL
The fusion gene was cloned into E. coli BL21 (DE3) in 600 mL Terrific Broth supplemented with 10 mg/L chloramphenicol at 30°C for 48 hours. When the absorbancy of A600 reached 0.8, 1 mL culture was aseptically removed as the sample, and 0.3 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mmol/L. Expression of RGD4C-βL was monitored after 0, 1, 3, and 5 hours at 25°C and 225 r/min. The supernatant was collected after centrifugation at 4°C and 11,000×g for 5 minutes. The supernatant and the precipitate were collected for 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Cell extracts were purified with the Ni-NTA purification system, using our previously reported method.4
Catalytic activity assay and plasma stability
The catalytic activity of RGD4C-βL was monitored by adding 90 μL nitrocefin solution at different concentrations (in phosphate-buffered saline [PBS] containing 1% dimethyl sulfoxide [DMSO]) into 10 μL fusion protein solution (in PBS); the absorbance at 490 nm was immediately recorded and then monitored every 12 seconds for 5 minutes. Hydrolysis of nitrocefin resulted in an increase in absorbance, and the kinetic constants of RGD4C-βL toward nitrocefin were determined by measuring nitrocefin hydrolysis in the range of 60 to 160 μg/mL with a fusion protein concentration of 1 μg/mL. Kinetic constants were calculated using Lineweaver–Burk plots. A plasma stability assay was carried out, and according to the activity measurement, the fusion protein was added to human plasma at 1 μg/mL and incubated at 37°C for 14 days. A sample was removed every 48 hours over that period, and the catalytic activity was monitored as described above. Each experiment was repeated thrice, and the results were averaged.
In vitro immunogenicity assay
The immunogenicity of RGD4C-βL was determined using two types of immune cells, human peripheral blood mononuclear cells (PBMC) and mouse splenocytes, by proliferation methods. Endotoxin in the RGD4C-βL samples was detected and removed, using the endotoxin detection and removal kits, until the endotoxin content was <0.1 EU/mL.
PBMC from community donors were separated by density centrifugation and cultured at 106/mL in X-vivo medium, in 96-well plates. Then, 10 μL fusion protein solution in PBS was added to 90 μL of the cell solution to a final concentration of 10 μg/mL, and the cells were incubated at 37°C for 48 hours. Following this, 10 μL CCK-8 working solution was added, and the absorbance at 450 nm was measured. Each PBMC culture was tested in replicates of at least six wells; control cultures contained no added protein, and lipopolysaccharide (LPS), at the same concentration, was added to the positive control culture.
C57 mice (6–8 weeks of age) were sacrificed by cervical dislocation. The spleen was extracted aseptically and ground to prepare a cell suspension in PBS. The suspension was then treated with erythrocyte lysis buffer, and cultured at 2 × 106/mL in Roswell Park Memorial Institute (RPMI)-1640 medium, in 96-well plates. Then, 10 μL fusion protein solution in PBS, at a concentration of 10 μg/mL, was added to 90 μL cell solution, and the cells were incubated at 37°C for 48 hours. Next, 10 μL Celltiter-Glo™ Luminescent Cell Viability Assay (Promega Corporation, Fitchburg, WI, USA) was added, and following incubation for 10 minutes, cell proliferation was monitored by bioluminescence. A positive response was observed when the proliferative response reached a stimulation index (average experimental cpm/average control cpm) of ≥2. A parametric, two-tailed Student’s t-test was performed. Significance was determined at P<0.05.
Immunofluorescence staining
NCI-H460 cells (4×105) were cultured overnight in 24-well plates. The cells were washed with PBS and fixed with 10% alcohol for 10 minutes at room temperature. After washing three times with PBS, the cells were stained with 20 μg/mL RGD4C-βL for 80 minutes at 4°C. The cells were viewed under a phase contrast microscope and a digital fluorescence microscope.
In vitro cytotoxicity
The C-Mel prodrug was required to determine the in vitro cytotoxicity of RGD4C-βL. A monolayer of NCI-H460 cells was cultured in RPMI-1640 medium containing 10% fetal bovine serum. The cells were seeded into 96-well plates at 5,000 cells/well and allowed to adhere for 24 hours at 37°C. RGD4C-βL was added at a final concentration of 2 μM, and following incubation for 60 minutes on ice, the cells were washed with fresh medium three times. C-Mel, at different concentrations ranging from 0.4–200 μM, was added, and following incubation for 4 hours, the cells were washed thrice and incubated for a further 12 hours. Then, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent was added to each well, and survival was measured after 4 hours, based on absorbance at 490 nm. Each data point was measured in triplicate. RGD4C-βL, melphalan, and C-Mel, at several concentrations, were coincubated with cells for 4 hours, washed thrice, and incubated for a further 12 hours, and the MTT assay was performed to determine their cytotoxicity, respectively. The entire experiment was performed three times and similar results were obtained each time.
Results
Characterization of RGD4C-βL
The expression of RGD4C-βL in E. coli was identified by SDS-PAGE (Figure 1A). RGD4C-βL was partially soluble, and the expression level of the protein was acceptable. RGD4C-βL was purified by stepwise affinity chromatography, followed by ion-exchange chromatography. These purification procedures resulted in a final yield of approximately 9 mg purified RGD4C-βL/L, and the purity was not less than 90%. The RGD4C-βL protein formed a homodimer under nonreducing conditions. Most of the dimer was reduced to a monomer, and a single band at 42 kDa was shown on reduced SDS-PAGE after treatment with DTT (Figure 1B).
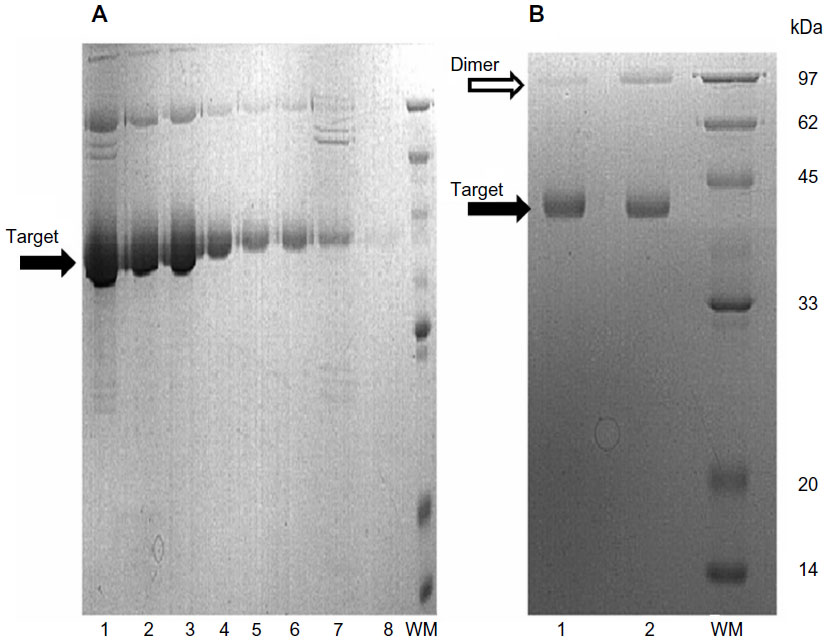 | Figure 1 Purification and characterization of RGD4C-βL. |
Catalytic activity assay and plasma stability
To investigate the utility of RGD4C-βL, we studied its catalytic activity and stability in a simulated in vivo environment by measuring the hydrolysis kinetics of the chromogenic antibiotic nitrocefin. The kinetic parameters, maximal initial velocity (Vmax), Michaelis-Menten constant (Km) and turnover number (Kcat) were 0.84 mmol · L−1 · min−1, 0.65 mmol · L−1, and 5,887.18 per second respectively. These parameters had the same order of magnitude as commercially available β-lactamase (data not shown). Analysis of the plasma stability showed that the fusion protein was very stable in human plasma, with >60% of the activity remaining after 14 days incubation in human plasma at 37°C (Figure 2).
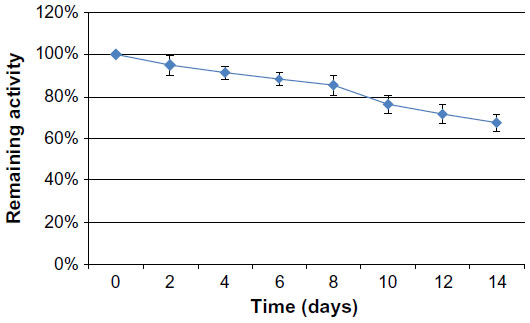 | Figure 2 Stability of RGD4C-βL in human plasma at 37°C. |
In vitro immunogenicity assay
To study the immunogenicity of RGD4C-βL, we used a PBMC proliferation assay and a mouse splenocyte proliferation assay. LPS was used as a positive control in both assays. Unfractionated PBMC from eight donors were incubated with RGD4C-βL for 48 hours. Based on the CCK-8 assay, the experimental group showed no significant difference compared with the blank control, while the positive control showed significant differences among the eight donors (Figure 3A). In the mouse splenocyte proliferation assay, the stimulation index (SI) values of the fusion protein in five tests were less than 2, while that of LPS was significantly higher, indicating the low immunogenicity of RGD4C-βL (Figure 3B).
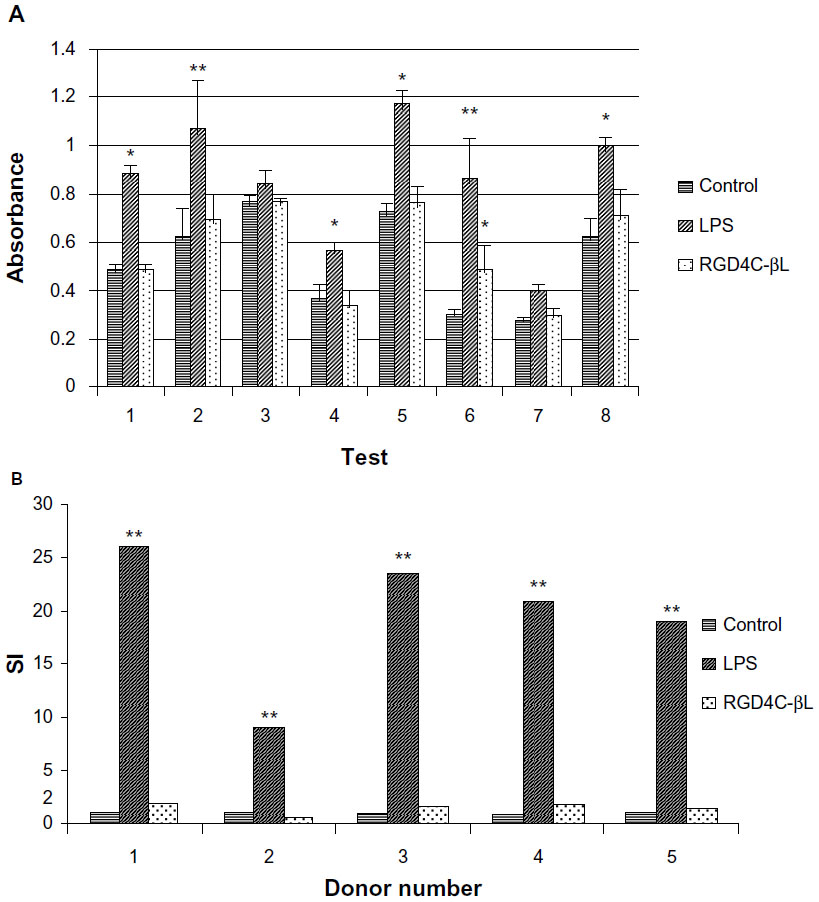 | Figure 3 (A) Human PBMC proliferation assay. In eight tests, the RGD4C-βL groups showed no significant differences compared with the vehicle control groups (P>0.05), and the LPS groups were significantly higher than the control groups (P<0.05), with the exception of two donors (No 3 and No 7). (B) Mouse splenocyte proliferation assay. In five tests, the SI of the RGD4C-βL groups was lower than 2, and of the LPS groups was significantly higher (>2). A positive response was observed if the proliferative response reached an SI (average experimental cpm/average control cpm) of ≥1.99. |
Immunofluorescence staining
To examine the binding effect of RGD4C-βL, the fusion protein was directly labeled with FITC. The results showed that RGD4C-βL retained the ability to bind with the integrin-positive cells (NCI-H460) (Figure 4).
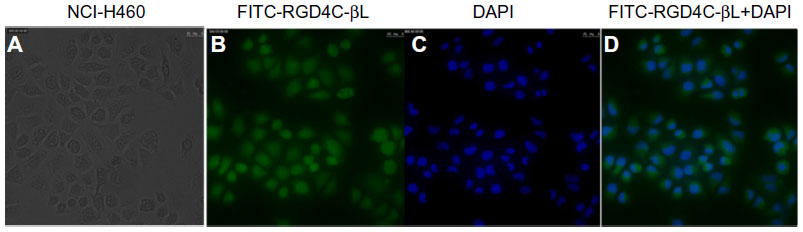 | Figure 4 Fluorescence imaging of NCI-H460 cells binding to FITC-RGD4C-βL. |
In vitro cytotoxicity
The sensitivity of cells to melphalan and the prodrug was determined using an MTT assay. In our studies, the prodrug was shown to be far less active than melphalan against the NCI-H460 cell line (Figure 5). However, the fusion protein was able to restore prodrug activity close to that of melphalan. The half maximal inhibitory concentration (IC50) for the combination of prodrug and 2 nM fusion protein was 18 μM. Another study has also reported the reversal of prodrug IC50 to that of the original drug.4 This showed that the fusion protein was able to activate the prodrug in vitro.
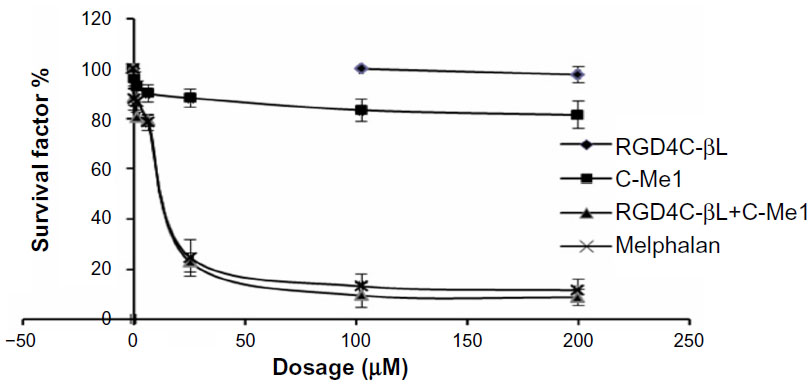 | Figure 5 In vitro activation of cephalosporin–melphalan by RGD4C-βL. |
Discussion
ADEPT systems are based on targeted delivery of catalytic enzymes that can hydrolyze a nontoxic prodrug to its active form. This site-specific activation of the prodrug results in a high local concentration of the chemotherapeutic agent in tumor tissues, while minimizing exposure of healthy tissues. ADEPT systems have been described based on a variety of different enzymes.7,8 The results of three clinical trials of ADEPT have been published.9,10 Encouraging tumor responses were observed in the first two trials, although accompanied by significant systemic toxicity. Immunogenicity seemed to be a key obstacle as all patients generated antibodies against the protein components of the treatment. Immune responses could not be prevented by cyclosporine administration and therefore limited the number of treatment cycles per patient. It is not known whether carboxypeptidase G2 contains T-cell epitopes.11 However, several other factors could have contributed to the immunogenicity observed in these ADEPT trials.12
The β-lactamase from Enterobacter cloacae was found to contain four cluster of differentiation (CD)4+ T-cell epitopes, as determined by a human cell-based proliferation assay. Two of the four epitopes were modified with single amino acid substitutions, which were selected based on reduced proliferative responses and acceptable functional activity and expression. Therefore, we hypothesized that the RGD4C/enzyme conjugate would be an effective option for ADEPT, due to the localization and reduced immunogenicity. In a previous study,6 we constructed a fusion protein that had a cyclic RGD4C peptide moiety at the NH2 terminus of β-lactamase, through a glycine-serine linker, and the results showed that the fusion protein, RGD4C-βL, had similar integrin αvβ3-binding affinity to that of the RGD4C peptide. In pursuing our goal of significantly reducing the risk that the fusion protein would generate an immune reaction in patients, a series of assays were employed to estimate the utility of RGD4C-βL.
Enzymatic activation of the prodrug, C-Mel, by the fusion protein was shown to occur at the same rate as that for β-lactamase. The fusion protein, when specifically bound to NCI-H460 cells, completely activated the nontoxic prodrug, and growth inhibition was similar to that obtained with melphalan. Fusion of RGD4C to β-lactamase had a reduced effect on the overall immunogenicity of the β-lactamase variant when tested using human PBMC and mouse splenocytes. Considering that the in vitro immune response to RGD4C-βL has already approached the limit of detection of available assays, further applications in therapy may not justify the risk of immune responses.13
We recognize several potential limitations of our study. First, our study only used an integrin-expressing NCI-H460 human lung cancer cell line to assess the tumor-inhibitory effect of RGD4C-βL combined with C-Mel. An integrin-positive tumor model, such as U87MG, is required to test the same fusion protein in future studies as U87MG tumors express higher levels of integrin than do NCI-H460 cells. Second, there was a lack of direct evidence to prove tumor uptake of the fusion protein, and biodistribution of RGD4C-βL in the tumor model is a critical factor contributing to cancer therapy.13,14 We labeled the fusion protein with the radionuclide 125I, and in future studies, single-photon emission computed tomography (SPECT) imaging could be carried out in an in vivo test.
In summary, we describe here the in vitro characterization of a RGD4C-βL fusion protein. Incorporation of cyclic RGD4C peptide to the NH2 terminus of β-lactamase had little or no effect on the catalytic effect of the enzyme. Immunogenicity, as assessed by immune cell proliferation, showed that RGD4C-βL did not cause significant human PBMC and mouse splenocyte proliferation compared with the LPS control group. The antitumor effect of RGD4C-βL combined with the C-Mel prodrug was similar to that of melphalan in integrin-positive cells. Furthermore, RGD4C-βL can be produced in acceptable yield as a soluble protein in E. coli and can be effectively recovered by affinity chromatography. An evaluation of the antitumor-effect and in vivo biodistribution of the fusion protein is currently in progress.
Acknowledgments
This project was supported by the National Natural Science Foundation of China (No 81301983) and the Development Fund of the Institute of Radiation Medicine, Chinese Academy of Medical Sciences (No SF1305).
Disclosure
The authors report no conflict of interest in this work.
References
Wang H, Chen K, Cai W, et al. Integrin-targeted imaging and therapy with RGD4C-TNF fusion protein. Mol Cancer Ther. 2008;7(5):1044–1053. | |
Tucker GC. Integrins: molecular targets in cancer therapy. Curr Oncol Rep. 2006;8(2):96–103. | |
Koivunen E, Wang B, Ruoslahti E. Phage libraries displaying cyclic peptides with different ring sizes: ligand specificities of the RGD-directed integrins. Biotechnology (N Y). 1995;13(3):265–270. | |
Kerr DE, Li Z, Siemers NO, Senter PD, Vrudhula VM. Development and activities of a new melphalan prodrug designed for tumor-selective activation. Bioconjug Chem. 1998;9(2):255–259. | |
Goding JW. Conjugation of antibodies with fluorochromes: modifications to the standard methods. J Immunol Methods. 1976;13(3–4):215–226. | |
Wang H, Shi PJ, Wu MF, Li N, Zhou XL, Fan FY. Construction, expression and functional characterization of the β-lactamase with αv integrin ligands. Protein Pept Lett. 2010;17(12):1562–1565. | |
Schellmann N, Deckert PM, Bachran D, Fuchs H, Bachran C. Targeted enzyme prodrug therapies. Mini Rev Med Chem. 2010;10(10):887–904. | |
Bagshawe KD. Targeting: the ADEPT story so far. Curr Drug Targets. 2009;10(2):152–157. | |
Sharma SK, Bagshawe KD, Begent RH. Advances in antibody-directed enzyme prodrug therapy. Curr Opin Investig Drugs. 2005;6(6):611–615. | |
Mayer A, Francis RJ, Sharma SK, et al. A phase I study of single administration of antibody-directed enzyme prodrug therapy with the recombinant anti-carcinoembryonic antigen antibody-enzyme fusion protein MFECP1 and a bis-iodo phenol mustard prodrug. Clin Cancer Res. 2006;12(21):6509–6516. | |
Abraham S, Guo F, Li LS, et al. Synthesis of the next-generation therapeutic antibodies that combine cell targeting and antibody-catalyzed prodrug activation. Proc Natl Acad Sci U S A. 2007;104(13):5584–5589. | |
Kaliyaperumal A, Jing S. Immunogenicity assessment of therapeutic proteins and peptides. Curr Pharm Biotechnol. 2009;10(4):352–358. | |
Brinks V, Jiskoot W, Schellekens H. Immunogenicity of therapeutic proteins: the use of animal models. Pharm Res. 2011;28(10):2379–2385. | |
Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J. 2012;14(2):296–302. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.