Back to Journals » Drug Design, Development and Therapy » Volume 8
Characterization and evaluation of amine-modified graphene amphotericin B for the treatment of visceral leishmaniasis: in vivo and in vitro studies
Authors Mudavath SL ![]() , Talat M, Rai M, Srivastava O, Sundar S
, Talat M, Rai M, Srivastava O, Sundar S ![]()
Received 13 March 2014
Accepted for publication 16 April 2014
Published 4 September 2014 Volume 2014:8 Pages 1235—1247
DOI https://doi.org/10.2147/DDDT.S63994
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Shyam Lal Mudavath,1 Mahe Talat,2 Madhukar Rai,1 Onkar Nath Srivastava,2 Shyam Sundar1
1Infectious Disease Research Laboratory, Department of Medicine, Institute of Medical Sciences, Banaras Hindu University, Varanasi, India; 2Nanoscience Unit, Department of Physics, Banaras Hindu University, Varanasi, India
Abstract: Amphotericin B (AmB) has been the first-line treatment for visceral leishmaniasis (VL), a neglected protozoan disease, especially in regions like Bihar, India, where resistance to antimonials is widespread. However, adverse drug reactions are a major limiting factor. We evaluated a novel formulation of AmB conjugated to amine-modified graphene (f-Gr) for safety and efficacy over conventional AmB. The f-Gr was prepared in a gentle one-step process of noncovalent (amine) functionalization with the help of amino acid L-cysteine. This f-Gr was further conjugated to AmB by peptide bond. The conjugate (f-Gr-AmB) was characterized by Raman spectroscopy, Fourier transform infrared spectroscopy, scanning electron microscopy, and transmission electron microscopy. f-Gr-AmB was found to exhibit lesser cytotoxicity toward J774A.1 cells than AmB, and did not induce any hepatic or renal toxicity in Swiss albino mice. In vitro antileishmanial assay in J774A.1 cells showed significantly enhanced efficacy of f-Gr-AmB over AmB. Furthermore, percentage inhibition of amastigote replication in a hamster model of VL was significantly higher in the f-Gr-AmB treated group (87.8%) compared to the AmB group (70.4%). These results suggest that f-Gr-AmB could be a safe and effective alternative to conventional AmB in the treatment of VL.
Keywords: antileishmanial, efficacy, cytotoxicity, macrophage, Raman spectroscopy, amastigote
Introduction
Visceral leishmaniasis (VL) is one of the most neglected tropical diseases caused by intracellular protozoan parasites of the Leishmania donovani complex. VL is the second most common cause of mortality and fourth most common cause of morbidity due to tropical diseases, causing 20,000 to 40,000 deaths annually.1 The treatment options for VL are highly circumscribed, and if untreated, the disease most often culminates in death.2,3 Amphotericin B (AmB) is used as a first-line drug for VL in India and constitutes an alternate treatment for patients resistant to antimonials elsewhere. However, it has major drawbacks in terms of protracted hospitalization and significant nephrotoxicity.4,5 In spite of the fact that lipid formulations of AmB exhibit better therapeutic indexes, their use is throttled by higher costs and their intravenous route of administration.6,7
The variable efficacy of certain antileishmanial drugs and significant toxicity of others has been a motivation to explore novel therapeutic strategies.8 One area of concern is the localization of the parasite within the lysosomes of reticuloendothelial cells, which hampers the access and thus bioavailability of antileishmanial drugs.9 Hence, the development of delivery systems that can target macrophages intracellularly is crucial, and could potentially lead to new treatment paradigms for VL with improved therapeutic efficacy and reduced toxicity.10 Nanomaterials have proved to be valuable as vectors for intracellular drug delivery. They fall into a size range close to that of proteins and other biological macromolecules, which makes it possible for them to interact with and enter cells.11 Previously, we described that nanoparticles of AmB have significantly greater efficacy than conventional AmB, with an improved safety profile.12 Subsequently, we also reported functionalized carbon nanotubes (f-CNTs) as an effective nanovector for AmB for oral administration in the treatment of VL.13,14
Graphene and its derivatives are now at the forefront of nearly every rapidly developing field of science and engineering, including biomedical applications such as biosensor development, imaging, drug delivery, bacterial inhibition, and photothermal therapy.15–19 The fascinating physicochemical properties and low cost, scalable production coupled with an intrinsic biocompatibility, and facile biofunctionalization make graphene an attractive nanoscaffold for drug delivery.20,21 Functionalized graphene oxide has been shown to be a promising nanovector for the efficient delivery of drugs into cells. Despite the great application potential, it is important to mention that graphene itself possesses zero band gap as well as inertness to reaction. The π-π stacking between graphene sheets (GS) results in the formation of multilayers. Pristine GS are hydrophobic in nature, and tend to aggregate in solution, which is driven by enhanced van der Waals forces; this feature gives rise to technical difficulties during application.22 This is one of the reasons that an increasing number of reports are available on functionalization of graphene and its derivatives. Thus, functionalization of GS is important for their future applications. Similar to the drug loading on CNTs, the graphene surface with delocalized p-electrons can be utilized for effective loading of drugs via π-π stacking. GS as a drug carrier are interesting because both sides of a single sheet could be accessible for drug binding. The extremely large surface area of graphene (~2,630 m2/g), with every atom exposed on its surface, allows for ultra-high drug loading efficiency on graphene as compared to CNT, the surface area of which is almost half that of graphene.23
However, the growing concern about cytotoxicity of graphene oxide has drawn researchers to look for alternate derivatives.24–26 Amine-functionalized graphene was shown to be a safer alternative to graphene oxide, with potential for biomedical applications.27 Hence, in the present study we explore the efficacy and toxicity of a novel AmB formulation as a conjugate (f-Gr-AmB) with amine-modified graphene (f-Gr) in the treatment of VL.
Materials and methods
Parasites for infection and cell lines
The J774A.1 macrophage cell line (obtained from the National Centre for Cell Science, Pune, India) was used as a cellular host. World Health Organization (WHO) reference strain of L. donovani LEM 138 (MHOM/IN/00/DEVI) was used for raising infection. Parasites were cultured in a supplemented Roswell Park Memorial Institute (RPMI) 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA), as described previously.28
Animals
Male Syrian golden hamsters, Mesocricetus auratus (45–50 g), were procured from the animal house facility of the Central Drug Research Institute (CDRI), Lucknow, India for studying in vivo antileishmanial activity, and male Swiss albino mice (30–40 g) for in vivo toxicity studies were purchased from Central Animal Facility, Institute of Medical Sciences (IMS), Banaras Hindu University (BHU), Varanasi, India. All experiments were performed using protocols approved by the Central Animal Ethics Committee (CAEC), IMS, BHU (CAEC number Dean/2014/CAEC/615). The guidelines of the Council for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Environment and Forests, Government of India were followed.
Synthesis of GS
The GS were synthesized by thermal exfoliation of graphite oxide (GO). GO was prepared using graphite powder according to a slightly modified Staudenmaier’s method.29 In a typical experiment, graphite powder (<50 μm, 1 g) was treated with a strong oxidizing solution of 18 mL concentrated analytical research grade H2SO4 (Sigma-Aldrich Co, St Louis, MO, USA), 9 mL concentrated analytical research grade HNO3 (Sigma-Aldrich Co), and 11 g potassium chlorate (less than 99% pure; Merck and Co, Inc., Whitehouse Station, NJ, USA) at room temperature for 5 days under magnetic stirring conditions. As-obtained GO solution was washed with distilled water and 10% HCl solution to remove sulfate and other ion impurities. Chemical purification was done to remove impurities that exist or are intercalated between the various graphite planes. The GO powder was dried at 80°C under vacuum. As-prepared GO (~200 mg) was thermally exfoliated by rapid heating at 1,050°C for 30 seconds under an argon atmosphere. The product was isolated by filtration, washed copiously with water, and vacuum dried.
Amine functionalization of GS
GS were functionalized with the amino acid L-cysteine in double distilled water. A mass of 25 mg of GS was added to 250 mL double distilled water and sonicated for 1.5 hours at room temperature to obtain a homogeneous solution. Then, 0.1 M L-cysteine (in double distilled water) was added to the solution and sonicated for 30 minutes, followed by 3 hours of constant magnetic stirring. The f-Gr so obtained was thoroughly washed with double distilled water in a centrifuge at 10,000 rpm for 10 minutes, and the solution phase was discarded. This washing was repeated five times to remove any unbound L-cysteine. f-Gr was dried in ambient atmosphere and stored at 4°C. Since this method is gentle and does not require any harsh chemicals such as acids, it is preferred for targeted drug delivery, biosensing, and other applications using enzymes.30–32
Attachment of AmB to f-Gr
f-Gr was dispersed in deionized water, and AmB powder was dissolved in dimethyl sulfoxide. The AmB solution was mixed with f-Gr solution separately and sonicated in an ultrasonication bath at room temperature for about 24 hours, followed by ~4 hours of magnetic stirring. The solution was centrifuged and washed with double distilled water approximately three to four times to remove unbound AmB. By comparing the optical density (OD) of known concentrations of AmB to the OD of unattached, dissolved AmB remaining in the supernatant of f-Gr-AmB, AmB loaded to f-Gr was calculated as
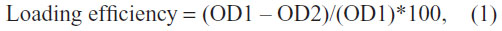
where OD1 = A409 of AmB (1 mg/mL), and OD2 = A409 of unattached AmB from the supernatant.
Characterization techniques
The ultrastructures of graphene and its derivatives (GS, f-Gr, and f-Gr-AmB) were examined by scanning electron microscope (SEM; a Philips XL 20; Koninklijke Philips NV, Amsterdam, the Netherlands or a Quanta 200; FEI, Hillsboro, OR, USA) and transmission electron microscope (TEM; Technai 20 G2, FEI). Functionalization of AmB was established by Fourier transform infrared (FTIR) spectroscopy using a Spectrum 100 spectrometer (PerkinElmer, Waltham, MA, USA) with the help of universal attenuated total reflectance attachment. Attachment of AmB to f-Gr was established by Raman spectroscopy, using an HR-800 Raman spectrometer for argon ion laser (λ=514 nm; Horiba Jobin Yvon, Kyoto, Japan).
In vitro cytotoxicity testing
The cytotoxicity of f-Gr-AmB, f-Gr, and AmB was assessed as described previously.12 J774A.1 macrophage cells were plated in a 96-well microplate at a density of 2.5×104cells/well. Then, the cells were incubated in triplicate with varying concentrations of f-Gr-AmB (0.005–0.64 μg/mL), f-Gr (0.0625–8.0 μg/mL), or AmB (0.005–0.64 μg/mL) for 72 hours at 37°C, 5% CO2; untreated cells served as a control. Cytotoxicity was assessed using the colorimetric MTT (3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium-bromide) reduction assay. The 50% cytotoxicity concentration (CC50) was calculated from the graph of ODs plotted against drug concentration, taking OD of the control well as a measure of 100% survival.
In vitro antileishmanial activity against intracellular amastigotes
The J774A.1 macrophage cells were seeded at a density of 5×104 cells/well on 8-chamber Lab Tek tissue culture slides (USA Scientific, Inc., Ocala, FL, USA) and were allowed to adhere for 2 hours in a CO2 incubator with 5% CO2 at 37°C. The wells were then washed twice with serum-free RPMI 1640 medium to remove non-adherent macrophages. Further, adhered macrophages were infected with metacyclic stage parasites of L. donovani LEM 138, maintaining a Leishmania: macrophage ratio of 10:1 in a 200 μL final solution of a complete RPMI 1640 medium supplemented with 15% (v/v) heat inactivated fetal bovine serum (Thermo Fisher Scientific) and 50 mg/L gentamicin overnight. After 24 hours, free promastigotes were washed with serum-free medium, and infected macrophages were incubated with AmB (0.005–0.02 μg/mL), f-Gr (0.005–0.02 μg/mL), and f-Gr-AmB (0.00125–0.005 μg/mL) in duplicate for 72 hours with 5% CO2 at 37°C, except in the control well (without drug), followed by methanol fixing for 1 minute and staining with giemsa (Qualigens, Mumbai, India). At least 100 macrophage nuclei were counted per well for calculating the percentage of infected macrophages and the number of amastigotes per 100 macrophages. The IC50 (concentration of drug that inhibits 50% of L. donovani amastigotes) of each drug was calculated, as described previously.12
In vivo antileishmanial studies
Hamsters are an optimal model of L. donovani infection, as they mimic several clinical and immunopathological aspects of human VL and can withstand repeated spleen biopsies to check parasite burden. Twenty-five male hamsters (4–6 weeks of age) were infected by intracardiac injection of 1×107 promastigotes of L. donovani LEM 138. After 30 days, infection was confirmed by giemsa staining of splenic smears from four randomly selected animals. AmB, f-Gr, and f-Gr-AmB were reconstituted for in vivo administration in 1× phosphate buffered saline (PBS) at a concentration of 1.5 mg/mL. The treatment and control groups consisted of 4–6 animals in each group, and treatment animals were treated intraperitoneally with AmB (n=10), f-Gr (n=10), and f-Gr-AmB (n=12) at a dose of 5 mg/kg body weight for 5 consecutive days, while the control group (n=8) received an equal volume of PBS. Autopsies were conducted on day 7 post-treatment. The weight of the spleen was measured immediately after autopsy, and dabbed imprints on glass slides were prepared. The parasite burden, percentage of inhibition of splenic parasite load, and the percentage of suppression of parasite replication were calculated.13 This experiment was repeated with 25 male hamsters for reproducibility.
In vivo toxicity studies
The in vivo toxicity of f-Gr-AmB, f-Gr, and AmB was assessed in 40 Swiss albino mice (25±5 weeks of age) with a 5-day course of daily intraperitoneal injection using 5 mg/kg, 10 mg/kg, and 20 mg/kg dose regimens in 12 mice with 4 mice for each concentration. Aspartate transaminase (AST), alanine aminotransferase (ALT), urea, and creatinine levels were used as biochemical markers to evaluate hepatotoxicity and nephrotoxicity. The control group, comprising 4 mice, was injected with PBS. After a 5-day course, the mice were euthanized, blood was drawn, and serum was separated. Levels of ALT, AST, urea, and creatinine were estimated to assess renal and hepatic function using commercially available kits (Autozyme GPT; Accurex Biomedical Pvt. Ltd., Mumbai, India. Enzopak SGOT; Reckon Diagnostics Pvt. Ltd., Gujart, India. Lyphozyme Urea Berthelot; Beacon Diagnostics Pvt. Ltd., Gujarat, India. Autozyme creatinine; Accurex Biomedical Pvt. Ltd., Mumbai, India).
Statistical analysis
GraphPad Prism version 5 was used to calculate the CC50 and IC50 values. An unpaired t-test (two-tailed) was applied to determine the significance of the differences between the cytotoxicity and antileishmanial activity of the two drugs, AmB and f-Gr-AmB.
Results
Schematic representations of functionalization of GS and attachment of AmB are shown in Figure 1. Representative TEM images of GS, f-Gr, and f-Gr-AmB samples are shown in Figure 2. The TEM image of GS reveals a wrinkled paper-like structure, which appears to be very thin (Figure 2A). The Figure 2A inset is the selected area electron diffraction pattern of GS, showing clear diffraction spots. The diffraction spots were indexed to hexagonal graphite crystal structure. In contrast, the TEM image of f-Gr shows a smoothened surface, which is due to the functionalization by L-cysteine (Figure 2B). The image of Figure 2C shows AmB deposited on f-Gr, and appears as a cloudy shadow. The selected area electron diffraction pattern of the inset in Figure 2C demonstrates the well-textured and crystalline nature of f-Gr-AmB, which supports the conclusion that AmB is attached.
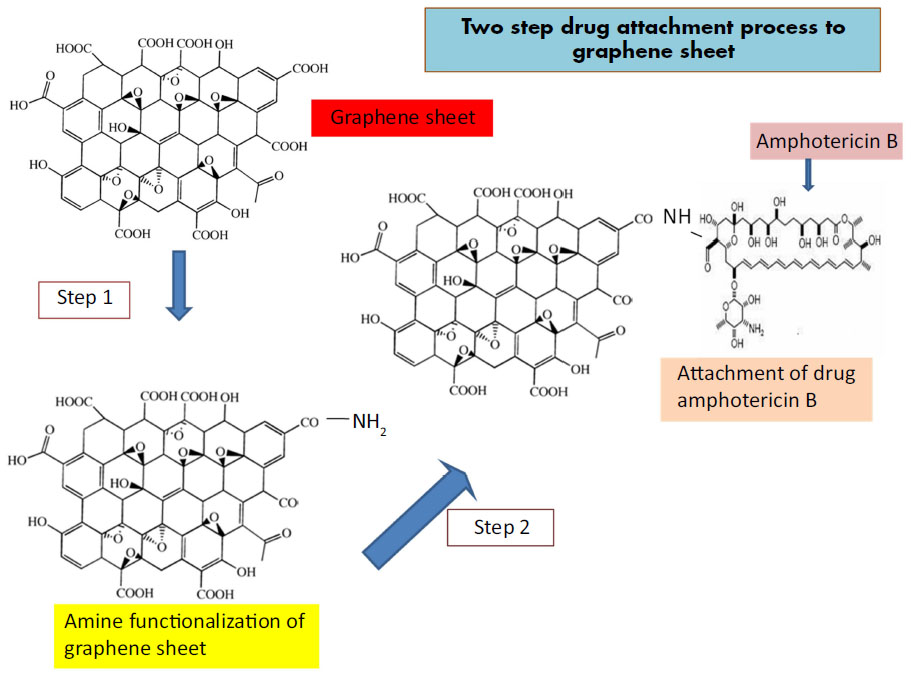 | Figure 1 Schematic representation of amphotericin B attachment to functionalized graphene sheet. |
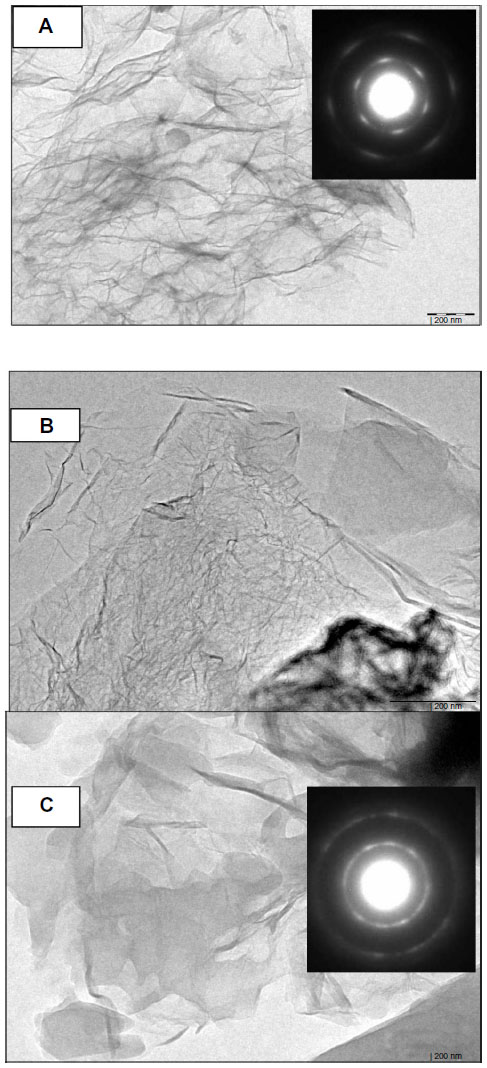 | Figure 2 TEM images of GS (A), f-Gr (B), and f-Gr-AmB (C). |
The surface morphologies of GS, f-Gr, and f-Gr-AmB were examined by SEM in secondary electron imaging mode. The random orientation and wavy appearance of exfoliated GO is seen from the SEM images. The top views of f-Gr clearly illustrate the typically flake-like structure with slightly scroll-edged shapes (Figure S1A). It is clearly visible from Figure S1B that the small L-cysteine molecules are closely anchored on the surface of GS and held noncovalently (marked with the arrow). Figure S1C presents the SEM image of f-Gr-AmB. It obviously shows that a layer of AmB was formed homogeneously like a blanket (rectangular area) on the f-Gr surface, confirming that the AmB molecule is uniformly attached on the GS.
FTIR analysis of GS, f-Gr, and f-Gr-AmB
A representative FTIR spectrum of GO, f-Gr, and f-Gr-AmB is shown in Figure S2A–C. It may be mentioned here that GS prepared from thermal exfoliation of GO at 1,050°C for 30 seconds invariably contains some oxygen functionalities. These are C=O, COOH, and OH functionalities, which are revealed by FTIR. C=C stretching at 1,558 cm−1 is attributed to the vibration of the carbon skeleton of GS; C=O stretching occurring at 1,748 cm−1 is also recorded. The observed peak in Figure S2A at 3,411 cm−1 corresponds to hydroxyl (–OH) stretching and vibration. In addition, the peaks at 1,398 and 1,120 cm−1 are related to C–OH (hydroxyl) and C–O (epoxy), respectively. This observation confirms the presence of oxygen containing functional groups, eg, C=O, C–OH, and C–O on the GS. It is generally recognized that the hydroxyl and epoxy groups are present above and below the basal planes, while the carboxylic groups are bound to the edge of GS.33 In f-Gr (Figure S2B), an additional peak is observed at 3,016 cm−1, which is due to the stretching vibrations of an alkane group, while the presence of a peak at 1,475 cm−1 can be assigned to N–H stretching vibrations of the amine group, and the 1,385 cm−1 peak is due to the C–N stretching vibrations. In Figure S2C, the peak at 1,902 cm−1 is due to the C–O stretching of f-Gr. The peak at 1,019 cm−1 is associated with the stretching vibration mode of the hydroxyl group of the carboxylic group present in AmB, which reacts with the amine group of the f-Gr. A new peak at 1,609 cm−1 represents amide carbonyl and results from the C=O stretching along with the N–H deformation mode.34
Raman spectra of GS and f-Gr-AmB
Figure 3A shows the Raman spectra of GS. The characteristic peak at 1,308 cm−1 of D (disorder) peak, 1,589 cm−1 of G (graphite) peak, and 2D peak at 2,661 cm−1 confirm the formation of GS.35 The intensity of D/G ratio is found to be ~0.82. Figure 3B shows the Raman spectra of AmB attached to f-Gr. Along with the almost eliminated D peak at 1,381 cm−1 and the G peak at 1,565 cm−1, two more peaks are appearing in the spectra, ie, at 1,012 cm−1 and at 1,156.78 cm−1. These are most probably due to the AmB attachment with f-Gr. A huge red shift of 25 cm−1 in the G peak is due to the attachment of AmB to the surface of f-Gr. The decreased value of the ID/IG ratio, ie, a decrease of ~0.404, is due to the reduced intensity of D peak and the sharp G peak, which signify that the AmB attached on the f-Gr is decreasing the defects of the system. It can be assumed that due to a masking effect of the AmB, the defects of the f-Gr are reduced, which is apparently visible in the spectra. So, it can be concluded that AmB is effectively attached on the surface, or most probably on the edges, of the f-Gr.
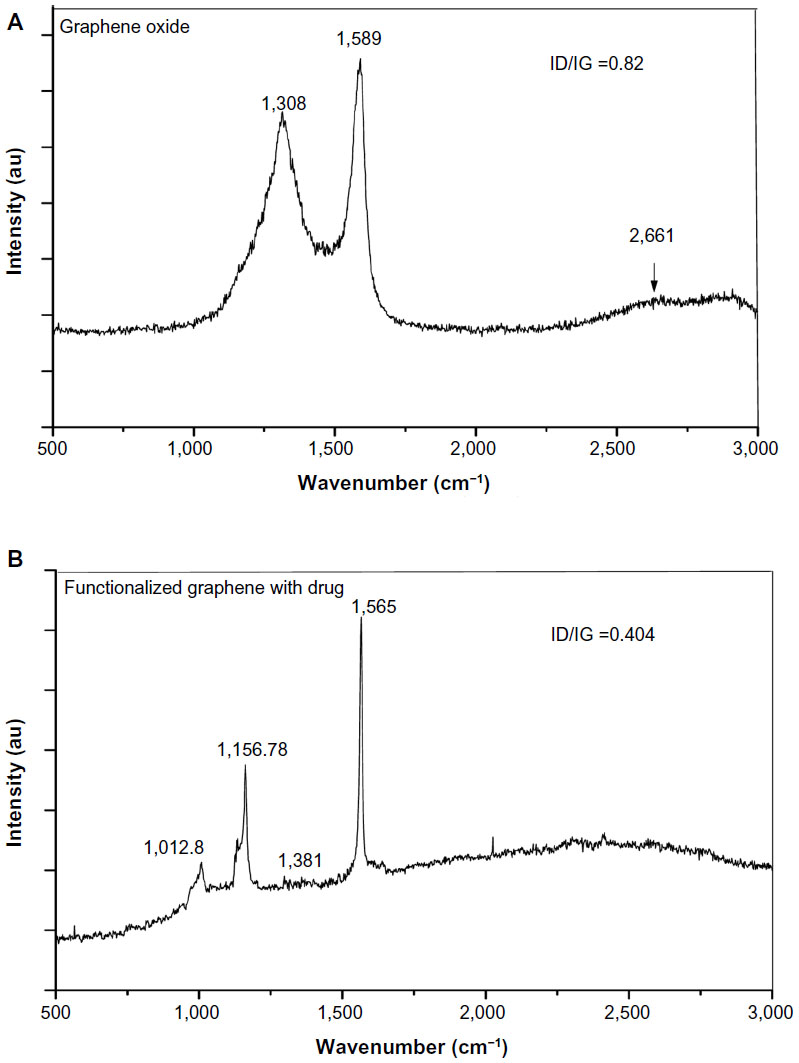 | Figure 3 Raman spectra of GS (A) and f-Gr-AmB (B). |
The OD of a known concentration of AmB (1 mg/mL) and unattached AmB from the supernatant of f-Gr-AmB, were 3.392 and 0.868 at 409 nm, respectively. The loading efficiency of 74.39% was consistent with no observable f-Gr-AmB precipitation during 24 hours of storage at room temperature.
f-Gr-AmB showed better antileishmanial activity without an increased cytotoxicity toward macrophages
Taking into account the improved antileishmanial activity of AmB, as a result of conjugation to f-Gr, we looked into whether this improvement is associated with increased toxicity for macrophages. The CC50 of the compounds against the J774A.1 cell line were: AmB, 0.375±0.06 μg/mL; f-Gr-AmB, 0.52±0.02 μg/mL; and f-Gr, 4.28±1.27 μg/mL (P=0.04). This CC50 of f-Gr-AmB is far higher than the IC50 (0.003 μg/mL) against intramacrophage amastigotes. It is important to note that f-Gr-AmB does not show any significant toxic effects in vitro on J774A.1 macrophage cells at antileishmanial concentrations. These data represent the mean ± standard deviation of two experiments performed in triplicate.
f-Gr-AmB showed greater inhibition of intramacrophage amastigotes
Amastigotes, the infective form of Leishmania, reside and replicate in the reticuloendothelial macrophages. Reduction in the number of intracellular amastigotes of infected macrophages is indicative of better antileishmanial activity of a drug formulation. The antileishmanial activity of the compounds was: AmB, IC50 =0.027±0.004 μg/mL; and f-Gr-AmB, IC50 =0.003±0.0004 μg/mL. The IC50 of f-Gr-AmB was nine times more than AmB against intramacrophage amastigotes (P=0.003). This gives substance to the conclusion that f-Gr-AmB is a better antileishmanial formulation compared to conventional AmB.
f-Gr-AmB showed improved antileishmanial activity in the hamster model of VL
For analysis, the outcome from both the sets of in vivo experiments was conflated to yield a mean value. No significant difference was observed in the weight of both control and treatment group hamsters. There was a substantial improvement in the antileishmanial activity of f-Gr-AmB compared to conventional AmB. The percentage inhibition of parasites in the spleens of f-Gr-AmB treated Leishmania-infected hamsters (87.8%) was higher when compared with AmB treated Leishmania-infected hamsters (70.4%) (P=0.002; Table 1). In contrast, f-Gr showed a negligible antileishmanial activity (4.96%). There was a more significant reduction in the parasite burden in the f-Gr-AmB group (5.0±1.2 LDU) than in the AmB treated group (16.9±2.5 LDU) (P=0.005; Table 1). The percentage suppression of parasite replication of f-Gr-AmB treated Leishmania-infected hamsters (90.3%) was higher when compared with AmB treated Leishmania-infected hamsters (75.9%) (P=0.001; Table 1). Furthermore, there was a greater reduction in the spleen size of f-Gr-AmB treated hamsters (2.6 cm) than in infected controls (3.4 cm), suggestive of a decrease in parasite burden (Figure 4).
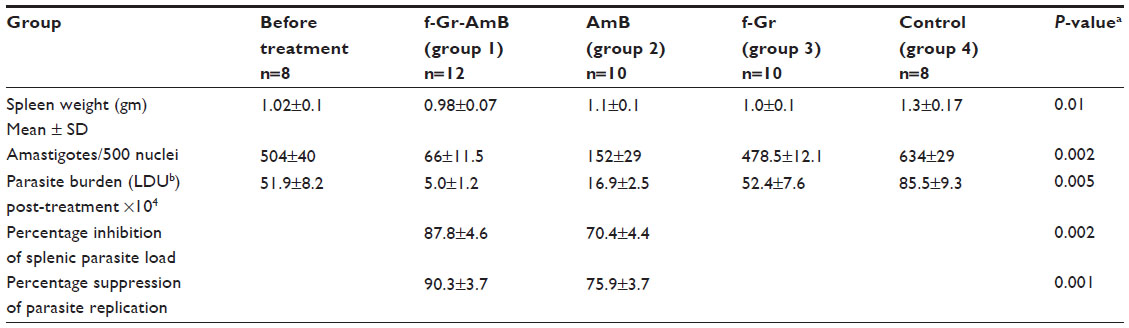 | Table 1 In vivo efficacy studies in Syrian golden hamsters |
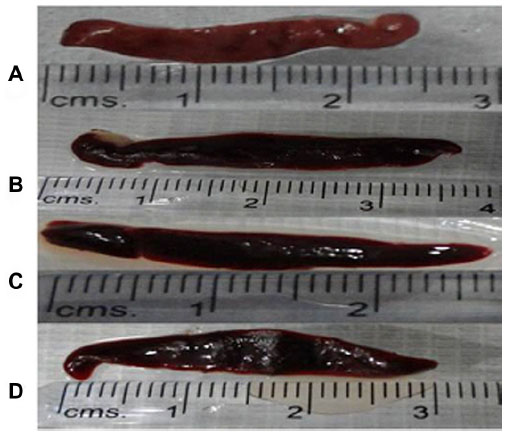 | Figure 4 Spleen sizes of hamsters of different experimental groups. |
f-Gr-AmB did not induce hepatorenal toxicity in Swiss albino mice
Adverse drug reactions are major limiting factor of AmB.36 Hence, we looked for the potential in vivo toxicity of the formulations in Swiss albino mice. No obvious signs of toxic effects for AmB, f-Gr, and f-Gr-AmB injected mice at a dose of up to 20 mg/kg within 5 days were noted. Neither significant body weight drop nor death was noted in any treatment group. No noticeable difference was observed from blood serum biochemistry of renal and hepatic enzymes, urea (range, 13.4–18.5 mg/dL), creatinine (range, 0.4–0.56 mg/dL), AST (range, 31.7–52.2 IU/L), or ALT (range, 33.8–46.3 IU/L) between the control and treatment groups at all doses, and all were within the normal reference range (Table 2).
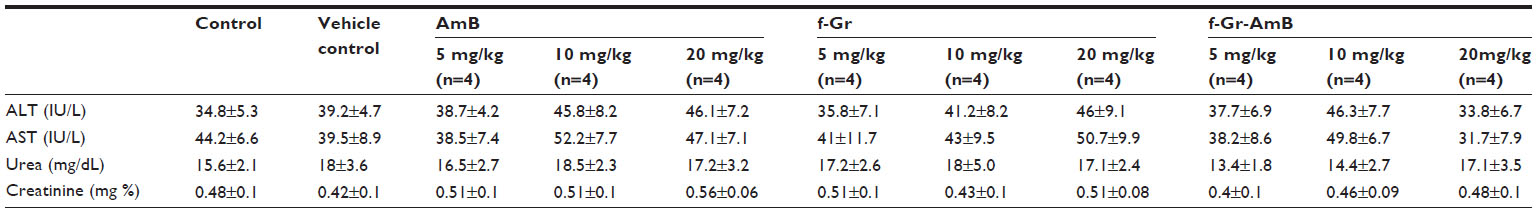 | Table 2 In vivo biochemical parameters for liver and kidney functions in Swiss albino mice |
Discussion
The present study was designed to evaluate graphene-based drug delivery of AmB for its toxicity, antileishmanial efficacy, and cost effectiveness. Drug delivery is one of the most extensively explored applications of carbon nanomaterials in biomedicine.37 During attachment, the carboxylic group of AmB reacts with the amine group of f-Gr, forming a stable f-Gr-AmB conjugate. Being a covalent bond, the drug is strongly bonded to the f-Gr and does not dissociate during delivery of the drug AmB.
The f-Gr-AmB conjugate exhibited significantly enhanced activity against intracellular amastigotes of L. donovani in a macrophage model with 9-fold improvement in IC50 values over conventional AmB. This may be attributed to more efficient intracellular uptake of f-Gr-AmB by macrophages, and hence better delivery of AmB to amastigotes.
These results were corroborated in a golden hamster model of L. donovani infection, where significantly greater inhibition was observed with f-Gr-AmB. Treatment of infected hamsters with f-Gr-AmB induced a remarkable decrease in parasitic load and amelioration of VL pathology compared to AmB treated hamsters. This improved efficacy was comparable to that observed with f-CNT conjugated and liposomal formulations of AmB, which suggests a general correlation between enhanced intracellular delivery and effectiveness of AmB.
A serious limitation of AmB in the treatment of VL has been its toxicity. In recent years various safer, lipid formulations of AmB (AmBisome, Abelcet, and Amphotec) have been commercialized, albeit at considerably increased expense. Carbon-based nanomaterials as scaffolds for AmB delivery are much more cost effective.20 However, cytotoxicity may set a substantial limitation when harnessing nanomaterials for biological settings. Both CNTs as well as some derivatives of graphene, particularly graphene oxide, have raised concerns of cytotoxicity and lack of hemocompatibility.26 Amine functionalization of graphene has been shown to be a safe alternative, and was thus employed in this study. Hence, quite predictably, the CC50 of f-Gr-AmB against J774A.1 cells was significantly more compared to that of AmB, with no evidence of cytotoxicity at clinically relevant concentrations. This was further affirmed by the minimal or no hepatic and renal toxicity in vivo.
In conclusion, treatment with f-Gr-AmB resulted in an improved therapeutic index between IC50 (0.003±0.0004 μg/mL) and CC50 (0.52±0.02 μg/mL), hence increasing its safety profile and antileishmanial activity over conventional AmB. To the best of our knowledge, this is the first report demonstrating the potential use of f-Gr as a vector for delivery of AmB. Our data evince the ascendancy of f-Gr-AmB over AmB, suggesting the potential of f-Gr as a novel nanovector for AmB in the treatment of VL. Further studies are ongoing to compare f-Gr-AmB efficacy with that of liposomal AmB and to scrutinize its potential for oral administration. Despite these facts, better understanding of quantitative pharmacokinetics, biodistribution, in vivo behavior, as well as long term toxicology are acutely crucial and require further research.
Acknowledgments
We would like to thank Dr Vijay Kumar Prajapati, Dr Paresh Kulkarni, and all research scholars of the Infectious Disease Research Laboratory, BHU for their kind help during the study, and Dr K Awsathi, Dr S Awasthi, and Dr H Raghuvanshi for the fruitful discussion on graphene synthesis. Shyam Lal Mudavath gives thanks to the University Grants Commission (UGC), New Delhi for providing a Junior Research Fellowship, and Mahe Talat is grateful to the Department of Science and Technology’s (DST’s) Women Scientists Scheme-A (WOS-A) for support.
This work was supported by DST (Unit on Nanoscience and Nanotechnology [UNANST], BHU) and the National Institute of Allergy and Infectious Diseases, National Institutes of Health (Tropical Medicine Research Center [TMRC] grant number P50AI074321). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Alvar J, Vélez ID, Bern C, et al. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 2012;7(5):e35671. | |
Thakur CP, Pandey AK, Sinha GP, Roy S, Behbehani K, Olliaro P. Comparison of three treatment regimens with liposomal amphotericin B (AmBisome) for visceral leishmaniasis in India: a randomized dose-finding study. Trans R Soc Trop Med Hyg. 1996;90(3):319–322. | |
Sundar S, Chatterjee M. Visceral leishmaniasis – current therapeutic modalities. Indian J Med Res. 2006;123(3):345–352. | |
Olliaro PL, Guerin PJ, Gerstl S, Haaskjold AA, Rottingen JA, Sundar S. Treatment options for visceral leishmaniasis: a systematic review of clinical studies done in India, 1980–2004. Lancet Infect Dis. 2005;5(12):763–774. | |
Sundar S, Singh A, Agarwal D, Rai M, Agrawal N, Chakravarty J. Safety and efficacy of high-dose infusions of a preformed amphotericin B fat emulsion for treatment of Indian visceral leishmaniasis. Am J Trop Med Hyg. 2009;80(5):700–703. | |
Egger SS, Meier S, Leu C, et al. Drug interactions and adverse events associated with antimycotic drugs used for invasive aspergillosis in hematopoietic SCT. Bone Marrow Transplant. 2010;45(7):1197–1203. | |
Singh RK, Pandey HP, Sundar S. Visceral leishmaniasis (kala-azar): challenges ahead. Indian J Med Res. 2006;123(3):331–344. | |
Jain K, Jain NK. Novel therapeutic strategies for treatment of visceral leishmaniasis. Drug Discov Today. 2013;18(23–24):1272–1281. | |
Chellat F, Merhi Y, Moreau A, Yahia L. Therapeutic potential of nanoparticulate systems for macrophage targeting. Biomaterials. 2005;26(35):7260–7275. | |
Kansal S, Tandon R, Dwivedi P, et al. Development of nanocapsules bearing doxorubicin for macrophage targeting through the phosphatidylserine ligand: a system for intervention in visceral leishmaniasis. J Antimicrob Chemother. 2012;67(11):2650–2660. | |
Hillaireau H, Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66(17):2873–2896. | |
Manandhar KD, Yadav TP, Prajapati VK, et al. Antileishmanial activity of nano-amphotericin B deoxycholate. J Antimicrob Chemother. 2008;62(2):376–380. | |
Prajapati VK, Awasthi K, Gautam S, et al. Targeted killing of Leishmania donovani in vivo and in vitro with amphotericin B attached to functionalized carbon nanotubes. J Antimicrob Chemother. 2011;66(4):874–879. | |
Prajapati VK, Awasthi K, Yadav TP, Rai M, Srivastava ON, Sundar S. An oral formulation of amphotericin B attached to functionalized carbon nanotubes is an effective treatment for experimental visceral leishmaniasis. J Infect Dis. 2012;205(2):333–336. | |
Yang M, Yao J, Duan Y. Graphene and its derivatives for cell biotechnology. Analyst. 2013;138(1):72–86. | |
Hu W, Peng C, Luo W, et al. Graphene-based antibacterial paper. ACS Nano. 2010;4(7):4317–4323. | |
Akhavan O, Ghaderi E, Rahighi R. Toward single-DNA electrochemical biosensing by graphene nanowalls. ACS Nano. 2012;6(4):2904–2916. | |
Robinson JT, Tabakman SM, Liang Y, et al. Ultrasmall reduced graphene oxide with high near-infrared absorbance for photothermal therapy. J Am Chem Soc. 2011;133(17):6825–6831. | |
Kuila T, Bose S, Khanra P, Mishra AK, Kim NH, Lee JH. Recent advances in graphene-based biosensors. Biosens Bioelectron. 2011;26(12):4637–4648. | |
Sanchez VC, Jachak A, Hurt RH, Kane AB. Biological interactions of graphene-family nanomaterials: an interdisciplinary review. Chem Res Toxicol. 2012;25(1):15–34. | |
Novoselov KS, Fal’ko VI, Colombo L, Gellert PR, Schwab MG, Kim K. A roadmap for graphene. Nature. 2012;490(7419):192–200. | |
Rodriguez-Pérez L, Herranz MA, Martin N. The chemistry of pristine graphene. Chem Commun. 2013;49(36):3721–3735. | |
Zhu Y, Murali S, Cai W, et al. Graphene and graphene oxide: synthesis, properties, and applications. Adv Mater. 2010;22(35):3906–3924. | |
Shen H, Liu M, He H, et al. PEGylated graphene oxide-mediated protein delivery for cell function regulation. ACS Appl Mater Interfaces. 2012;4(11):6317–6323. | |
Sun X, Liu Z, Welsher K, et al. Nano-graphene oxide for cellular imaging and drug delivery. Nano Res. 2008;1(3):203–212. | |
Singh SK, Singh MK, Nayak MK, et al. Thrombus inducing property of atomically thin graphene oxide sheets. ACS Nano. 2011;5(6):4987–4996. | |
Singh SK, Singh MK, Kulkarni PP, Sonkar VK, Grácio JJ, Dash D. Amine-modified graphene: thrombo-protective safer alternative to graphene oxide for biomedical applications. ACS Nano. 2012;6(3):2731–2740. | |
Maurya R, Mehrotra S, Prajapati VK, Nylén S, Sacks D, Sundar S. Evaluation of blood agar microtiter plates for culturing leishmania parasites to titrate parasite burden in spleen and peripheral blood of patients with visceral leishmaniasis. J Clin Microbiol. 2010;48(5):1932–1934. | |
Staudenmaier L. Verfahren zur darstellung der graphitsäure. Berichte der Deutschen Chemischen Gesellschaft [A method for representation of graphite acid. Reports of the German Chemical Society]. 1898;31(2):1481–1487. German. | |
Besteman K, Lee JO, Wiertz FG, Heering HA, Dekker C. Enzyme-coated carbon nanotubes as single-molecule biosensors. NanoLett. 2003;3(6):727–730. | |
Rana VK, Choi MC, Kong JY, et al. Synthesis and drug-delivery behavior of chitosan-functionalized graphene oxide hybrid nanosheets. Macromol Mater Eng. 2011;296(2):131–140. | |
Shan C, Yang H, Han D, Zhang Q, Ivaska A, Niu L. Water-soluble graphene covalently functionalized by biocompatible poly-L-lysine. Langmuir. 2009;25(20):12030–12033. | |
Schniepp HC, Li JL, McAllister MJ, et al. Functionalized single graphene sheets derived from splitting graphite oxide. J Phys Chem B. 2006;110(17):8535–8539. | |
Kaushik A, Khan R, Solanki PR, et al. Iron oxide nanoparticles-chitosan composite based glucose biosensor. Biosens Bioelectron. 2008;24(4):676–683. | |
Dresselhaus MS, Jorio A, Hofmann M, Dresselhaus G, Saito R. Perspectives on carbon nanotubes and graphene Raman spectroscopy. Nano Lett. 2010;10(3):751–758. | |
Sundar S, More DK, Singh MK, et al. Failure of pentavalent antimony in visceral leishmaniasis in India: report from the center of the Indian epidemic. Clin Infect Dis. 2000;31:1104–1107. | |
De Jong WH, Borm PJ. Drug delivery and nanoparticles: Applications and hazards. Int J Nanomedicine. 2008;3(2):133–149. |
Supplementary materials
 | Figure S1 SEM images of GS (A), f-Gr (B), and f-Gr-AmB (C). |
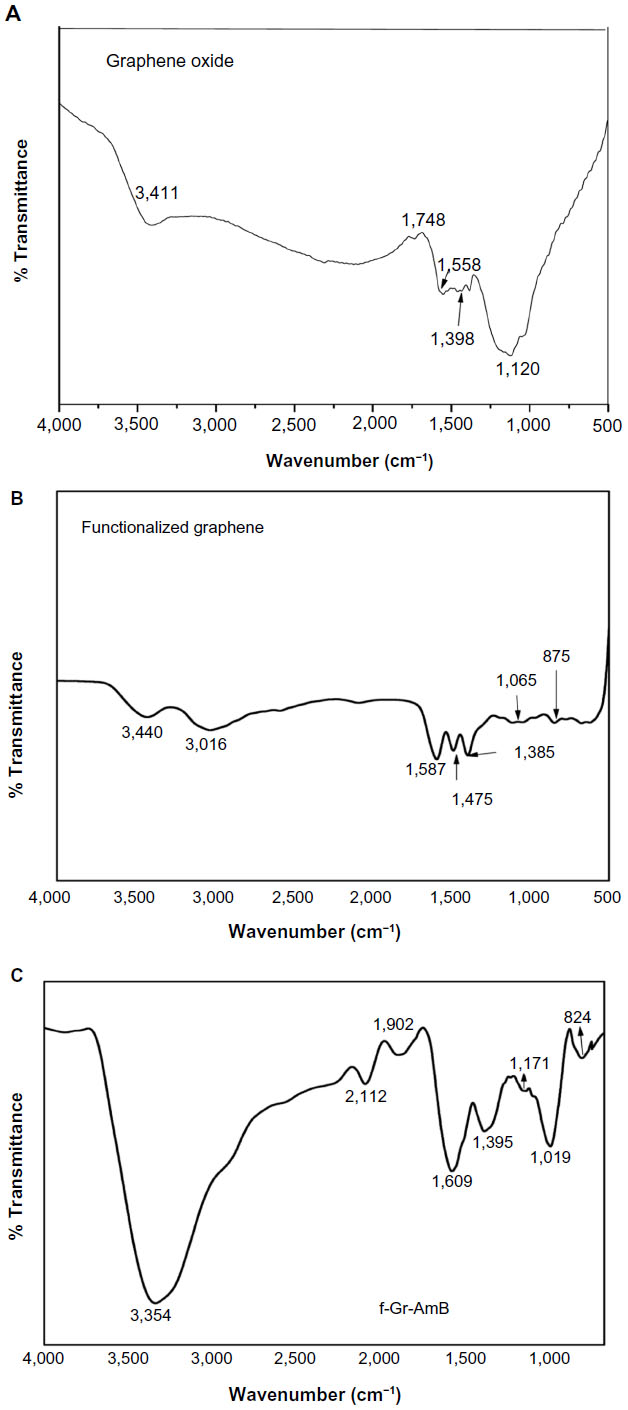 | Figure S2 FTIR spectra of GS (A), f-Gr (B), and f-Gr-AmB (C). |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.