Back to Journals » International Journal of General Medicine » Volume 15
Chaperone-Mediated Autophagy and Its Implications for Neurodegeneration and Cancer
Received 8 April 2022
Accepted for publication 9 June 2022
Published 15 June 2022 Volume 2022:15 Pages 5635—5649
DOI https://doi.org/10.2147/IJGM.S368364
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Masresha Ahmed Assaye,1,2 Solomon T Gizaw2
1Department of Internal Medicine, School of Medicine, College of Health Sciences, Addis Ababa University, Addis Ababa, Ethiopia; 2Department of Medical Biochemistry, School of Medicine, College of Health Sciences, Addis Ababa University, Addis Ababa, Ethiopia
Correspondence: Masresha Ahmed Assaye, Tel +251-911347186, Email [email protected]
Abstract: Proteostasis, also known as protein homeostasis, is critical for cell survival. Autophagy is a cellular process that degrades and recycles damaged or long-lived proteins, misfolded proteins, and damaged or abnormal organelles in order to preserve homeostasis. Among the three forms of autophagy, chaperone-mediated autophagy (CMA) is distinct from macroautophagy and microautophagy; it does not require the formation of vacuoles and only degrades selected individual proteins. CMA helps to maintain cellular homeostasis by regulating protein quality, bioenergetics, and substrate-associated cellular processes at the right moment. This pathway’s dysfunction has been linked to several diseases and disorders. Neurodegenerative diseases and cancer have received the most attention. In various neurodegenerative disorders, especially in their later stages, CMA activity declines. CMA has been shown to act as a tumor suppressor in cancer by destroying specific tumor promoters. Once a tumor has grown, it also helps tumor survival and the metastatic cascade. The presence of changes in CMA in these diseases disorders raises the idea of targeting CMA to restore cellular homeostasis as a potential therapeutic method. Manipulation of CMA activity may be effective therapeutic strategies for treating these diseases. Therefore, in this paper; we introduce the basic processes, regulatory mechanisms, and physiological functions of CMA; evidences supporting the role of impaired CMA function in neurodegeneration and cancer; and the potential of how targeting CMA could be a promising therapeutic method for the two diseases.
Keywords: chaperone, autophagy, chaperone-mediated autophagy, lysosome, cancer, neurodegeneration, therapy
Introduction
Cellular protein homeostasis, also referred to as proteostasis, is essential for cellular function and survival. It is based on a delicate balance of protein synthesis and breakdown.1 The ubiquitin-proteasome system (UPS) and the autophagy-lysosome pathway (ALP) are important systems found in almost all cell types that mediate the degradation of intracellular proteins into their constitutive amino acids.2
Autophagy is a highly conserved mechanism for delivering cytoplasmic components for lysosomal degradation in order to maintain the homeostatic balance of cellular protein and organelle synthesis, degradation, and recycling.3 Autophagy is classified into three types: macroautophagy (MA), microautophagy (MA), and chaperone-mediated autophagy (CMA).4,5
Chaperone-mediated autophagy (CMA) is a type of autophagy that is specialized in protein degradation and is based on the individual translocation of a cargo protein across the lysosomal membrane.6 It is distinguished by its selectivity and the fact that, unlike the other two lysosomal pathways, it does not require vesicle formation; substrate proteins appear to pass the lysosomal membrane directly to enter the lysosomal lumen.7 Individual proteins that are degraded by this autophagic pathway are chosen based on a recognition KFERQ-like consensus motif. This permits specific proteins to be removed without interfering with surrounding proteins, making CMA an effective method for the breakdown of damaged or aberrant proteins and surplus subunits of multi-protein complexes.8 Furthermore, because of its selectivity, CMA can play a regulatory role in processes like proteostasis, cellular energetics, and immune system activity by helping to modulate intracellular levels of enzymes, transcription factors, and cell maintenance proteins.6,8
Malfunctioning of CMA plays a key role in an increasing number of severe human disorders.9 The most emphasis has been paid to neurodegenerative illnesses and cancer, and there has been more experimental evidence of CMA involvement in these diseases.10,21 CMA activity diminishes in a variety of neurological disorders, particularly in their later stages, and increases in a variety of malignancies and immunological diseases.10 Several lines of evidence suggest that reversing altered CMA could be beneficial to disease processes and that relevant substances could be used as therapeutic candidates for disease treatment.7,10 A thorough understanding of CMA and its regulation is essential for determining how to target CMA for illness treatment.10 Therefore, this review aims to assess chaperone-mediated autophagy and its implications for neurodegeneration and cancer.
Molecular Chaperones
The term “molecular chaperone” appeared first in 1978 in a paper from the laboratory of Ron Laskey to describe a nuclear protein required for the correct assembly of nucleosomes from histones and DNA in extracts of amphibian eggs. Nucleosome assembly represents the major chaperoning function of nucleoplasmin.11 Molecular chaperones (or chaperones) are proteins that interact with, stabilize, or assist another protein in achieving its native and functionally active shape without being present in the final structure.12 They are a type of protein that aids in protein folding and protects cells from various stresses caused by the disruption of proteins’ native three-dimensional structures. They are proteins that detect and bind to the exposed hydrophobic surfaces of non-native proteins in a noncovalent contact to prevent irreversible aggregation.13 They are a sort of protein that helps maintain protein homeostasis or proteostasis in the cells of all living things.14
Functions of Chaperones
Chaperones are highly conserved molecular machines that regulate protein homeostasis in cells (proteostasis). They promote de novo protein folding and maturation, protein translocation, protein-complex assembly and disassembly, protein disaggregation and refolding, and protein degradation across species.15 They have long been known to (i) aid protein folding from unfolded or partially folded states, promoting the formation and maintenance of multisubunit complexes; (ii) mediate protein degradation via the UPS or ALS systems; (iii) inhibit protein aggregation by binding to fully or partially unfolded states; and (iv) promote the disassembly of unwanted aberrant protein aggregates. In addition to these four well-established activities, evidence is developing that molecular chaperones can interact with protein aggregates (macromolecular structures generated by aberrant protein self-assembly) and lower their toxicity without stimulating their deconstruction.16
Families of Chaperones
Molecular chaperones are a large and diverse group of proteins that share the property of assisting non-covalent folding and unfolding, and the assembly and disassembly, of other macromolecular structures, but are not permanent components of these structures when they are performing their normal biological function.11 Nucleoplasmins, chaperonins, and heat shock proteins (Hsp) are some of the proteins that make up molecular chaperones.16 Heat shock proteins account for a large number of them (HSP). These proteins were given the term heat shock proteins, and it was later shown that their expression is raised in response to a variety of stressors, including infection, inflammation, exercise, hunger, and hypoxia.12,15 The molecular mass of mammalian HSPs has been used to classify them into families. HSP100, HSP90, HSP70, HSP60, and tiny HSPs (molecular weight between 12 and 43 kDa) are the most common heat shock proteins (HSPs) (Table 1). HSP70 is the most common and studied type of the HSP family. It is used for folding of newly synthesized proteins, refolding of misfolded proteins, disaggregation, membrane translocation, endocytosis, and degradation of terminally misfolded proteins.15 The HSP70 family of chaperones is composed of at least 13 members, including stress-induced proteins such as HSPA1A [HSP70-1/HSP72; 72 kDa; 641 amino acid (aa) residues], and members that are not stress-inducible, such as HSPA5 (GRP78/Bip/Mif-2; 78 kDa; 654 aa), HSPA8 (HCC70/HSP73/HSP71; 73 kDa; 646 aa) and HSPA9 (mtHSP70/HSP75/GRP75/mortalin/; 75 kDa; 679 aa). In the large HSP70 family, the HSPA8 chaperone plays a major role in the protein quality control system. It acts as a folding catalyst of proteins ensuring the re-folding of misfolded conformers and as a controller targeting proteins for subsequent degradation.17 The small Hsp (Hsp10 and Hsp40) are denominated co-chaperones. Families of co-chaperones modulate the activity of the main chaperones by regulating their ATPase cycle or the recognition, binding, or release of chaperone substrates.15 HSP60 and small HSPs, also known as chaperonins, are major players in folding, although HSP70 may also participate in this process. Chaperonins are divided into Group I or Group II chaperonins. Class I chaperonins are found in bacteria, mitochondria, and chloroplasts, forming heptameric ring structures, and associate with smaller Hsp10-proteins (GroES in bacteria). The GroEL system in Escherichia coli and mitochondrial Hsp60, both of which are necessary for maintaining cellular protein homeostasis, are examples of Group I chaperonins. Eukaryotes and archaebacteria microbes both have Class II chaperonins in their cytoplasm. Because they have a built-in lid, they do not need co-chaperonins to work. The eukaryotic TriC/CCT machinery (TCP-1 ring complex/chaperonin containing TCP-1 complex), which is made up of eight subunits, and the thermosome in archaebacteria are examples of type II chaperonin.17 Both classes share the same functional feature: they entirely encapsulate their respective substrates in their folding chambers and urge the substrate protein towards its native fold in an energy-dependent manner.13,18
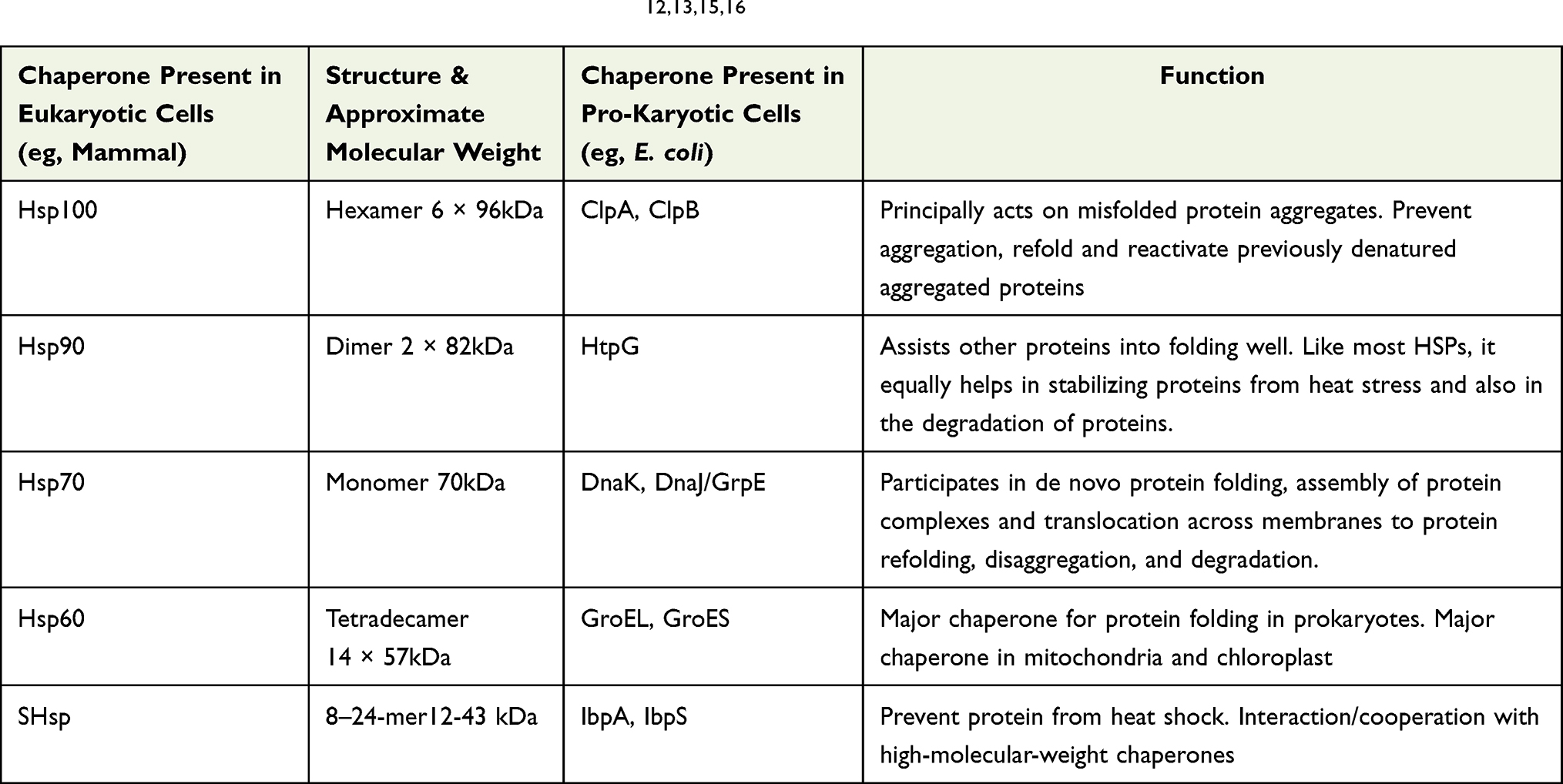 |
Table 1 Major Classes of Heat Shock Proteins (HSPs)12,13,15,16 |
Autophagy
The term autophagy [self (auto)-eating (phagy)] was first introduced in 1963 by Christian de Duve, who discovered the involvement of lysosomes in the autophagy process.19 It is a highly conserved cellular process that performs “housekeeping” functions in normal physiological processes, such as the removal of long-lived, harvested, and misfolded proteins, as well as damaged organelles. Under cellular stress conditions, such as malnutrition, hypoxia, oxidative stress, pathogen infections, radiation, or cancer medication, on the other hand, there is an increase in the degree of autophagy, which leads to adaptation and cell survival.20 Chaperone-mediated autophagy (CMA), microautophagy, and macroautophagy are the three types of autophagy (Figure 1). Instead of trafficking membrane vesicles, cargo is preferentially identified by a chaperone protein and subsequently internalized into the lysosome for destruction in CMA. This pathway is highly selective for a subset of cytosolic soluble proteins, but it is incapable of degrading organelles. The substrate is captured immediately by lysosomal membrane invagination and rearrangement in microautophagy. It is a mostly non-selective procedure that allows lysosomes to directly take up and break down cytosolic cargo.21,22 However, an exceptive form of microautophagy known as endosomal microautophagy has been discovered to have a selective degradation system for substrates (e-MI). Both CMA and e-MI require a pentapeptide motif related to KFERQ in substrate proteins for binding to HSC70. In contrast to CMA, selective e-MI does not need LAMP2A and protein unfolding. Consequently, CMA targets and degrades substrate proteins, making it quite different from the other two autophagic pathways.22 In macroautophagy, the whole regions of the cytosol are sequestered and delivered to lysosomes for degradation. It includes different stages, including phagophore initiation, vesicle nucleation, vesicle elongation, and autophagosome fusion with lysosomes. Macroautophagy can be both a selective and nonselective (bulk) kind of autophagy. It is a nonselective degrading system for the most part.21,23 Bulk cytoplasm is randomly swallowed by a phagophore in non-selective macroautophagy. Selective macroautophagy is a type of autophagy that particularly targets damaged or superfluous organelles such as mitochondria (mitophagy), peroxisomes (pexophagy), ribosomes (ribophagy), aggresomes (aggrephagy), and so on.21
 |
Figure 1 Three forms of autophagy. There are three types of autophagy: macroautophagy, chaperone-mediated autophagy and microautophagy. (A) Macroautophagy: during the process of macroautophagy substrate proteins and organelles are sequestered by autophagosome. Fusion of lysosome with the autophagosome to form the autolysosome is crucial process for degradation. (B) During chaperone-mediated autophagy, proteins carrying the pentapeptide KFERQ-like sequence are recognized by the Hsc70 chaperone, which then associates with the integral lysosome membrane protein LAMP-2A, triggering its oligomerization. This event leads the translocation of the bound protein into the lysosome interior through a process that requires Hsc70. (C) Microautophagy: it is the process where lysosomes directly engulf cytosolic components via lysosomal membrane invagination or protrusion without prior formation of an autophagosome. Created with BioRender.com. |
Chaperone‑Mediated Autophagy (CMA)
Chaperone-mediated autophagy (CMA) is a type of autophagy in which a chaperone-dependent selective mechanism delivers and degrades cellular components. It is a lysosome-based catabolic process that occurs to maintain cellular homeostasis by catabolic lysis of excess or superfluous soluble cytosolic proteins. It can be initiated in response to a variety of stress circumstances in order to restore cellular balance.10 The cargo selectivity of this lysosomal route is its most distinguishing trait. Selectivity is driven by the presence of a CMA-targeting motif, a pentapeptide sequence sharing biochemical similarity to KFERQ in the substrate proteins.9,24 Unlike macro- and microautophagy, CMA is engaged in cargo delivery and does not need the creation of intermediate vesicles, membrane fusion, or any type of membrane deformation. It is a mechanism that does not rely on vacuoles.22 CMA is well suited for eliminating misfolded, oxidized, or damaged cytosolic proteins under both healthy and pathological settings because of its highly selective mechanism.4
Basic Components of CMA Machinery
CMA pathway mainly includes the CMA receptor: lysosome-associated membrane protein type 2A (LAMP2A), the chief chaperone: heat shock-cognate protein of 70 kDa (Hsc70) in cytoplasm and lysosome, and substrates: KFERQ-like consensus motif-contained cytosolic proteins.10
Substrate-CMA Motif Protein (Pentapeptide Motif KFERQ)
CMA degrades proteins that have been targeted to lysosomes by hsc70, a cytosolic chaperone that recognizes and binds a pentapeptide motif (KFERQ-like) in the substrate protein.25 Such sequences can be found in 40% of cytosolic proteins. It is possible that posttranslational changes such as deamination can result in KFERQ-like sequences in some proteins.3
Cytosolic and Lysosomal Chaperones
HSC70 remains the only chaperone proved to directly bind the KFERQ-like motif. It binds hydrophobic regions to assist in protein folding. In contrast, HSC70 binds KFERQ-like motifs to target proteins for degradation via CMA.26 Cytosolic Hsc70 uses the KFERQ motif to identify a substrate protein and delivers it to lysosomes for breakdown. HSC70 belongs to the Hsp70 protein family and it has a constitutive expression, participating principally in the CMA pathway.27 Hsc70 not only directs the CMA substrate to the lysosomal membrane, where it may interact with the CMA receptor, but it also helps the substrate unfold, which is necessary for the protein’s translocation across the membrane.28 Other co-chaperones, such as Hsc40 and Hsc90, can interact with Hsc70 and help in this process. This lysosomal hsc70 (lys-hsc70) is the defining property of the CMA-capable fraction of cellular lysosomes. The heat shock protein of 90-kDa, or hsp90, is a second chaperone that can be found on both sides of the lysosomal membrane. The proportion of hsp90 found in the cytoplasm is proposed to participate in substrate protein unfolding.29
LAMP2A
Lysosome-associated membrane protein type 2A (LAMP2A) is one of the three splicing variants of the LAMP2 gene, which contains a cytosolic tail that differs from the other isoforms and is crucial for CMA to occur.6 It is responsible for substrate binding and internalization to lysosomes, and thus, the lysosomal expression level of LAMP2A is a rate-limiting factor for CMA.3
Lysosomes
Lysosomes are membrane-bound organelles that play a role in a variety of cell functions, including cytoplasmic waste degradation, cell signalling, energy metabolism, secretion, and plasma membrane repair.1 Lysosomes, as CMA’s final destination, keep their lumen at a very acidic pH of 5.1–5.5 in order to digest transported contents and subsequently induce efflux of digested metabolites, recycling crucial digested components. Both soluble lysosomal hydrolases (eg, proteases, lipases, glycosidases, and nucleases) and lysosomal membrane proteins (eg, lysosome associated membrane proteins) are necessary for lysosome function.3
The Process of CMA
CMA process begins with the heat-shock protein of 70 kDa (Hsc70), a cytosolic member of the Hsp70 chaperone family, recognizing cytosolic proteins with the loose pentapeptide motif KFERQ (Figure 2). Many co-chaperones are involved in this process, including Hsp40, Hsp90, and Hip.23 Once the substrate proteins are bound by hsc70, they are directed to the surface of a fraction of lysosomes that are involved in the autophagic pathway. Substrates are translocated to the lysosomal membrane surface after binding to the Hsc70 complex, where the single-span lysosomal receptor LAMP2A can bind the substrate protein with its 12 amino acid tail exposed in the cytoplasm.1,8,9 LAMP2A is in charge of both substrate binding and lysosome internalization. LAMP2A levels and conformation state are necessary for this mechanism to perform properly. LAMP2A levels and conformational status are crucial, and this is a rate-limiting phase in the process that can be changed through synthesis, degradation, and redistribution.3
 |
Figure 2 The process of CMA: (1) recognizing substrate proteins and targeting them to lysosome; (2) binding and unfolding substrate proteins; (3) multimerization of LAMP-2A and HSP90 binds to LAMP2A to stabilize it while it organizes into higher molecular weight complexes (4) translocation into lysosomes, substrate translocation mediated by lysosomal HSC70 (lys-HSC70) (5) degradation by lysosome hydrolytic enzymes (6) once substrate translocation is complete, LAMP2A dissociates into monomers in a process dependent on cytosolic HSC70. Created with BioRender.com. |
At the lysosomal membrane, LAMP2A exists as a monomer and forms a multimeric complex with other proteins. For substrate translocation into the lumen, multimerization of LAMP2A to create a 700 kDa protein complex, a transmembrane protein channel, is required.2 CMA substrates bind to monomeric LAMP2A at the lysosomal membrane, which causes LAMP2A to multimerize, resulting in a 700-kDa complex necessary for substrate translocation into the lysosome. As the substrate unfolds, LAMP2A multimerization promotes substrate affinity and likely prevents aggregation.3,26 This protein complex allows the substrate protein to enter the lysosome lumen, where it is degraded by hydrolytic enzymes. The substrate can bind to the receptor while still folded, but it must unfold to cross the lysosomal membrane. This process is presumably mediated by hsc70 and some of its co-chaperones, and it is completed before the LAMP-2A complex is fully assembled.8 The presence of a luminal chaperone, lys-Hsc70, inside the lysosome is also essential for complete substrate internalization (Rios et al, 2021).30 As LAMP-2A moves from monomers to multimers, a lysosomal version of hsp90 maintains its stability.30 The substrate is digested into amino acids by lysosomal hydrolytic enzymes also known as cathepsins once it reaches the lysosomal matrix. Lys-hsc70 (lysosomal HSC70) causes LAMP2A to disassemble from its multimeric form into its monomeric form, allowing the next substrate protein to bind to LAMP2A in a new cycle.22 Subsequently, Lys-Hsc70 (lysosomal Hsc70) and EF1α (Elongation Factor 1α) induces LAMP2A multimer to disassemble into the monomeric form so that a new degradation cycle can start with binding to the monomeric LAMP2A.4,23 CMA activation does not require de novo synthesis of LAMP2A because its lysosomal abundance can be modified by changing its stability, organization and dynamics at the lysosomal membrane. These events are tightly regulated by lysosome-associated proteins such as the GFAP/EF1α pair and the mTORC2/AKT1/PHLPP1 axis.25
Physiological Roles of CMA
Regardless of cell type, CMA contributes to two general functions, energy homeostasis and protein quality control. While energy homeostasis is achieved through recycling amino acids, the latter, protein quality control, is by removing damaged and prone-to-aggregate proteins (Cuervo and Wong, 2014).8,31 CMA also participates in cell type-specific actions that are dependent on the substrate protein degraded by this pathway.10,33
CMA has specialized functions in distinct cell types, frequently connected to the degradation of a specific protein or collection of proteins in these cells, in addition to its broad functions shared by all cells. CMA is involved in the presentation of cytoplasmic peptides by class II molecules in professional antigen-presenting cells, which contributes to organism autoimmunity. These cells’ CMA rates and antigen presentation are reduced when hsc70 and hsp90 are manipulated.33 By degrading inactive versions of the transcription factor MEF2D (myocyte enhancer factor 2D) in neurons, CMA enhances neuronal survival.8,31 Inactivated molecules of the myocyte enhancer factor 2D (MEF2D), a transcription factor needed for neuronal survival, accumulate in neurons when CMA is compromised, making cells more vulnerable to stresses.33 The transcription factor Pax2 is degraded by CMA in the kidney, which is critical for tubular cell growth control.8 The degradation of the paired box gene 2 (Pax2), a protein crucial for cell proliferation and differentiation, is hampered by malfunctioning CMA in kidney epithelial cells. In these cells, failure to destroy Pax2 causes aberrant kidney cell proliferation and hypertrophy.31
Major Mechanisms That Regulate CMA
Regulation of CMA via LAMP2A
The CMA receptor, LAMP2A, is the primary target for CMA regulation because its level on the lysosomal membrane directly correlates with CMA activity at any given time. It is altered by de novo synthesis, degradation (turnover), and redistribution.3 Changes in levels and dynamic change of LAMP2A from monomer to oligomer to degradation at the lysosomal membrane regulate CMA flux (that is, the rate at which cargo is degraded).26 As such, up-regulation of CMA degradation in response to prolonged starvation, oxidative stress or inhibition of other proteolytic pathways is accompanied by an increase in Lamp2a levels and Lamp2a-positive lysosomes. Conversely, reducing Lamp2a levels results in a decrease in CMA activity.34
Regulation of CMA via HSC70
Dependence on the LAMP2A, discussed above, is the best criteria to determine whether protein degradation occurs via CMA. However, HSC70 requires LAMP2A for lysosomal docking in CMA.26 The presence of this chaperone on the lumenal side of the membrane is required for full substrate translocation. Although the processes controlling this increase are still unknown, its distribution and level directly reflect the CMA activity and status.10,28
Post-translational modifications, such as phosphorylation, acetylation, or even ubiquitination, within incomplete motifs can confer specific properties, recreating a complete KFERQ-like motif. Not only inside the motif, but also outside it, by changing the protein’s conformation, post-translational modifications can reveal or hide an existing one. As a result, CMA substrates vary greatly.6,26 CMA targeting of proteins with canonical motifs can also be modulated by post-translational modifications outside the motif, which facilitate conformational changes that expose or mask the motif. Hypoxia-inducible factor 1 (HiF1), a validated CMA substrate, for example, is CMA-dependently degraded only when it is ubiquitylated on Lys63 by the E3 ubiquitin-protein ligase STUB1 (REF.29). Acetylation of mammalian Ste20-like kinase 1 (MST1; also known as STK4) in a residue far from the canonical CMA motif, on the other hand, prevents lysosomal degradation, and HSC70 can only bind the canonical motif after deacetylation.26
Regulation of CMA via Signal Pathways
There are several signalling pathways regulating CMA activity, including the calcineurin–NFAT pathway in CMA activation in T cells, PARα signalling in CMA inhibition, and the TOR complex 2 (TORC2)-AKT1-PHLPP1 axis in CMA inhibition. Endoplasmic reticulum (ER) stress is another factor that can activate CMA.1,10 A lysosome-associated form of the GFAP (glial fibrillary acidic protein) and EF1α also modulated CMA activity in a GTP-dependent manner in response to both starvation and oxidative stress.4
Regulation of CMA by NFAT and Calcium Signalling
The calcineurin–NFAT route, which is needed for CMA activation in T cells and also gave unique insights into CMA activation in response to oxidative stress, was the first signaling mechanism found in the activation of CMA. The transcription factor NFAT1 binds to many putative NFAT1-binding sites in the lamp2 proximal promoter region. During T cell activation, the production of reactive oxygen species (ROS) stimulates the nuclear translocation of NFAT1 and the consequent increase in lamp2a expression.26
Regulation of CMA by TORC2-AKT1-PHLPP1 Axis
CMA has been found to be negatively regulated by target of rapamycin complex 2 (mTORC2).1 The dynamics of LAMP2A assembly into the translocation complex involves mTOR complex 2 or TORC2, the kinase AKT, and the phosphatase PHLPP1. TORC2 provides a continuous inhibitory regulation on CMA through phosphorylation of AKT, that is, the kinase of glial fibrillary acidic protein (GFAP) at the lysosomal membrane Continuous GFAP phosphorylation makes LAMP2A assembly/disassembly from the translocation complex constitutively slow. When maximal CMA activation is required, TORC2ʹs inhibitory effect must be released, which is accomplished by recruiting PHLPP1 to the lysosomal membrane in a Rac-1-dependent manner.26 In an acute liver failure model, for example, activation of the PI3K/Akt/mTOR pathway is associated with a decrease in LAMP2A and Hsc70.1
Regulation via Retinoic Acid Receptor Alpha (RARα)
Signalling mediated by the RARα has been shown to negatively regulate the transcription of CMA components, including LAMP2A (Lioa et al, 2021).4 Genetic and chemical blockage of RARα demonstrates that not only LAMP2 but also proteins facilitate LAMP2A trafficking to lysosomes. RARα inhibitors hold great promise in the systemic restoration of impaired CMA in ageing.26
Regulation of CMA by ER Stress
CMA and p38 mitogen-activated protein kinase (P38MAPK, MK14) activation are triggered by ER stress. They show that a PERK-dependent (PKR-like ER protein kinase) recruitment of mitogen-activated protein kinase kinase 4 (MKK4) and subsequent activation of a P38MAPK pool at the lysosomal membrane occurs when ER stress is induced in various ways. LAMP2A is phosphorylated at Thr211 and Thr213 when P38MAPK is activated. LAMP2A’s stability and oligomerization at the lysosomal membrane are both improved by this phosphorylation. ERICA stands for “ER-stress-induced CMA,” and it indicates an exquisite link between ER-stress and CMA.35 ER stress causes a P38MAPK-dependent dual phosphorylation of LAMP2A, which activates LAMP2A, which is known as the ERICA pathway.1
Pathogenic Implication of CMA
With CMA being shown to participate in an increasing number of cellular processes, it is not surprising that there is a growing recognition that CMA dysfunction contributes to disease.26 Increasing evidence has shown that a malfunction of CMA plays a key role in several human disorders, especially age-associated disorders, when CMA is deregulated by both genetic and environmental factors. CMA activity has been linked to diseases in both reduced and increased forms, emphasizing the significance of strict regulation of CMA activity. Neurodegenerative diseases and cancer have gotten the greatest attention among them.22,26 CMA can interfere with the disease process by degrading disease-associated proteins. The first link between CMA dysfunction and a human disease was Parkinson’s disease. CMA failure has since been linked to the development of an expanding array of neurodegenerative diseases.26 CMA’s altered activity has been linked to an increase in disease pathogenesis. CMA activity decreases in a variety of neurological disorders, particularly in their later stages, and increases in a variety of malignancies and immunological diseases.10
CMA and Neurodegenerative Diseases
It is widely accepted that dysregulation of the proteolytic pathways is implicated in the onset and/or development of neurodegenerative diseases.33 Several neurodegenerative disorders are characterized by abnormal build-up of misfolded and aggregated proteins in neurons. Neurons are post-mitotic cells that require a high-performance protein breakdown system to cope with the stress. Unfolded or damaged proteins are prone to aggregate if not eliminated, which is a common hallmark of many neurodegenerative disorders.1 There is mounting evidence that dysregulation of the CMA pathway plays a critical role in neurodegeneration.27 CMA aids in the degradation of several proteins that have a proclivity to aggregate, and as a result, a decrease in CMA activity results in the accumulation of toxic aggregates. Diseases have been linked to both an increase and a decrease in CMA activity. In general, there is a failure of the proteolytic systems in neurodegenerative pathologies, resulting in the accumulation of deleterious protein transformation.4,22 Neurodegenerative diseases such as Alzheimer’s (AD), Parkinson’s (PD) and Huntington’s (HD) all share a common pathogenic feature: aggregated protein deposits in specific brain regions.36 Pathogenic protein accumulation in the form of insoluble inclusions is a common factor underlying all of these diseases. They become much more resistant to CMA-mediated degradation once they form insoluble inclusions. These aggregates can frequently cause a “clogging or blockage effect” at the lysosomal membrane, making them toxic by inhibiting the CMA-mediated degradation of other cytosolic substrate proteins.24,33,37 Several pathogenic proteins of these diseases have been identified as the substrates of CMA, such as alpha-synuclein (α-syn), PARK7/DJ-1 (Parkinsonism-associated deglycase), LRRK2 (leucine-rich repeat serine/threonine-protein kinase 2) and UCHL1 (ubiquitin carboxyl terminal hydrolase isozyme L1) in PD, tau protein and RCAN1 (regulator of calcineurin) in AD and HTT (huntingtin) in HD. These proteins contain KFERQ motifs in their amino acid sequences and have been proved to be CMA substrates (Table 2). A common feature of these neurodegeneration-related proteins is that the unmodified protein behaves as a conventional CMA substrate in the majority of cases, whereas the pathogenic variants are targeted to lysosomes but fail to degrade and inhibit CMA.26
 |
Table 2 Clinical, Pathological and Biochemical Features of Neurodegenerative Disease and CMA4,10,37 |
Parkinson’s Disease (PD)
Following Alzheimer’s disease, Parkinson’s disease (PD) is the second most common type of neurodegenerative disease (AD). It was the first neurodegenerative disorder associated with CMA.6 It is distinguished by extensive and progressive loss of dopaminergic (DA) neurons in the substantia nigra, as well as α-synuclein protein aggregation in Lewy bodies. Except for a 5–10% familial component, Parkinson’s disease is mostly sporadic.6,38 There is a growing body of evidence linking CMA to Parkinson’s disease from genetic, in vitro, and in vivo studies. CMA dysregulation is linked to the onset or progression of Parkinson’s disease (PD).27 LAMP2A levels in early-stage Parkinson’s disease brains are reduced even before α-syn accumulation, indicating that CMA dysfunction occurs early in Parkinson’s disease pathogenesis.26 Its major pathogenic proteins in both familial and sporadic Parkinson’s disease, α-syn and LRRK2, have been identified as CMA substrates.10
Alpha-synuclein (α-syn) is the main constituent of Lewy bodies. α-syn levels or conformations are critical to PD pathogenesis. α-syn levels or conformations are important in the pathogenesis of Parkinson’s disease. CMA was found to be involved in α-syn degradation in various neuronal cell lines and primary cultures of cortical and midbrain neurons.27 CMA has been shown to degrade α-Syn in both neuronal cultures and animal models (Wu et al, 2018).36 Although the CMA process efficiently degrades wild-type α-syn, mutations in α-syn are poorly degraded.10
Several protein mutations associated with hereditary forms of Parkinson’s disease have been shown to inhibit CMA. In addition to the damage caused by protein aggregate accumulation, CMA inhibition may impair cellular protection pathways due to the accumulation of key CMA substrates. Some mutant forms of α-Syn have been shown to have an effect on CMA via increased binding capacity to Lamp2a. In these conditions, not only is α-Syn translocation and degradation in lysosomes hampered, but other substrates’ access to Lamp2a is also hampered. Similarly, mutant forms of the proteins leucine-rich repeat kinase 2 (LRRK2) and ubiquitin C-terminal hydrolase L1 (UCH-L1), which are frequently associated with Parkinson’s disease, block CMA by inhibiting the translocation complex.34 Mutation of the Leucine-rich repeat kinase 2 (LRRK2) genes is the most common familial cause of PD which causes autosomal dominant PD. LRRK2 is found in membrane microdomains, multivesicular bodies, and autophagic vesicles, and it is also a CMA substrate.31 The CMA machinery recognizes both wild-type and mutant LRRK2. The mutant, on the other hand, has been shown to inhibit the dynamic assembly of the CMA translocation complex at the lysosome membrane, causing the CMA process to be disrupted.1 Mutant forms of the ubiquitin carboxyl-terminal esterase L1 (UCHL-1) protein, which is related to Parkinson’s disease, have also been shown to interact abnormally with CMA components.31 The involvement of CMA in PD pathogenesis is supported not only by genetic evidence but also by changes in CMA parameters in PD. In postmortem brain samples from Parkinson’s disease patients, both hsc70 and lamp2A were found to be down-regulated in the substantia nigra pars compacta and amygdala when compared to healthy controls.39
Alzheimer’s Disease (AD)
Alzheimer’s disease (AD) is the most common neurodegenerative disorder, characterized by pathological aggregation of amyloid-(A) and hyperphosphorylated tau (hp-tau) proteins in the form of senile plaques (SPs) and neurofibrillary tangles (NFTs), respectively.6 CMA activity has been shown to decrease in Alzheimer’s disease, and several AD-associated proteins have been shown to interact with CMA.10 RCAN1 and Tau protein, as well as their degradation via CMA, were found to be reduced in Alzheimer’s disease patients.4 Mutant forms of tau protein, like PD-related proteins, are targeted to lysosomes but fail to reach the lysosomal lumen.26
Tau is a key component of neurofibrillary tangles in Alzheimer’s disease patients’ brains, which begin in memory-related areas and spread throughout the rest of the brain. CMA can degrade normal tau, but mutant tau can bind to LAMP2A and disrupt its lysosomal membrane translocation. When CMA is blocked, neurofibrillary tangles increase consistently.10 CMA also aids in the degradation of other proteins associated with neurodegeneration, such as RCAN1. RCAN1 was degraded by the CMA pathway due to two KFERQ-like motifs discovered in the protein.26 Regulator of calcineurin 1 (RCAN1), identified in Down syndrome, has been implicated in the AD pathogenesis. RCAN1 expression increases in AD brains possibly by disruption of the CMA process. Mechanism study shows that RCAN1 protein is a typical substrate for CMA, and inhibition of RCAN1 degradation in cells reduces calcineurin-NFAT activity. Amyloid precursor protein (APP) can be processed to produce β-amyloid (Aβ).10 APP with KFERQ-like motif at its C terminus can produce Aβ (β-amyloid peptide), which is another key pathogenic molecule associated with AD.4 APP is a single-span membrane protein and, as such, is not a candidate for CMA degradation. However, APP undergoes physiological cleavage to release a short cytosolic stub where, coincidentally, the KFERQ motif is located. Although direct evidence for its CMA degradation is still missing, mutation of this motif led to cytosolic accumulation of this small, highly toxic peptide.26,27
Huntington’s Disease (HD)
HD is a neurodegenerative disorder characterized by uncontrollable movement, dementia, and emotional instability.27 A mutation causes an expansion of the polyglutamine (polyQ) sequence in the N-terminus of the Huntington (Htt) protein, which causes this disease. A mutation in the gene causes the protein to clump together. The main HD pathology in striatal and cortical neurons is the accumulation of mutant Htt due to aberrant degradation. KFERQ-like motifs have been discovered in Htt exon 1 fragments that can be targeted for CMA-mediated degradation by Hsc70. As a result, wild-type Htt is a CMA substrate, whereas mutant Htt protein inhibits its uptake and is difficult to degrade by lysosomes.10
HD is the only neurodegenerative disease in which LAMP2A and HSC70 expression levels are elevated.6 Interestingly, components of CMA (LAMP2A and Hsc70) are upregulated in early-stage HD cellular and animal models. However, LAMP2A levels decrease as the disease progresses. The early increase in CMA components could be a compensatory response to huntingtin mutant MA inhibition in order to restore proteolytic homeostasis. However, the decrease in lysosome LAMP2A levels suggests that CMA function is lost in the late phase of HD. Further research into the biphasic change in CMA activity should provide important information on how CMA may be dynamically regulated in neurons and diseases under stress conditions.1 The therapeutic applicability of mutant huntingtin re-routing towards CMA may be limited by the fact that the normal age-related decline in CMA activity is accelerated in the Huntington disease brain.26
CMA and Cancer
The altered activity of CMA is closely linked to the pathogenesis of various cancers.10 It has bidirectional roles in cancer, depending on the situation. CMA was initially linked to pro-tumorigenic functions because upregulation of this autophagy pathway is linked to positive modulation of tumor cell survival and growth. CMA, on the other hand, has an antitumor function in non-tumorigenic cells, preventing malignant transformation.22
CMA and Tumor Promotion
After malignant transformation, CMA becomes extremely active in order to maintain critical pro-oncogenic functions. Once a tumor forms, it aids in tumor survival and the metastatic cascade. CMA up-regulation has been linked to a variety of cancer cells and tumors as a means of promoting cell survival.22 Changes in CMA activity have been studied for various types of cancer. For example, studies in several cancer cell lines, including lung (A549, H460), breast (MCF7), liver (HUH7), epithelial (Saos 2 osteosarcoma), stomach, colon, uterus, and ovary, have concluded that CMA activity is constitutively upregulated in cancer cells. Taking into account the various cancer cell lines investigated, it was possible to conclude that CMA is found to be upregulated 2.8-fold with respect to non-oncogenic control cells.30 CMA activity has been reported to be upregulated in most cancer cell lines, and increased LAMP2A expression levels have been observed in a wide range of human tumors.3 CMA is activated in a variety of cancers, including breast, colorectal, gastric, and liver cancer, indicating that LAMP2A overexpression promotes tumor growth and metastasis.41 CMA up-regulation may play a role in cancer pathology development. CMA’s role in promoting cancer cell survival has been linked to several mechanisms, including preventing cells from entering an apoptotic state, providing general protection against oxidative stress, and protecting pro-oncogenic proteins from degradation by other pathways. CMA has also been implicated in tumour resistance to therapy. Macrophages, one of the main cell types present in the tumour microenvironment, have been reported to activate CMA in other tumour resident cells, through the release of IL-17, promoting cell survival and chemo resistance. Induction of CMA in macrophages promotes their activation to aid in tumor growth.25,34 Recent studies showed that many tumor suppressors can serve as substrates of CMA, and CMA promotes cancer by degradation of these tumor suppressors, mainly including ATG5, BBC3, cyclin D1, HMGB1, MST1, N-CoR p21Cip1/WAF1, p53, p300/CBP PED and RND3.10 Cancer cells often depend on anaerobic glycolysis for energy production even in the presence of oxygen known as the Warburg effect. Functional CMA is required to sustain the Warburg effect (aerobic glycolysis).32 CMA-mediated degradation of glycolytic enzymes such as acetylated pyruvate kinase PKM2 is linked to the Warburg effect, which is the metabolic shift to glycolysis, an essential energy source for tumor growth and proliferation.3 Additionally, CMA indirectly reduces p53 levels, thus releasing its transcriptional inhibition on key glycolytic enzymes and resulting in higher glycolytic rates.33
CMA and Metastasis
During metastasis, cancer cells leave the primary tumor to nucleate and form new tumors in other organs or tissues. Metastasis takes place in several steps: local invasion, intravasation, survival in the circulation, extravasation and colonization. CMA may contribute to tumor metastasis since blockage of CMA in lung cancer cells markedly decreases their metastatic potential by reducing migration and resistance to anoikis.25 Correlation between CMA activity and metastasis was noticed in breast cancer.42 Inhibition of chaperone mediated autophagy reduces tumor growth and metastasis and promotes drug sensitivity in colorectal cancer.41 Moreover, CMA degrades the multifunctional protein HSD17B4, which modulates the properties for the invasion and migration of cells.20 In addition, inhibition of CMA in lung cancer cells markedly decreased their metastatic potential by reducing migration and resistance to anoikis (a form of programmed cell death that occurs in tumor cells when they were detached from the surrounding extracellular matrix).43
Anti-Tumor Functions of CMA
Although the dependence of cancer cells on CMA suggests a pro-oncogenic function for CMA, the effect of CMA in normal cells seems to be quite the opposite, as it protects cells from the damage caused by extracellular and intracellular injuries, which, if allowed to accumulate could facilitate oncogenesis.8 Most studies support that CMA has an anti-oncogenic role in normal untransformed cells and prevents malignant transformation.26 Recent studies support that the antioncogenic capacity of CMA is, at least in part, due to its ability to reduce levels of pro-oncogenic proteins.25 CMA has been shown to play a role of tumor suppressor by degrading specific tumor promoters. For example, CMA inhibits cellular transformation, cell proliferation, and colony formation by degradation of Myc, inhibits lung cancer cell growth by degradation of MCL1, and degrades mutant p53.1 CMA may also inhibit cancer growth by degradation of many other tumor promoters such as AF1Q, Eps8, Galectin-3, HIF1α, Histones H3-H4, HK2, HSD17B4, MDM2, PKM2 and RhoH.10 CMA also has anti-tumor function by facilitating immunogenic apoptosis. CMA may also protect against malignant transformation by assuring genome stability through its role in efficient DNA repair (Figure 3).25
 |
Figure 3 Anti-oncogenic function of CMA turns pro-oncogenic in cancer cells. Studies in vitro and in vivo support an anti-oncogenic function for CMA in normal cells through a variety of mechanism (blue font). This could explain why conditions in which CMA activity are reduced. Reduced CMA has been shown to increase DNA damage and reduce proteostasis providing thus an environment favourable for malignant transformation. Right after transformation CMA activity is upregulated and remains constitutively active in most tumor cells. High CMA activity in cancer cells sustains different pro-oncogenic functions (red font). Created with BioRender.com. |
Targeting CMA for Neurodegenerative Diseases and Cancer Therapy
The connection between CMA and different human diseases has motivated a growing interest in understanding this fundamental cellular process and manipulating it for therapeutic purposes.10
Targeting CMA for Neurodegenerative Diseases Therapy
Studying the pathological protein along with the wild-type version and delineating the exact step affected in CMA would be a key in providing specific therapeutic interventions.26 There is a strong link between a decline of CMA and the abnormal aggregation of such proteins in various models of neurodegenerative diseases, suggesting that CMA is a new and promising target for treating these neurodegenerative disorders.10,29 In aging, the function of CMA is impaired, causing an inefficient stress response and the accumulation of damaged, oxidized or misfolded proteins, associated with aging-related diseases. Restoration of CMA activity can be a promising therapeutic approach for such human disease conditions in which the upregulation of CMA activity may result in promotion of the degradation of disease aggregation-prone proteins.10 Although there is no established agent for modifying CMA activity for clinical application, recent study has shown the potential therapeutic efficacy of specific CMA activation by regulatory steps and increasing level of LAMP2A receptor.3,44 Recent study has shown the potential therapeutic efficacy of specific CMA activation by retinoic acid receptor alpha (RARα) antagonists for mouse model of Parkinson disease.44 The lysosomal mTORC2/PHLPP1/Akt axis could become a target to restore CMA dysfunction in aging and disease.45 Controlling de novo synthesis, preventing its multimerization into lysosomes (possibly via HSP90 and/or other chaperones), and regulating the degradation rate of LAMP2A monomer units (for reuse) in lysosomes are all possibilities for targeting LAMP2A.46 Many studies have suggested that enhancing LAMP2A expression to upregulate the activity of CMA can be an important therapeutic target. A previous study stated that recombinant adeno-associated virus augmenting the LAMP2A level protected dopaminergic neurons in the substantia nigra from α-synuclein-induced degeneration. In addition, it has also been reported that various compounds, such as geldanamycin, 6-aminonicotinamide, glucose-6-phosphate dehydrogenase inhibitor, silymarin, chronic caffeine, manganese, trehalose, b-asarone, and other compounds extracted from natural medicinal plants, or even combination treatments with bortezomib and suberoylanilide hydroxamic acid (SAHA), can increase LAMP2A levels and activate the CMA pathway. However, these compounds are not able to specifically regulate the CMA pathway and have many other targets. Thus, it is important to develop selective CMA modulators that can be used to manage human diseases.46–48Overall; these findings strongly suggest that manipulation of CMA activity may be an effective therapeutic strategy for treating neurodegenerative diseases.1,10,38
Targeting CMA for Cancer Therapy
As mentioned above, CMA plays different roles in different diseases and even different stages of diseases. Mostly, enhanced CMA activity performs an anti-tumor role in normal physiological conditions. However, in most tumor cells, enhanced CMA is necessary for tumor growth. Therefore, it is important to accurately identify the role of CMA in disease progression.4 Understanding the mechanisms behind this functional switch of CMA from anti-oncogenic to pro-oncogenic is fundamental for targeting CMA in cancer treatment.25 The suppression of CMA by genetic or pharmacological methods enhances the sensitivity of anticancer therapy in diverse cancer cells. Diverse inhibitors of CMA can be used alone or in combination with anticancer drugs for anticancer therapy.20 CMA seems to be constitutively upregulated in multiple types of cancer cells and in tumors and its blockage in transformed cells has anticancer potential.25 Indeed, selective inhibition of CMA by knockdown of LAMP2A can reduce tumor growth. For example, inhibiting CMA activity in breast tumor cells can be exploited as a potential therapeutic application in the treatment of breast cancer.49 CMA inhibition in malignant cells lowers cellular proliferation and reduces tumorigenic and metastatic capacity. For example, blockage of CMA in cancer cells reduces tumor growth and metastasis by abrogating part of the Warburg effect and increasing the tumor susceptibility to cytotoxic agents.25 A research conducted on inhibition of chaperone‑mediated autophagy reduces tumor growth and metastasis and promotes drug sensitivity in colorectal cancer. The result showed that CMA is a promising predictor of chemosensitivity to 5‑FU treatment and anti‑CMA therapy may be a novel therapeutic option for patients with colorectal cancer.41 A similar study was also done and showed that inhibiting CMA activity in breast tumor cells can be exploited as a potential therapeutic application in the treatment of breast cancer.49 Considering the physiological anti-oncogenic role of CMA, there is general agreement that preventive interventions should aim at restoring CMA activity to that in healthy conditions. For example, genetic prevention of the age-dependent decrease in CMA in mouse liver has proven effective in slowing-down hepatic proteotoxicity and accumulation of intracellular damage.25,26
Conclusion
Increasing evidence has shown that a malfunction of CMA plays a key role in several human disorders. Among various disease conditions, neurodegenerative diseases and cancer have received the most attention for which stronger experimental evidence of CMA involvement has been obtained. Both diminished and enhanced CMA activities have been shown to associate with diseases, which highlights the importance of tight regulation of CMA activity. Dysregulation of the CMA pathway and/or reduced activity of CMA are highly associated with abnormal accumulation of misfolded and aggregated protein, which are the major hallmarks of neurodegenerative diseases. As a result, restoration of CMA activity can be a promising therapeutic approach for such human disease conditions in which the upregulation of CMA activity may result in the promotion of the degradation of disease aggregation-prone proteins. Modulation of CMA is highly promising from a therapeutic standpoint, based on the fact that blocking CMA in human tumors has been shown to reduce tumor development and metastasis. However, more research is needed to better understand and explain the role of CMA in the development and treatment of neurodegenerative disorders and malignancies.
Abbreviations
AD, alzheimer’s disease; ALP, autophagy lysosome pathway; AMPK, AMP-activated protein kinase; APP, amyloid precursor protein; ASN, α-synuclein; ATG5, autophagy-related gene 5; BCL2, B-cell lymphoma 2; CMA, chaperone mediated autophagy; e-MI, endosomal microautophagy; ER, endoplasmic reticulum; ERAD, ER-associated degradation; ERICA, ER-stress-induced CMA; GFAP, glial fibrillary acidic protein; HD, Huntington’s disease; HIF1, α hypoxia-inducible factor-1 α; HIF1α, hypoxia-inducible factor 1α; HIP, hsc70 interacting protein; HK2, hexokinase II; HMGB1, high-mobility group box 1; HOP, hsc70-hsp90 organizer protein; hsc70, heat shock cognate protein of 70 kDa; HSP, heat shock proteins; HSPA8, heat shock protein family a member 8; Htt, Huntington; IκBα, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha; LAMP-2A, lysosome associated membrane protein type 2A; LRRK2, leucine-rich repeat kinase 2; LRRK2, leucine-rich repeat serine/threonine-protein kinase 2; Lys-HSc70, lysosomal heat shock cognate 70 kDa; Lys-hsc70, lysosome-associated hsc70; MDM2, murine double minute; MEF2D, myocyte-specific enhancer factor 2D; MKK4, mitogen-activated protein kinase kinase 4; mTOR, mammalian target of rapamycin; ND, neurodegenerative disease; P38MAPK, P38 mitogen-activated protein kinase; PD, Parkinson’s disease; TORC2, target of rapamycin complex 2; UCHL1, ubiquitin carboxyl terminal hydrolase isozyme L1; UPR, unfolded protein response; UPS, ubiquitin-proteasome system.
Acknowledgments
The authors express their thanks to BioRender for providing them high-resolution figures with publication license. Figures were created with BioRender.com.
Funding
This work is not funded by any governmental or non-governmental agencies.
Disclosure
The authors report no conflict of interest in this work.
References
1. Li W, Nie T, Xu H, et al. Chaperone-mediated autophagy: advances from bench to bedside. Neurobiol Dis. 2019;122:41–48. doi:10.1016/j.nbd.2018.05.010
2. Cai Z, Zeng W, Tao K, et al. Chaperone-mediated autophagy: roles in neuroprotection. Neurosci Bull. 2015;31(4):452–458. doi:10.1007/s12264-015-1540-x
3. Hosaka Y, Araya J, Fujita Y, et al. Role of chaperone-mediated autophagy in the pathophysiology including pulmonary disorders. Inflamm Regan. 2021;41(1):1–10.
4. Liao Z, Wang B, Liu W, et al. Dysfunction of chaperone-mediated autophagy in human diseases. Mol Cell Biochem. 2021;476(3):1439–1454. doi:10.1007/s11010-020-04006-z
5. Saha S, Panigrahi DP, Patil S, et al. Autophagy in health and disease: a comprehensive review. Biomed Pharmacother. 2018;104:485–495. doi:10.1016/j.biopha.2018.05.007
6. Auzmendi-Iriarte J, Matheu A. Impact of chaperone-mediated autophagy in brain aging: neurodegenerative diseases and glioblastoma. Front Aging Neurosci. 2021;12:630743. doi:10.3389/fnagi.2020.63074
7. Xilouri M, Stefanis L. Chaperone mediated autophagy to the rescue: a new-fangled target for the treatment of neurodegenerative diseases. Mol Cell Neurosci. 2015;66:29–36. doi:10.1016/j.mcn.2015.01.003
8. Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24(1):92–104. doi:10.1038/cr.2013.153
9. Juste YR, Cuervo AM. Analysis of chaperone-mediated autophagy. Methods Mol Biol. 2019;1880:703–727. doi:10.1007/978-1-4939-8873-0_47
10. Li W, Dou J, Yang J, et al. Targeting chaperone-mediated autophagy for disease therapy. Curr Pharmaco Rep. 2018;4(3):261–275.
11. Ellis RJ. Assembly chaperones: a perspective. Phil Trans R Soc B. 2013;368(1617):3–7. doi:10.1098/rstb.2011.0398
12. Mrinal S, Kunal S, Subhankar M. Molecular chaperones: present scenario and future perspectives. J Pharm Biol Chem Sci. 2013;4(2):636–648.
13. Slavotinek AM, Biesecker LG. Unfolding the role of chaperones and chaperonins in human disease. Trends Genet. 2001;7(9):1–8.
14. Friesen EL, De Snoo ML, Rajendran L, et al. Chaperone-based therapies for disease modification in Parkinson’s disease. Parkinsons Dis. 2017:1–11. Doi:10.1155/2017/5015307
15. Shemesh N, Jubran J, Dror S, et al. The landscape of molecular chaperones across human tissues reveals a layered architecture of core and variable chaperones. Nat Commun. 2021;12(1):1–16. doi:10.1038/s41467-021-22369-9
16. Kumar CS, Mande SC, Mahajan G. Multiple chaperonins in bacteria novel functions and non-canonical behaviors. Cell Stress &chaperones. 2015;20(4):555–574. doi:10.1007/s12192-015-0598-8
17. Bonam SR, Ruff M, Muller S. HSPA8/HSC70 in immune disorders: a molecular rheostat that adjusts chaperone-mediated autophagy substrates. Cells. 2019;8(8):1–24. doi:10.3390/cells8080849
18. Ansari M, Mande SC, Glimpse A. Into the structure and function of atypical type i chaperonins. Front Mol Biosci. 2018;5(31):29–35. doi:10.3389/fmolb.2018.00031
19. Helgason G, Vignir H, Tessa L, et al. Role of autophagy in cancer prevention, development and therapy. Essays Biochem. 2013;55:133–151.
20. Yun CW, Jeon J, Go G, et al. The dual role of autophagy in cancer development and a therapeutic strategy for cancer by targeting autophagy. Int J Mol Sci. 2021;22(1):1–22. doi:10.3390/ijms2201017
21. Ho M, Patel A, Hanley C, et al. Exploiting autophagy in multiple myeloma. J Cancer Metastasis Treat. 2019;5(70):1–20.
22. Andrade-Tomaz M, de Souza I, Rocha CR, et al. The role of chaperone-mediated autophagy in cell cycle control and its implications in cancer. Cells. 2020;9(9):1–15. doi:10.3390/cells9092140
23. Tang Y, Wang XW, Liu ZH, et al. Chaperone-mediated autophagy substrate proteins in cancer. Oncotarget. 2017;8(31):51970–51985. doi:10.18632/oncotarget.17583
24. Kon M, Cuervo AM. Chaperone-mediated autophagy in health and disease. FEBS Lett. 2010;584(7):1399–1404. doi:10.1016/j.febslet.2009.12.025
25. Arias E, Cuervo AM. Pros and cons of chaperone-mediated autophagy in cancer biology. Trends Endocrinol Metab Trend. 2020;31(1):53–66. doi:10.1016/j.tem.2019.09.007
26. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19(6):365–381. doi:10.1038/s41580-018-0001-6
27. Alfaro IE, Albornoz A, Molina A, et al. Chaperone mediated autophagy in the crosstalk of neurodegenerative diseases and metabolic disorders. Front Endocrinol. 2019;9(778):1–7. doi:10.3389/fendo.2018.00778
28. Orenstein SJ, Cuervo AM. Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin. Cell Dev Biol. 2010;21(7):719–726.
29. Liu X, Huang S, Wang X, et al. Chaperone-mediated autophagy and neurodegeneration: connections, mechanisms, and therapeutic implication. Neurosci. 2015;31(4):407–415. doi:10.1007/s12264-015-1542-8
30. Rios J, Sequeida A, Albornoz A, et al. Chaperone mediated autophagy substrates and components in cancer. Fron Oncol. 2021;10(614677):1–9. doi:10.3389/fonc.2020.614677
31. Koga H, Cuervo AM. Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol Dis. 2011;43:29–37. doi:10.1016/j.nbd.2010.07.006
32. Tasset I, Cuervo AM. Role of chaperone-mediated autophagy in metabolism. FEBS. 2016;283(13):2403–2413. doi:10.1111/febs.13677
33. Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22(8):407–417. doi:10.1016/j.tcb.2012.05.006
34. Catarino S, Pereira P, Girao H. Molecular control of chaperone-mediated autophagy. Essays Biochem. 2017;61:663–674. doi:10.1042/EBC20170057
35. Robert G, Jacquel A, Auberger P. Chaperone-mediated autophagy and its emerging role in hematological malignancies. Cells. 2019;8(10):1–10. doi:10.3390/cells8101260
36. Wu MY, Song JX, Wang SF, et al. Selective autophagy: the new player in the fight against neurodegenerative diseases? Brain Res Bull. 2018;137:79–90. doi:10.1016/j.brainresbull.2017.11.009
37. Wang G, Mao Z. Chaperone-mediated autophagy: roles in neurodegeneration. Transl Neurodegener. 2014;3(1):1–7. doi:10.1186/2047-9158-3-20
38. Campbell P, Morris H, Schapira A. Chaperone-mediated autophagy as a therapeutic target for Parkinson disease. Expert Opin Ther Targets. 2018;22(10):823–832. doi:10.1080/14728222.2018.1517156
39. Sala G, Marinig D, Arosio A, et al. Role of chaperone-mediated autophagy dysfunctions in the pathogenesis of Parkinson’s disease. Front Mol Neurosci. 2016;9(157):1–7. doi:10.3389/fnmol.2016.00157
40. Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. 2003;4(1):49–60. doi:10.1038/nrn1007
41. Xuan Y, Zhao S, Xiao X. Inhibition of chaperone mediated autophagy reduces tumor growth and metastasis and promotes drug sensitivity in colorectal cancer. Mol Med Rep. 2021;23(5):1–8. doi:10.3892/mmr.2021.11999
42. Han Q, Deng Y, Chen S, et al. Downregulation of ATG5-dependent macroautophagy by chaperone-mediated autophagy promotes breast cancer cell metastasis. Sci Rep. 2017;7(1):1–11. doi:10.1038/s41598-017-04994-x
43. Kon M, Kiffin R, Koga H, et al. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. 2011;3(109):109–117. doi:10.1126/scitranslmed.3003182
44. Ho PW, Leung CT, Liu H, et al. Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone-mediated autophagy. Autophagy. 2020;16(2):347–370. doi:10.1080/15548627.2019.1603545
45. Arias E, Koga H, Diaz A, et al. Lysosomal mTORC2/PHLPP1/akt regulate chaperone-mediated autophagy. Mol Cell. 2015;59(2):270–284. doi:10.1016/j.molcel.2015.05.030
46. Bonam SR, Wang F, Muller S. Lysosomes as a therapeutic target. Nat Rev Drug Discov. 2019b;18(12):923–948. doi:10.1038/s41573-019-0036-1
47. Kanno H, Handa K, Murakami T, Aizawa T, Ozawa H. Chaperone-mediated autophagy in neurodegenerative diseases and acute neurological insults in the central nervous system. Cells. 2022;11(1205):1–18. doi:10.3390/cells11071205
48. Bonam SR, Tranchant C, Muller S. Autophagy-lysosomal pathway as potential therapeutic target in Parkinson’s disease. Cells. 2021;10(12):1–40. doi:10.3390/cells10123547
49. Saha T. LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy. 2012;8(11):1643–1656. doi:10.4161/auto.21654
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
VPS35-Retromer: Multifunctional Roles in Various Biological Processes – A Focus on Neurodegenerative Diseases and Cancer
Fan X, Xie Y, Cao S, Zhu L, Wang X
Journal of Inflammation Research 2025, 18:4665-4680
Published Date: 3 April 2025
Morin Mitigates Methamphetamine-Induced Neurotoxicity: Effects on Motor and Cognitive Function
Anyanwu GE, Umeano AV, Ojiakor VO, Katchy AU, Anyanwu CN, Fakorede S
Journal of Experimental Pharmacology 2025, 17:307-321
Published Date: 11 June 2025
Detection and Management of Invasive Mold Disease in Pediatric Hematological Cancer Patients
Bochennek K, Rohm T, Lehrnbecher T
Infection and Drug Resistance 2025, 18:6851-6863
Published Date: 23 December 2025
Vitamin D Receptor in Cancer: Biological Functions, Mechanistic Insights, and Clinical Relevance
Liang M, Yin S, Dai Y, Xu F, Chang B, Volarević S, Li X, Wu D, Li Z, Wang T
Cancer Management and Research 2026, 18:571200
Published Date: 8 January 2026