Back to Journals » Clinical Epidemiology » Volume 8
Challenges in translating end points from trials to observational cohort studies in oncology
Authors Ording A, Cronin-Fenton D ![]() , Ehrenstein V, Lash T
, Ehrenstein V, Lash T ![]() , Acquavella J, Rørth M, Sørensen HT
, Acquavella J, Rørth M, Sørensen HT ![]()
Received 8 October 2015
Accepted for publication 29 February 2016
Published 11 June 2016 Volume 2016:8 Pages 195—200
DOI https://doi.org/10.2147/CLEP.S97874
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lars Pedersen
Anne Gulbech Ording,1 Deirdre Cronin-Fenton,1 Vera Ehrenstein,1 Timothy L Lash,1,2 John Acquavella,1 Mikael Rørth,1 Henrik Toft Sørensen1
1Department of Clinical Epidemiology, Aarhus University Hospital, Aarhus, Denmark; 2Department of Epidemiology, Rollins School of Public Health, Emory University, Atlanta, GA, USA
Abstract: Clinical trials are considered the gold standard for examining drug efficacy and for approval of new drugs. Medical databases and population surveillance registries are valuable resources for post-approval observational research, which are increasingly used in studies of benefits and risk of new cancer drugs. Here, we address the challenges in translating endpoints from oncology trials to observational studies. Registry-based cohort studies can investigate real-world safety issues – including previously unrecognized concerns – by examining rare endpoints or multiple endpoints at once. In contrast to clinical trials, observational cohort studies typically do not exclude real-world patients from clinical practice, such as old and frail patients with comorbidity. The observational cohort study complements the clinical trial by examining the effectiveness of interventions applied in clinical practice and by providing evidence on long-term clinical outcomes, which are often not feasible to study in a clinical trial. Various endpoints can be included in clinical trials, such as hard endpoints, soft endpoints, surrogate endpoints, and patient-reported endpoints. Each endpoint has it strengths and limitations for use in research studies. Endpoints used in oncology trials are often not applicable in observational cohort studies which are limited by the setting of standard clinical practice and by non-standardized endpoint determination. Observational studies can be more helpful moving research forward if they restrict focus to appropriate and valid endpoints.
Keywords: endpoint determination, medical oncology, treatment outcome, neoplasms, research design
Introduction
Cancer is a global public health epidemic, strongly associated with reduced quality of life, disability, and premature death.1 The need for accessible and effective cancer prevention, screening, diagnostics, treatment, follow-up, and rehabilitation is growing, and promising therapies are continuously under development. Nonetheless, the success of such actions must be demonstrated before they are implemented in clinical practice.2,3
The goals of drug treatment in oncology, other than supportive care, are to prolong life and improve quality of life in cancer patients. Clinical trials are considered the gold standard for examining drug efficacy and for approval of new drugs by the regulatory agencies in the United States and Europe.4 In the United States, the emphasis is the overall survival, while the European Union allows approval of oncology treatments with progression-free survival as the primary endpoint and overall survival as the secondary endpoint.5,6 After authorization, regulatory agencies may require studies that identify, characterize, or quantify safety hazards to refine the safety profile of new drugs.5,6
Large databases have been established in many countries to measure national cancer incidence and outcomes, such as the Surveillance, Epidemiology and End Results database in the United States and nationwide cancer registries in Europe. Such databases are a valuable resource for observational cancer research,7–9 which are increasingly used in studies of cancer drug safety. Registry-based studies can examine the effect of drugs in real-world populations and compare outcomes to a comparison group.
Recent reviews highlight the challenges in, and importance of, applying clinically meaningful endpoints in cancer trials.10,11 Similar challenges exist in observational studies which have a different set of premises for their conduct.12 Here, we address the challenges in translating endpoints from oncology trials to observational studies.
Clinical trials versus observational cohort studies
The strengths and weaknesses of randomized clinical trials (RCTs) versus observational cohort studies have been discussed in detail elsewhere.12 Briefly, in RCTs, randomization will, on average, balance patient characteristics between trial arms receiving contrasted treatments, while blinding of the intervention assignment is expected to eliminate differential endpoint assessment.13 The practical weaknesses of RCTs include: high costs in terms of time and money, especially when examining rare, but important, endpoints; ethical infeasibility of an experiment in the absence of equipoise; and selected nature of the RCT participants, who often represent a narrow subset of the ultimate target population, based on strict inclusion and exclusion criteria, for example, stable health or younger age.4 Strict enrollment criteria combined with short-term follow-up may preclude evaluation of rarer side effects and treatment toxicities that, in some patient groups, outweigh the long-term benefit of the intervention. At the same time, for other than supportive care therapies, the long-term benefit of clinical oncology interventions is increased survival, and the increment for a successful cancer drug is usually defined as an added survival for a median of 3–6 months.14 After approval, benefit and risk should be evaluated in large, real-world observational studies,4,10 with a broader patient population than volunteers in a trial to examine the benefit-to-risk balance of new oncology treatments other than supportive care.
Observational studies are less expensive and time consuming than clinical trials and allow for long-term and complete follow-up when patient populations are identified in medical databases. There are three major weaknesses of the observational design. First, adequate confounding control requires excellent information on a sufficient set of confounders. Potential confounders are rarely known to the same extent in observational studies as in clinical trials. Second, recordings of endpoints in observational studies are restricted by clinical practice routine, in contrast to endpoints in clinical trials, which can be defined and ascertained by the researcher. Third, promising new treatments cannot be evaluated in observational studies ahead of regulatory approval, while post-approval observational studies can contribute by evaluating treatment effectiveness, safety, and off-label use of cancer drugs.15
The traditional RCT also meets its limitations when novel drugs present exceptional benefits in early clinical studies in cancers with no effective therapy, since equipoise is lost and randomization may be unethical. In such cases, external comparators, such as patients included in standard-of-care arms from recent trials, can be used and compared with a single-arm intervention study in an observational context. However, in single-arm trials, there is still no precise estimate of efficacy from a direct comparison in a clinical trial.
Registry-based cohort studies can examine rare endpoints that have been recorded in registries, and multiple endpoints at once, and use real-world health services. These studies may identify previously unrecognized safety issues. In contrast to RCTs, cohort studies often include a variety of patient types, including old and frail patients with comorbidity. An overview of strengths and weaknesses of clinical trials and observational cohort studies is presented in Table 1.
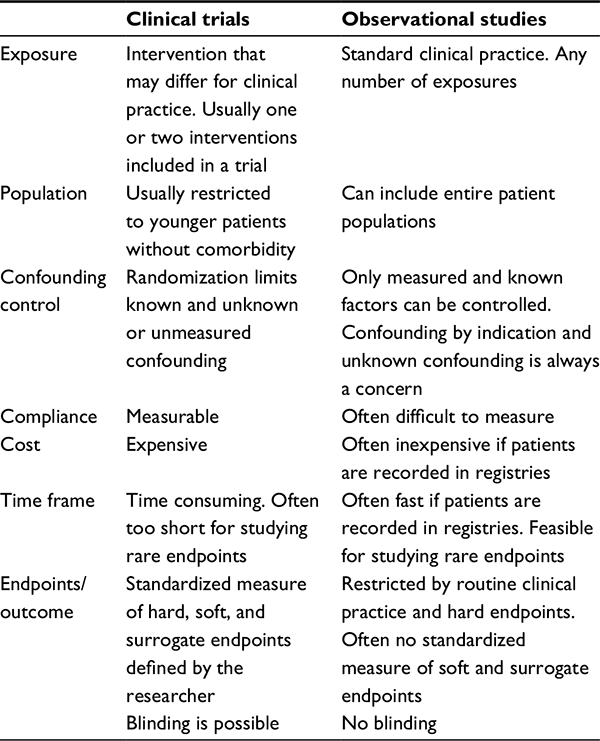 | Table 1 Characteristics of the clinical trial and registry-based cohort studies |
Types of endpoints
Major endpoints in oncology research can be defined as any state of disease, disease-related event, or death following the disease index outcome of interest, such as overall survival, progression-free survival, or adverse events. Research studies usually have one primary endpoint corresponding to the main research question, and one or more secondary endpoints to answer more specific questions, for example, assess outcomes in patient subgroups. A study can include hard endpoints and soft endpoints. Hard endpoints can easily be measured directly and unambiguously, such as death from any cause, whereas soft endpoints rely on subjective assessment by patients or researchers. In RCTs, endpoints are defined a priori, whereas the observational study can include endpoints defined a priori or a posteriori during or after the conduct of the study.
Candidate endpoints for oncology studies should be adequate, valid, and reproducible, and reflect the clinical benefit of treatment interventions for patients. The endpoint definition must be adapted to the cancer type and associated treatment modalities, although controversy often surrounds what constitutes a “clinically meaningful” endpoint.14
Overall survival and cause-specific mortality
As most cancers are life threatening, overall survival is a major clinically relevant hard endpoint in cancer drug studies in the adjuvant and advanced disease setting, and the strongly preferred endpoint adequate for approval of oncology drugs in the United States.6 Overall survival, defined as time to death from any cause, is easy to measure and is not subject to misclassification arising from clinical interpretation. Cause of death, on the other hand, is a soft endpoint relying on subjective assessment, and is therefore subject to potential (but usually non-differential) misclassification even when an expert panel is used to assess cause of death in clinical trials.16
Improvement in overall survival and cancer-specific survival can be interpreted as convincing evidence for treatment efficacy. For cancers with relatively good prognosis, demonstrating evidence of treatment efficacy may require a substantial number of study participants and long-term follow-up to obtain a sufficient number of deaths to estimate statistically stable associations. To reduce the observation time required to accrue a sufficient number of deaths, one may include high-risk patients with expected low survival time, or use temporally more proximal surrogate endpoints for overall and cause-specific survival.
Surrogate endpoints
Surrogate endpoints can be defined as markers intended to act in place of a clinical endpoint, such as prostate-specific antigen as a marker of biochemical failure.17,18 Surrogates are used to obtain an estimate of treatment effect on survival without having to observe deaths. Prolonged progression-free survival as a surrogate for overall survival is allowed by the European Medicines Agency as a primary outcome in oncology trials, if overall survival is reported as a secondary endpoint.5
Surrogate endpoints have two noteworthy strengths. First, they allow earlier assessment of treatment efficacy, and are therefore less affected by disease management subsequent to drug intervention, whereas more distal endpoints, such as death, can be severely impacted by post-intervention treatment and care after the intervention under study. Second, they reduce the sample size and observation time in studies of underlying clinical endpoints that are more distal in time.19 In order for a surrogate endpoint to be valid, it should correlate with the underlying distal clinical endpoint and with the intervention, and optimally, mediate the entire association between the intervention and the clinical endpoint.19
Surrogate endpoint data are generally of high quality in clinical trials with standardized endpoint assessment, but data on surrogate endpoints of the same quality are rarely available in observational cohort studies in databases.
Other surrogate endpoints for survival and tumor processes used in the adjuvant setting of drug efficacy include disease recurrence. For example, disease-free survival is calculated based on the proportion of individuals without recurrence and death over a period of time. If recurrence-/disease-free survival predicts death over a period of time, it can act as a surrogate for survival. For examining drug effectiveness in an observational study, assessing recurrence rather than survival or mortality shows the effect of the drug on the cancer rather than the general health effects associated with the drug. In the advanced disease setting, the response rate is often used, as it refers to the proportion of study participants with a response to treatment, measured as a predefined reduction in tumor burden.20 Another endpoint that allows for assessment of early treatment benefit in advanced-disease cancer trials is progression-free survival. This concept can, for example, be defined as time from intervention to tumor progression or death from any cause.20
Patient-reported endpoints
Patient-reported outcome measures are increasingly being applied in oncology.21 Patient-reported endpoints provided directly by the patient, such as quality of life, symptoms, or pain, can be measured with standardized scales.22 Thus, measurable patient-reported endpoints can be easily included in clinical trials.21,23 In observational cohort studies, patient-reported endpoints are rarely available from medical databases, so they must also be collected directly from patients using the standardized scales. However, since RCTs include volunteer patients, their results may not be comparable to those of patients attending routine clinical practice.
Challenges with patient-reported endpoints are a potential lack of generalizability between patient populations, and that benefits to subgroups of patients can be masked by overall assessment of an entire study population.22 Patient assessment may also vary across the disease trajectory, and disease progression or treatment toxicities may affect changes in quality of life or pain rather than a study intervention. Patient-reported endpoints are, however, imperative in studies when incremental gain in survival associated with novel drugs is small compared with existing drugs.20 A treatment benefit should be accompanied by acceptable quality of life, alleviation of pain, or reduced toxicities compared with existing therapies. Safety, efficacy, and patient tolerance should be the foremost concern but viewed in the context of escalating health care costs. To optimize economic efficiency, pharmacoeconomic studies are increasingly used in determining health care policies by tracking changes in utilization and spending with the introduction of novel oncology drugs.24
Clinical practice challenges in standardized endpoint determination
In clinical practice as well as in clinical trials, a cancer patient may quit pharmacological treatment, switch drugs, stop any further treatment, or if there is symptomatic benefit, continue with the same drug. These various clinical pathways complicate assessing the effect of an initial treatment intervention on overall survival. Disease recurrence or progression is often documented in medical records by clinical examinations, biochemical tests, or radiological imaging, all of which may be subjectively interpreted and may not be reproducible. In clinical trials, on the other hand, disease recurrence or progression is often objectively defined by radiological imaging alone with the assessment blinded to the intervention. However, time to disease recurrence and time to progression are continuous endpoints and therefore depend on the timing of assessment during the disease course, and the cancer growth rate,20 which affects both clinical trials and observational cohort studies. Many trials use standardized criteria defined by the Response Evaluation Criteria in Solid Tumors25 guidelines to measure response to treatment, but these criteria hold no measure of direct clinical benefit.26 Improved response rates do not always translate into longer overall and progression-free survival because a high response rate is required to induce a measurable impact on survival.27 Cohort studies relying on medical databases usually lack detailed clinical information on treatment response, recurrence, or progression; such endpoints can be obtained from medical records or patient examination, which is time consuming and expensive. Validated algorithms that capture disease recurrence and treatment lines from medical databases have therefore been developed.28,29 Patients are managed in standard clinical care, which is not readily comparable to standard endpoints assessed in clinical trials. Accordingly, using validated algorithms to identify time to disease recurrence has limitations, at least with respect to the timing of the endpoint determination, which may relate to the timing of follow-up visits in clinical practice. In clinical trials, using death or time to subsequent treatment as distal and proximal endpoints, respectively, may therefore improve translation of findings from clinical trials to those of observational cohort studies better than time to progression or progression-free survival,30 and allow for better comparison of study results across study designs.
Controversies in surrogate endpoint definitions
Commonly used endpoints for treatment benefit are recurrence-free survival, disease-free survival, relapse-free survival, and progression-free survival, and their respective inverse measures, time to recurrence, time to relapse, and time to progression. No standardized concept exists for defining these endpoints, and it is unclear from the name of the endpoint which components are included.31–33 The “survival” endpoints often include death as a component in the definition, whereas the “time to” endpoints treat death as a censoring event, but there are several exceptions in the literature.
For example, in breast cancer trials of adjuvant hormonal therapy with disease-free survival as the endpoint, some studies include local, regional, and distant metastasis and contralateral breast cancer in the definition;34–38 few studies include ipsilateral and contralateral carcinoma in situ;35,36 others include all-cause mortality34,36,37 and second primary (non-breast) cancers.34 Similar controversies exist for other cancer sites.39 To overcome some of these challenges in breast cancer trials, the Standardized Definitions for Breast Cancer Clinical Trial Endpoints in the Adjuvant Setting system proposed a standardized definition of endpoints in adjuvant clinical trials.40 Since then, several researchers have called for appropriate endpoint definitions in oncology research,10,11,31,41 but these attempts have not been widely accepted.
Progression-free survival as an endpoint in cancer trials has been heavily debated,31,42–44 and remains controversial for several reasons. First, progression-free survival is not a meaningful measure of clinical benefit for patients for whom a measure of symptom control or quality of life may be more relevant, especially if it is not a reliable surrogate for overall survival. For example, improvement in progression-free survival and an associated decrease in patient distress is not always a desirable outcome when treatment toxicities are instant, serious, or unacceptable, for example, severe disabling neuropathy. Second, the exact timing of disease progression is never known and depends on the timing of the clinical assessment and measurement variability. Third, prolonged progression-free survival does not necessarily translate into increased overall survival.20,31,42–44 Fourth, progression-free survival is not a valid surrogate endpoint in all settings. For example, it may be a valid surrogate endpoint in advanced colorectal cancer but not in advanced breast cancer,45 and may only be a valid surrogate for overall survival in studies with median survival after disease progression of less than 1 year.46
Conclusion
Efficacy and effectiveness of novel cancer drugs is established through accumulating experimental and observational evidence. Clinical trials – experimental cohort studies – have the advantage of being well controlled by the researcher and allow for use of any appropriate clinically relevant proximal or distal endpoints. Observational cohort studies, on the other hand, can include a substantial number of patients with various characteristics, and a long-term follow-up – usually at a lower cost than clinical trials. Regardless of design, studies should always apply valid and reproducible endpoints to accurately examine treatment benefit. However, there is no international consensus definition for soft endpoints.
In summary, the observational cohort study complements the clinical trial by examining the effectiveness of drug interventions applied in clinical practice, and by examining long-term clinical outcomes, which are often not feasible to study in a clinical trial. A clinical trial can include any predefined endpoint, whereas the observational study is restricted by standard clinical care, which is not always comparable to standard endpoint assessment in clinical trials. Observational studies can be more helpful moving research forward if they restrict focus to appropriate and valid endpoints.
Acknowledgments
This work was supported by the Program for Clinical Research Infrastructure established by the Lundbeck Foundation and the Novo Nordisk Foundation. The Department of Clinical Epidemiology, Aarhus University Hospital, receives funding for other studies from companies in the form of research grants to (and administered by) Aarhus University. None of these studies has any relation to the present study.
Disclosure
The authors declare no conflicts of interest in this work.
References
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | ||
Pignatti F, Gravanis I, Herold R, Vamvakas S, Jonsson B, Marty M. The European Medicines Agency: an overview of its mission, responsibilities, and recent initiatives in cancer drug regulation. Clin Cancer Res. 2011;17(16):5220–5225. | ||
Martell RE, Sermer D, Getz K, Kaitin KI. Oncology drug development and approval of systemic anticancer therapy by the U.S. Food and Drug Administration. Oncologist. 2013;18(1):104–111. | ||
Booth CM, Tannock IF. Randomised controlled trials and population-based observational research: partners in the evolution of medical evidence. Br J Cancer. 2014;110(3):551–555. | ||
European Medicines Agency. Guideline on the evaluation of anticancer medicinal products in man. EMA/CHMP/205/95/Rev. 4. 2012. | ||
U.S. Department of Health and Human Services. Food and Drug Administration. Guidance for industry. Clinical trial endpoints for the approval of cancer drugs and biologics. 2007. | ||
Warren JL, Klabunde CN, Schrag D, Bach PB, Riley GF. Overview of the SEER-medicare data: content, research applications, and generalizability to the United States elderly population. Med Care. 2002;40(8 Suppl):IV-3-18. | ||
Coleman MP, Forman D, Bryant H, et al. Cancer survival in Australia, Canada, Denmark, Norway, Sweden, and the UK, 1995-2007 (the International Cancer Benchmarking Partnership): an analysis of population-based cancer registry data. Lancet. 2011;377(9760):127–138. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Wilson MK, Collyar D, Chingos DT, et al. Outcomes and endpoints in cancer trials: bridging the divide. Lancet Oncol. 2015;16(1):e43–e52. | ||
Wilson MK, Karakasis K, Oza AM. Outcomes and endpoints in trials of cancer treatment: the past, present, and future. Lancet Oncol. 2015;16(1):e32–e42. | ||
Sorensen HT, Lash TL, Rothman KJ. Beyond randomized controlled trials: a critical comparison of trials with nonrandomized studies. Hepatology. 2006;44(5):1075–1082. | ||
Greenland S. Randomization, statistics, and causal inference. Epidemiology. 1990;1(6):421–429. | ||
Ellis LM, Bernstein DS, Voest EE, et al. American Society of Clinical Oncology perspective: raising the bar for clinical trials by defining clinically meaningful outcomes. J Clin Oncol. 2014;32(12):1277–1280. | ||
Booth CM, Tannock IF. Evaluation of treatment benefit: randomized controlled trials and population-based observational research. J Clin Oncol. 2013;31(26):3298–3299. | ||
Rampatige R, Mikkelsen L, Hernandez B, Riley I, Lopez AD. Systematic review of statistics on causes of deaths in hospitals: strengthening the evidence for policy-makers. Bull World Health Organ. 2014;92(11):807–816. | ||
Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69(3):89–95. | ||
D’Amico AV, Moul JW, Carroll PR, Sun L, Lubeck D, Chen MH. Surrogate end point for prostate cancer-specific mortality after radical prostatectomy or radiation therapy. J Natl Cancer Inst. 2003;95(18):1376–1383. | ||
Prentice RL. Surrogate and mediating endpoints: current status and future directions. J Natl Cancer Inst. 2009;101(4):216–217. | ||
Dancey JE, Dodd LE, Ford R, et al. Recommendations for the assessment of progression in randomised cancer treatment trials. Eur J Cancer. 2009;45(2):281–289. | ||
Basch E, Snyder C, McNiff K, et al. Patient-reported outcome performance measures in oncology. J Oncol Pract. 2014;10(3):209–211. | ||
Trask PC, Hsu MA, McQuellon R. Other paradigms: health-related quality of life as a measure in cancer treatment: its importance and relevance. Cancer J. 2009;15(5):435–440. | ||
Bottomley A, Jones D, Claassens L. Patient-reported outcomes: assessment and current perspectives of the guidelines of the Food and Drug Administration and the reflection paper of the European Medicines Agency. Eur J Cancer. 2009;45(3):347–353. | ||
Siegel JE, Torrance GW, Russell LB, Luce BR, Weinstein MC, Gold MR. Guidelines for pharmacoeconomic studies. Recommendations from the Panel on Cost Effectiveness in Health and Medicine. Panel on Cost Effectiveness in Health and Medicine. Pharmacoeconomics. 1997;11(2):159–168. | ||
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. | ||
George SL. Response rate as an endpoint in clinical trials. J Natl Cancer Inst. 2007;99(2):98–99. | ||
Torri V, Simon R, Russek-Cohen E, Midthune D, Friedman M. Relationship between response and survival in patients with advanced ovarian cancer. J Natl Cancer Inst. 1992;84(11):899–900. | ||
Lash TL, Riis AH, Ostenfeld EB, Erichsen R, Vyberg M, Thorlacius-Ussing O. A validated algorithm to ascertain colorectal cancer recurrence using registry resources in Denmark. Int J Cancer. 2015;136(9):2210–2215. | ||
Bikov KA, Mullins CD, Seal B, Onukwugha E, Hanna N. Algorithm for identifying chemotherapy/biological regimens for metastatic colon cancer in SEER-medicare. Med Care. 2015;53(8):e58–e64. | ||
Nava F, Tramacere I, Fittipaldo A, et al. Survival effect of first- and second-line treatments for patients with primary glioblastoma: a cohort study from a prospective registry, 1997-2010. Neuro Oncol. 2014;16(5):719–727. | ||
Saad ED, Katz A. Progression-free survival and time to progression as primary end points in advanced breast cancer: often used, sometimes loosely defined. Ann Oncol. 2009;20(3):460–464. | ||
Mathoulin-Pelissier S, Gourgou-Bourgade S, Bonnetain F, Kramar A. Survival end point reporting in randomized cancer clinical trials: a review of major journals. J Clin Oncol. 2008;26(22):3721–3726. | ||
Nout RA, Fiets WE, Struikmans H, Rosendaal FR, Putter H, Nortier JW. The in- or exclusion of non-breast cancer related death and contralateral breast cancer significantly affects estimated outcome probability in early breast cancer. Breast Cancer Res Treat. 2008;109(3):567–572. | ||
Breast International Group (BIG) 1-98 Collaborative Group; Thurlimann B, Keshaviah A, Coates AS, et al. A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N Engl J Med. 2005;353(26):2747–2757. | ||
Goss PE, Ingle JN, Martino S, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med. 2003;349(19):1793–1802. | ||
Baum M, Budzar AU, Cuzick J, et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet. 2002;359(9324):2131–2139. | ||
Coombes RC, Hall E, Gibson LJ, et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N Engl J Med. 2004;350(11):1081–1092. | ||
Jakesz R, Jonat W, Gnant M, et al. Switching of postmenopausal women with endocrine-responsive early breast cancer to anastrozole after 2 years’ adjuvant tamoxifen: combined results of ABCSG trial 8 and ARNO 95 trial. Lancet. 2005;366(9484):455–462. | ||
Chua YJ, Sargent D, Cunningham D. Definition of disease-free survival: this is my truth-show me yours. Ann Oncol. 2005;16(11):1719–1721. | ||
Hudis CA, Barlow WE, Costantino JP, et al. Proposal for standardized definitions for efficacy end points in adjuvant breast cancer trials: the STEEP system. J Clin Oncol. 2007;25(15):2127–2132. | ||
Moossdorff M, van Roozendaal LM, Schipper RJ, et al. Inconsistent selection and definition of local and regional endpoints in breast cancer research. Br J Surg. 2014;101(13):1657–1665. | ||
Booth CM, Eisenhauer EA. Progression-free survival: meaningful or simply measurable? J Clin Oncol. 2012;30(10):1030–1033. | ||
Stewart DJ. Before we throw out progression-free survival as a valid end point…. J Clin Oncol. 2012;30(27):3426–3427. | ||
Gill S, Berry S, Biagi J, et al. Progression-free survival as a primary endpoint in clinical trials of metastatic colorectal cancer. Curr Oncol. 2011;18 Suppl 2:S5–S10. | ||
Saad ED, Katz A, Hoff PM, Buyse M. Progression-free survival as surrogate and as true end point: insights from the breast and colorectal cancer literature. Ann Oncol. 2010;21(1):7–12. | ||
Broglio KR, Berry DA. Detecting an overall survival benefit that is derived from progression-free survival. J Natl Cancer Inst. 2009;101(23):1642–1649. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.