")
Back to Journals » OncoTargets and Therapy » Volume 11
CEP55 promotes the proliferation, migration and invasion of esophageal squamous cell carcinoma via the PI3K/Akt pathway
Authors Jia Y , Xiao Z, Gongsun X, Xin Z, Shang B, Chen G, Wang Z, Jiang W
Received 21 March 2018
Accepted for publication 7 June 2018
Published 20 July 2018 Volume 2018:11 Pages 4221—4232
DOI https://doi.org/10.2147/OTT.S168861
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Yang Jia, Zhaohua Xiao, Xin Gongsun, Zhongwei Xin, Bin Shang, Gang Chen, Zhou Wang, Wenpeng Jiang
Department of Thoracic Surgery, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, China
Background: Centrosomal protein 55 (CEP55) is an important prognostic biomarker that plays an essential role in the proliferation, migration and invasion of multiple tumors. We aimed to investigate the prognostic value of CEP55 in pN0 esophageal squamous cell carcinoma (ESCC) and explore its biological function in ESCC cells.
Methods: We used immunohistochemistry and Western blot analysis to detect the expression of CEP55 in ESCC. Furthermore, both in vitro and in vivo assays were used to determine the effect of CEP55 on malignant behavior in ESCC cells.
Results: As expected, we found that CEP55 was overexpressed in ESCC. Univariate and multivariate analyses demonstrated that patients with CEP55 overexpression had a poor prognosis. Additionally, the abilities of proliferation, migration and invasion of cells, as well as the epithelial–mesenchymal transition markers, were all altered with the changed CEP55 expression levels in ESCC cells. Further study elucidated that CEP55 facilitated ESCC via the PI3K/Akt pathway. Blockade of this pathway markedly attenuated CEP55-mediated proliferation, migration, invasion and epithelial–mesenchymal transition of ESCC cells.
Conclusion: Oncogenic CEP55 correlates with a poor prognosis by regulating tumor cell proliferation, migration and invasion via the PI3K/Akt pathway. It can serve as a promising prognostic biomarker and therapeutic target of pN0 ESCC after Ivor-Lewis esophagectomy.
Keywords: CEP55, proliferation, migration, invasion, esophageal squamous cell carcinoma, PI3K/Akt pathway
Introduction
Esophageal carcinoma (EC) is one of the most common aerodigestive tract malignant tumors and is the sixth leading cause of mortality.1,2 Esophageal squamous cell carcinoma (ESCC) may account for 90% of EC in the high-risk areas, especially in some areas of China.3 Despite significant improvements in diagnostic techniques and therapeutic modalities, the prognosis of individuals with ESCC remains poor.4 According to the National Comprehensive Cancer Network guidelines, ESCC patients without lymph node metastasis (pN0) undergoing complete tumor resection may not receive adjuvant therapy. However, the 5-year survival rate of ESCC patients in this stage is only ~70%.5 Additionally, patients in the same stage tend to have obviously different survival statuses. Thus, we should further stratify patients based on their differential prognosis and then provide individualized treatment to improve the overall survival (OS).
Proliferation, migration and invasion are the important biological characteristics in most malignancies that affect patient prognosis. Centrosomal protein 55 (CEP55), also known as FLJ10540, C10orf3 and URCC6, is the latest identified member of the centrosome- and midbody-associated protein family, and it mainly participates in the process of cytokinesis.6 Accumulating evidence has shown that CEP55 was overexpressed in multiple tumors.7–16 Interestingly, it was identified as a prognostic signature, and its overexpression was significantly correlated with the poor prognosis of patients with oral cavity squamous cell carcinoma, epithelial ovarian carcinoma, hepatocellular carcinoma, prostate cancer and pancreatic cancer, among others.7,11,13,14,16–20 In addition, some studies have demonstrated that CEP55 overexpression may promote the proliferation, migration and invasion of tumor cells.9,13,15 However, the prognostic value of CEP55 in patients with pN0 ESCC and its biological function in ESCC cells remain unclear.
The phosphatidylinositol-3-kinase (PI3K)/Akt pathway is an important signaling pathway that is implicated in multiple oncogenic processes, including cell proliferation, differentiation, apoptosis, epithelial–mesenchymal transition (EMT), migration and invasion.13,15,21–23 Some studies have shown that the PI3K/Akt pathway may interact with other molecules to modulate the biological behavior of ESCC cells.24–26 Additionally, it was reported to participate in the process of CEP55-mediated proliferation, migration, invasion and transformation in multiple tumors.9,13,15 However, whether the PI3K/Akt pathway is involved in CEP55-mediated malignant behavior and biological interactions of ESCC cells is not fully understood.
In this study, we demonstrated that CEP55 is overexpressed in ESCC. Furthermore, we elucidated that CEP55 promotes cell proliferation in vitro and in vivo, modulates cell invasion and migration, and induces ESCC cells to undergo EMT via the PI3K/Akt pathway. Our results confirmed that CEP55 may act as a promising prognostic marker in patients with pN0 ESCC after Ivor-Lewis esophagectomy with two-field lymphadenectomy. Additionally, CEP55 or the PI3K/Akt pathway may be the potential target for postoperative adjuvant treatment.
Patients and methods
Patient recruitment
Thirty pairs of frozen ESCC tissues and their corresponding noncancerous esophageal tissues (>5 cm from the margin of the tumor) were collected from Shandong Provincial Hospital Affiliated to Shandong University from December 2015 to May 2016. In addition, 195 formalin-fixed paraffin-embedded tumor specimens were harvested from patients who underwent Ivor-Lewis esophagectomy with two-field lymphadenectomy from January 2005 through December 2007.27 All patients achieved complete tumor resection (R0), and the number of lymph node dissections was >12 (12–28, average 17.6). Additionally, they were all pathologically diagnosed as pN0 ESCC, and preoperative neoadjuvant chemotherapy and postoperative adjuvant treatment were not imposed. Clinicopathologic data are listed in Table 1.
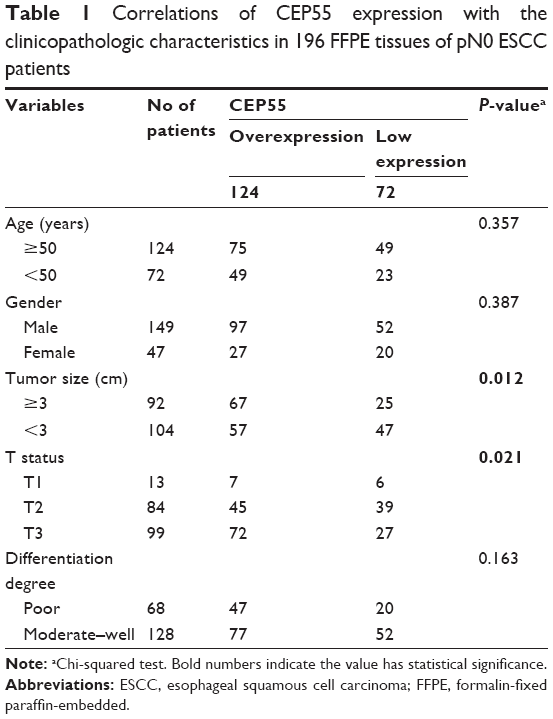 | Table 1 Correlations of CEP55 expression with the clinicopathologic characteristics in 196 FFPE tissues of pN0 ESCC patients |
This study was approved by the Ethics Committee of Shandong Provincial Hospital Affiliated to Shandong University in accordance with the ethical guidelines of the Declaration of Helsinki. Written informed consent was obtained from all participants.
Cell culture and transfection
Five established human ESCC cell lines (Eca109, KYSE150, KYSE450, EC9706 and TE-1) and one normal human esophageal epithelial cell line (HET-1A) were purchased from the Cell Bank of the Shanghai Institute in China. All cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific). Human CEP55 shRNA and overexpressing vector were chemically synthesized and packaged into lentiviruses (Genechem, Shanghai, China). Puromycin at 5 μg/mL was used to select the stable transfections for 1 week. Cells transfected with shRNA, scramble sequence, overexpression vector and control vector were separately labeled as shCEP55, Scramble, CEP55 and Vector. Cells without lentiviruses transfection were labeled as Blank control. LY294002, wortmannin and MK2206 (Selleckchem, Houston, TX, USA) were used to determine the involvement of the PI3K/Akt pathway in CEP55-mediated cell proliferation, migration and invasion.
Immunohistochemistry and immunohistochemical score (IHS)
We performed the streptavidin-peroxidase method to detect CEP55 expression in specimens.28 Rabbit anti-CEP55 monoclonal antibodies (Abcam Inc., Cambridge, MA, USA) were used at a dilution of 1:150. The secondary biotinylated antibody kit was purchased from ZSGB Biotech (Beijing, China). Two independent pathologists blinded to the clinical data were invited to evaluate the sections. The method and procedure of IHS can be referenced in our previous research.29
RNA extraction and quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the tissue specimens and cell lines. The procedures of RNA extraction and qRT-PCR were described in a previous study.28 The polymerase chain reaction primers were as follows: CEP55, 5′-TTGGAACAACAGATGCAGGC-3′ (forward primer) and 5′-GAGTGCAGCAGTGGGACTTT-3′ (reverse primer); β-actin, 5′-AGAGCCTCGCCTTTGCCGATCC-3′ (forward primer) and 5′-ATACACCCGCTGCTCCGGGTC-3′ (reverse primer).
Western blot analysis
Proteins were extracted from the tissue samples and cell lines, and the total protein concentration was determined using a bicinchoninic acid kit. Equal amounts of protein (40 μg) were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and were transferred onto polyvinylidene difluoride membrane filters (EMD Millipore, Billerica, MA, USA). Next, 5% non-fat dry milk was used to block nonspecific binding. The membranes were incubated with primary antibodies. Following washing, the membranes were incubated with the corresponding secondary antibodies conjugated with horseradish peroxidase. Finally, the protein levels were quantified using an enhanced chemiluminescence detection system (Amersham imager 600; General Electric, Fairfield, CT, USA). The antibodies used in this study were as follows: CEP55, N-cadherin, Vimentin and Snail (1:1,000; Abcam Inc.); E-cadherin (1:1,000; Affbiotech, Cincinnati, OH, USA); Claudin-1 and GAPDH (1:1,000; Proteintech, Chicago, IL, USA); Akt (1:1,000; Omimabs, Alhambra, CA, USA) and pAktS473 and pAktT308 (1:1,000; Cell Signaling Technology, Danvers, MA, USA).
Cell counting kit-8 and clonogenic assays
The cell counting kit-8 (CCK-8; Dojindo, Rockville, MD, USA) assay was performed to test the viability of cells with differential CEP55 expression. In total, 5×103 cells were seeded into 96-well plates in 100 μL of medium, followed by incubation with 10 μL of CCK-8 per well at 24, 48 and 72 hours for 1 hour. We used a microplate reader to measure the absorbance at 450 nm. For the clonogenic assay, cells were seeded into six-well plates at a density of 500 cells/well. The cell colonies were stained with hematoxylin after 2 weeks and the colonies that contained >50 cells were counted. The percentage of colony formation was calculated for each group. All these experiments were performed in triplicate.
Cell migration and invasion assays
For both migration and invasion assays, the cells were precultured in serum-free medium for 24 hours. The uncoated Transwells were used in the migration assay, while 40 μL of Matrigel (1:4; BD Biosciences San Jose, CA, USA) was precoated to the upper surface of the Transwells in the invasion assay. Next, 1.5×105 starved cells were seeded into the upper chamber of 24-well Transwells (8 mm pore size polycarbonate membrane; EMD Millipore) with 200 μL of FBS-free medium, and 600 μL of medium with 15% FBS was added to the lower chamber. The migration assay was incubated for 24 hours, and the invasion assay proceeded for 48 hours. Next, the cells on the lower surface of the membrane were fixed and stained, and the cells were counted from three independent visual fields.
Tumor xenografts in mice
Six-week-old BALB/c nude mice (five mice per group) were fed in a pathogen-free animal facility. In total, 2×106 cells suspended in 200 μL of PBS were injected subcutaneously into unilateral flanks. We used a Vernier caliper to measure the length (a) and width (b) of the xenografted tumors every 4 days for 24 days. The tumor volume (V) was calculated using the following formula: V=(a×b2)/2. The mice were sacrificed by cervical dislocation, and their tumors were weighed. The volume and weight of five tumors in each group were calculated and statistically analyzed. All procedures related to animal handling, care and treatment were performed in strict accordance with the recommendations of regulations on the management of experimental animals, which were approved by the State Council of the People’s Republic of China on October 31, 1988 and promulgated by Decree No 2 of the State Science and Technology Commission on November 14, 1988. Additionally, the protocol was approved by the Animal Ethics Committee of Shandong Provincial Hospital Affiliated to Shandong University.
Statistical analysis
The quantitative data were expressed as the mean ± SD. The differences between two groups were calculated using two-tailed Student’s test, and the differences among more than two groups were calculated by one-way analysis of variance. The chi-squared test was used to analyze the relationship between CEP55 expression and clinicopathologic variables. The survival curves were calculated by the Kaplan–Meier method. Univariate and multivariate analyses were performed to evaluate the prognostic factors. A statistically significant difference was represented by a two-tailed P-value <0.05. Statistical analysis of survival was performed using SPSS version 17.0 (SPSS Inc., Chicago, IL, USA).
Results
Expression of CEP55 in ESCC
The expression of CEP55 was investigated in 30 pairs of frozen ESCC tissues and corresponding noncancerous esophageal tissues using immunohistochemistry and Western blot analysis. Significant staining was readily detected in the cytoplasm of tumor cells (Figure 1Aa), while weak immunostaining was seen in noncancerous tissue (Figure 1Ab). Furthermore, the expression of CEP55 in all 30 tissue pairs was verified through Western blot analysis. Additionally, it demonstrated higher CEP55 expression in cancerous tissues than that in corresponding noncancerous tissues (CEP55/GAPDH: 0.71±0.14 vs 0.49±0.09, P<0.001; Figure 1Ba). The bands of Western blotting in eight representative pairs of tissues are shown in Figure 1Bb.
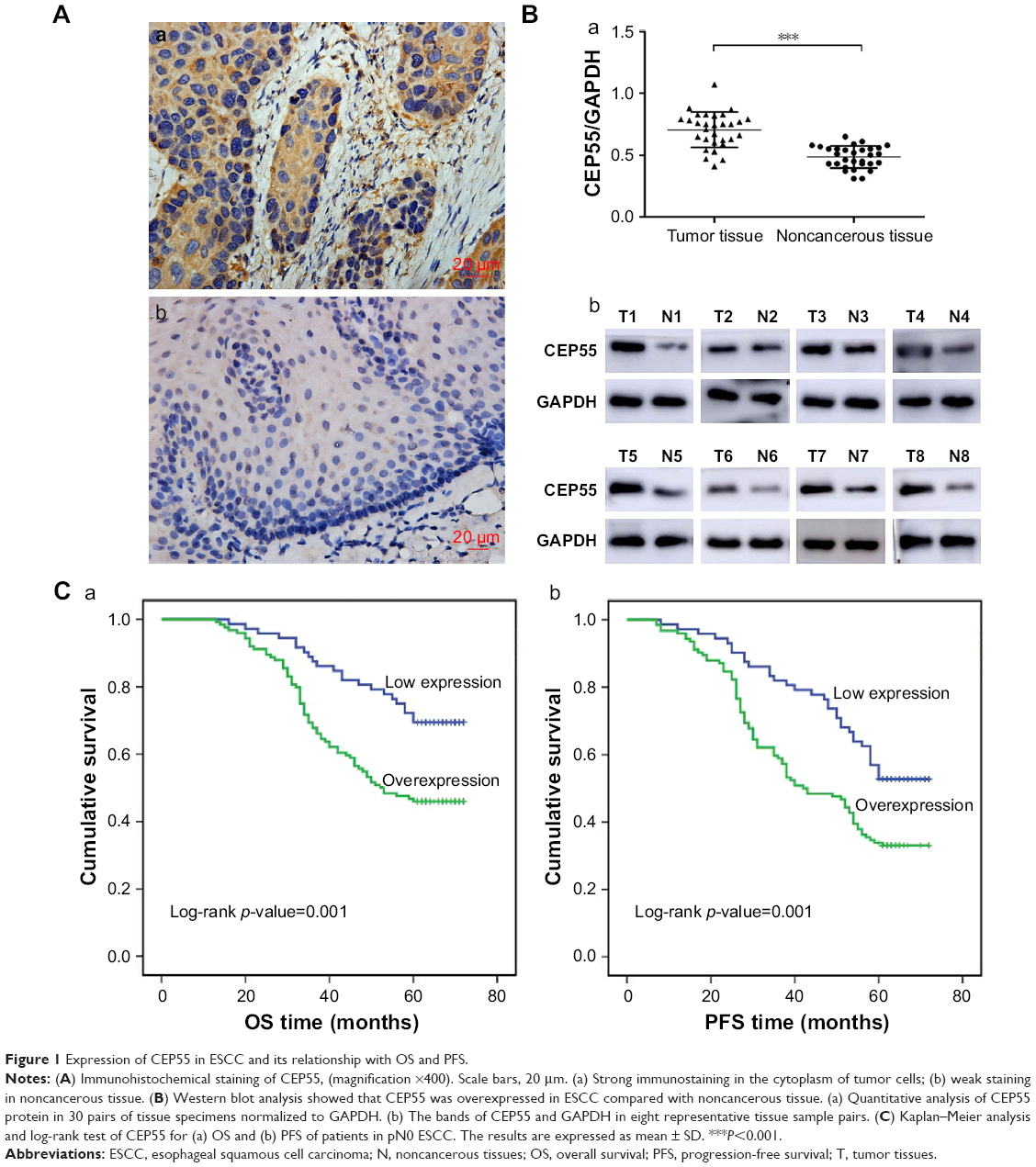 | Figure 1 Expression of CEP55 in ESCC and its relationship with OS and PFS. |
CEP55 overexpression may lead to a poor prognosis of pN0 ESCC
According to the IHS criteria, we divided all 196 pN0 ESCC patients into two groups: 124 cases (56.7%) were categorized as the overexpression group and 72 cases as the low expression group. Next, the clinicopathologic characteristics (including age, gender, tumor size, T status, degree of differentiation and CEP55 expression) were analyzed (Table 1). The chi-squared analysis demonstrated that the expression of CEP55 was positively associated with tumor size (P=0.012) and T status of the tumor (P=0.021). By contrast, there were no significant differences between CEP55 expression and age, gender or degree of differentiation.
In total, 107 of 196 pN0 ESCC patients were confirmed to have survived the disease. Seventy-nine cases were found to have a progression-free survival (PFS) status within 5 years after Ivor-Lewis esophagectomy with two-field lymphadenectomy. The OS and PFS times were 56.1±1.37 and 50.3±1.52 months, respectively. Kaplan–Meier analysis was used to calculate the effects of the patients’ clinicopathologic characteristics on the OS and PFS rates. The log-rank test showed that both the OS (69.4% vs 46.0%, P=0.001; Figure 1Ca; Table 2) and PFS (52.8% vs 33.1%, P=0.001; Figure 1Cb; Table 2) rates were significantly decreased for pN0 ESCC patients with CEP55 overexpression. Furthermore, multivariate analysis revealed that CEP55 overexpression was an independent prognostic risk factor for both OS (P=0.012; Table 2) and PFS (P=0.028; Table 2). As expected, T status was an important prognostic factor of tumor patients. The results of this study also suggested that advanced T status may lead to a poor prognosis of ESCC patients (P<0.001; Table 2).
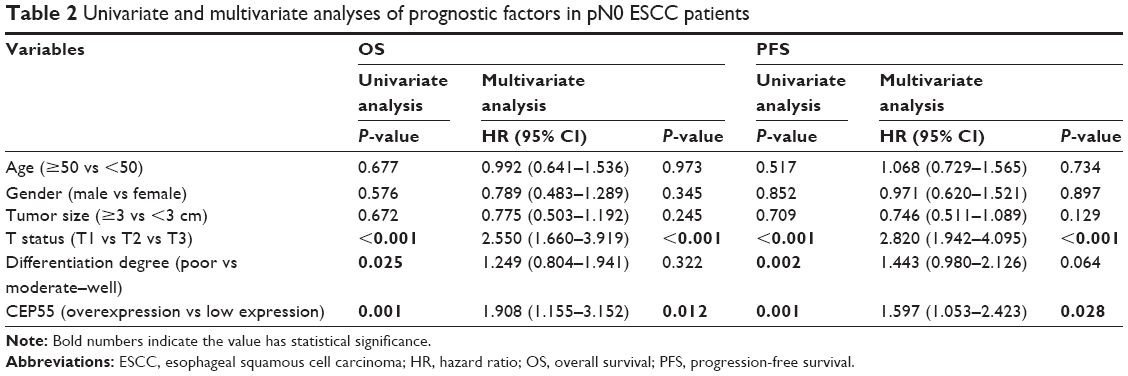 | Table 2 Univariate and multivariate analyses of prognostic factors in pN0 ESCC patients |
Cell transfection
The expression of CEP55 in five ESCC cell lines and HET-1A was detected via qRT-PCR and Western blot analysis. Compared with HET-1A, CEP55 was overexpressed in both the mRNA (Figure 2Aa) and protein (Figure 2Ab) levels in tumor cells, especially in KYSE450 cells. Thus, Eca109 and KYSE450 cells were used to knock down CEP55 with shRNA. Additionally, EC9706 cells were used to upregulate CEP55 with the overexpression vector. Compared with the Blank control and Scramble groups, cells transfected with shCEP55-1 and shCEP55-2 obviously downregulated CEP55 (P<0.05), especially shCEP55-1 (Figure 2Ba and Bb). Therefore, this sequence was selected for the subsequent experiments. Likewise, CEP55 was significantly upregulated in the CEP55 group compared with that in the Blank control and Vector groups (Figure 2Ba and Bc).
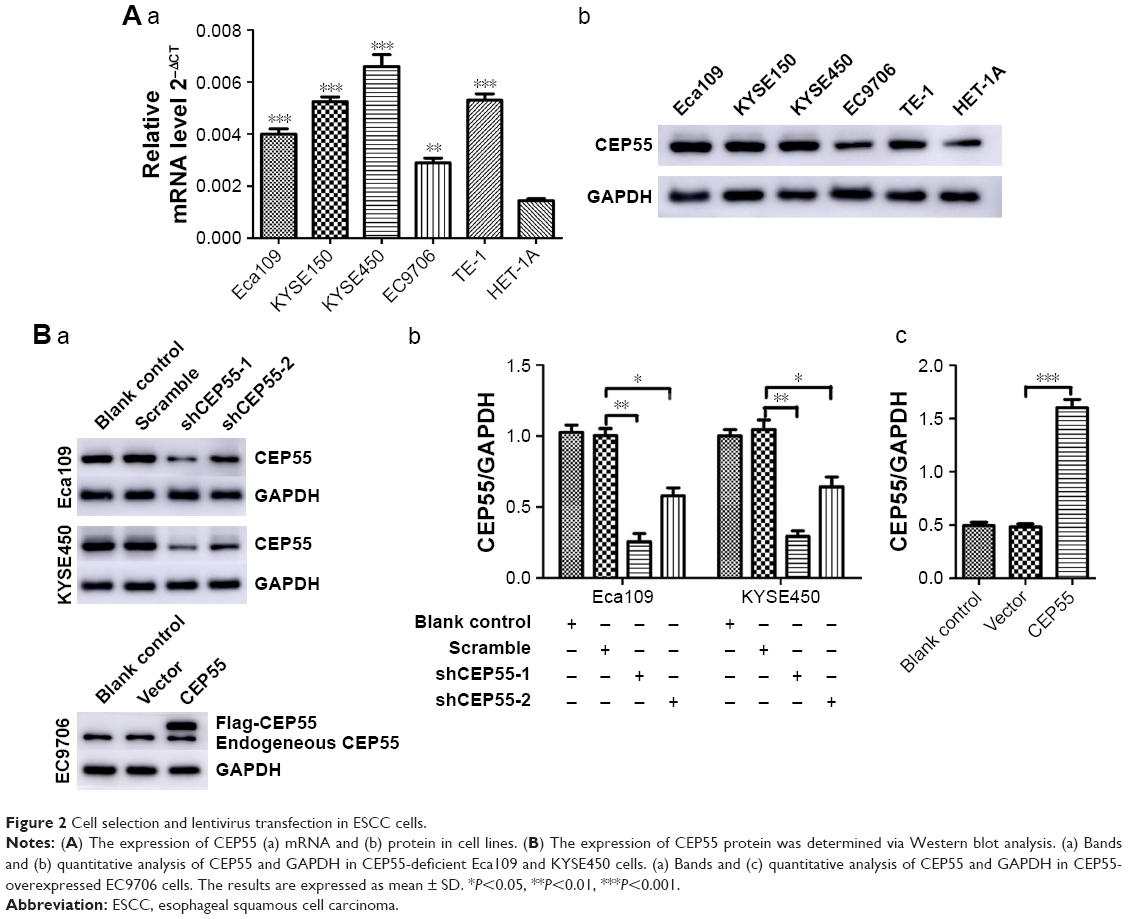 | Figure 2 Cell selection and lentivirus transfection in ESCC cells. |
CEP55 may promote the proliferation of ESCC cells
Both in vitro and in vivo assays were performed to explore the oncogenic role of ESCC. The CCK-8 assay showed that CEP55 depletion was markedly decreased, but CEP55 overexpression significantly increased the OD value at 450 nm at every interval examined (P<0.05; Figure 3Aa–c). Clonogenic assays showed that CEP55 knockdown abrogated the colony-formation ability of Eca109 and KYSE450 cells (P<0.001; Figure 3Ba and Bb). By contrast, CEP55 upregulation enhanced this ability of colony formation in EC9706 cells (P<0.01; Figure 3Bc and Bd). The pro-survival role of CEP55 in ESCC cells was also determined in vivo. Both the volume (P<0.001; Figure 3Ca) and weight of the tumors (1.87±0.31 vs 0.74±0.20 g, P<0.001; Figure 3Cb) were significantly attenuated in CEP55-deficient Eca109 cells. Conversely, CEP55 overexpression markedly increased both the volume (P<0.001; Figure 3Cc) and weight (1.02±0.24 vs 2.08±0.32 g, P<0.001; Figure 3Cd) of the xenografted tumors in EC9706 cells.
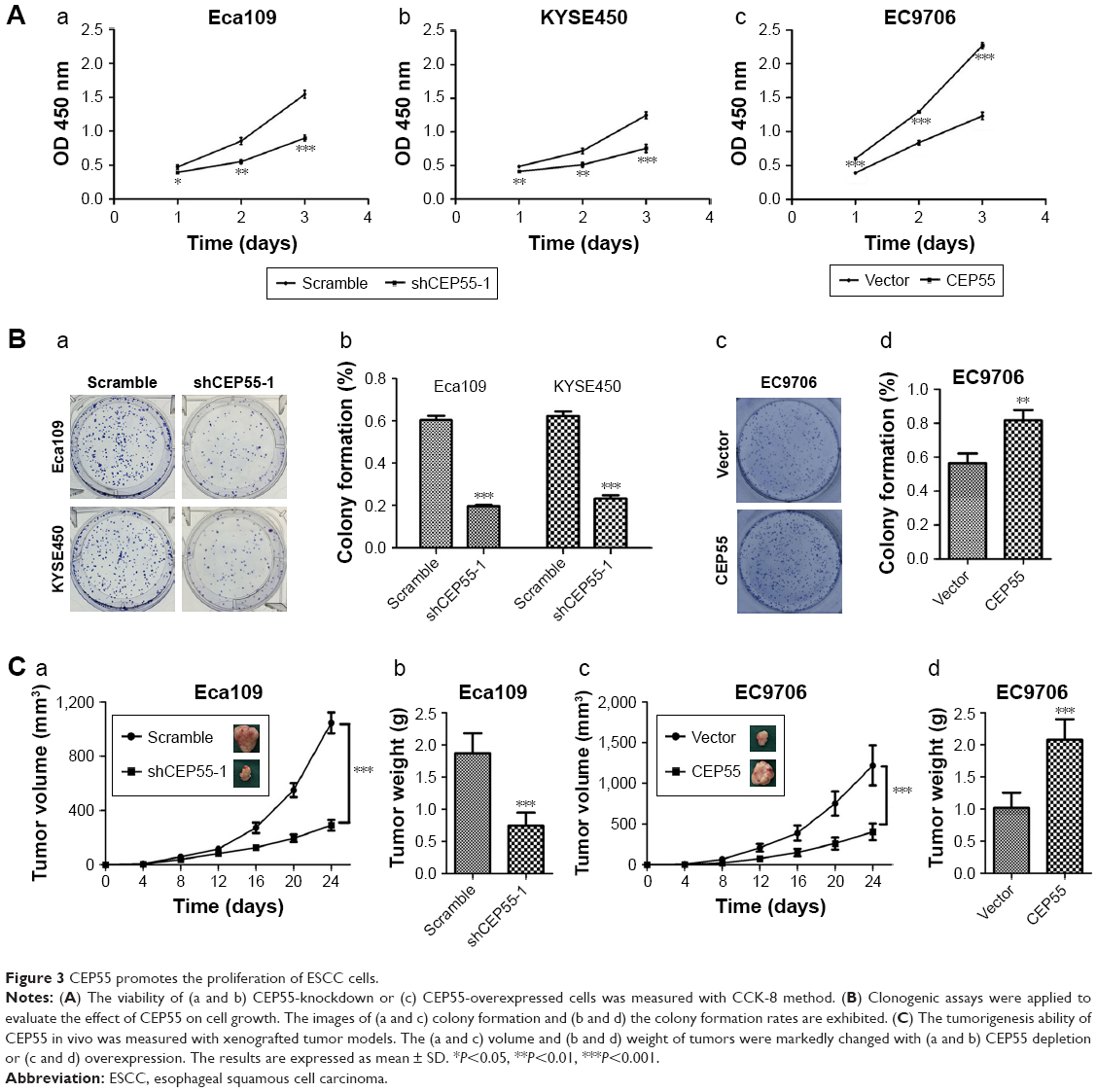 | Figure 3 CEP55 promotes the proliferation of ESCC cells. |
CEP55 may enhance the migration and invasion and mediate the EMT of ESCC cells
The effect of CEP55 on cell migration and invasion was investigated in ESCC. The Transwell assays indicated that CEP55 may enhance the migration and invasion of ESCC cells, showing that the number of migrated cells was markedly attenuated in CEP55-deficient Eca109 and KYSE450 cells (P<0.01; Figure 4Aa and Ab), but was significantly increased in CEP55-overexpressed EC9706 cells (P<0.01; Figure 4Ac and Ad). In addition, the invasive ability was altered with the changed expression of CEP55 in ESCC cells (P<0.01; Figure 4Ba–d).
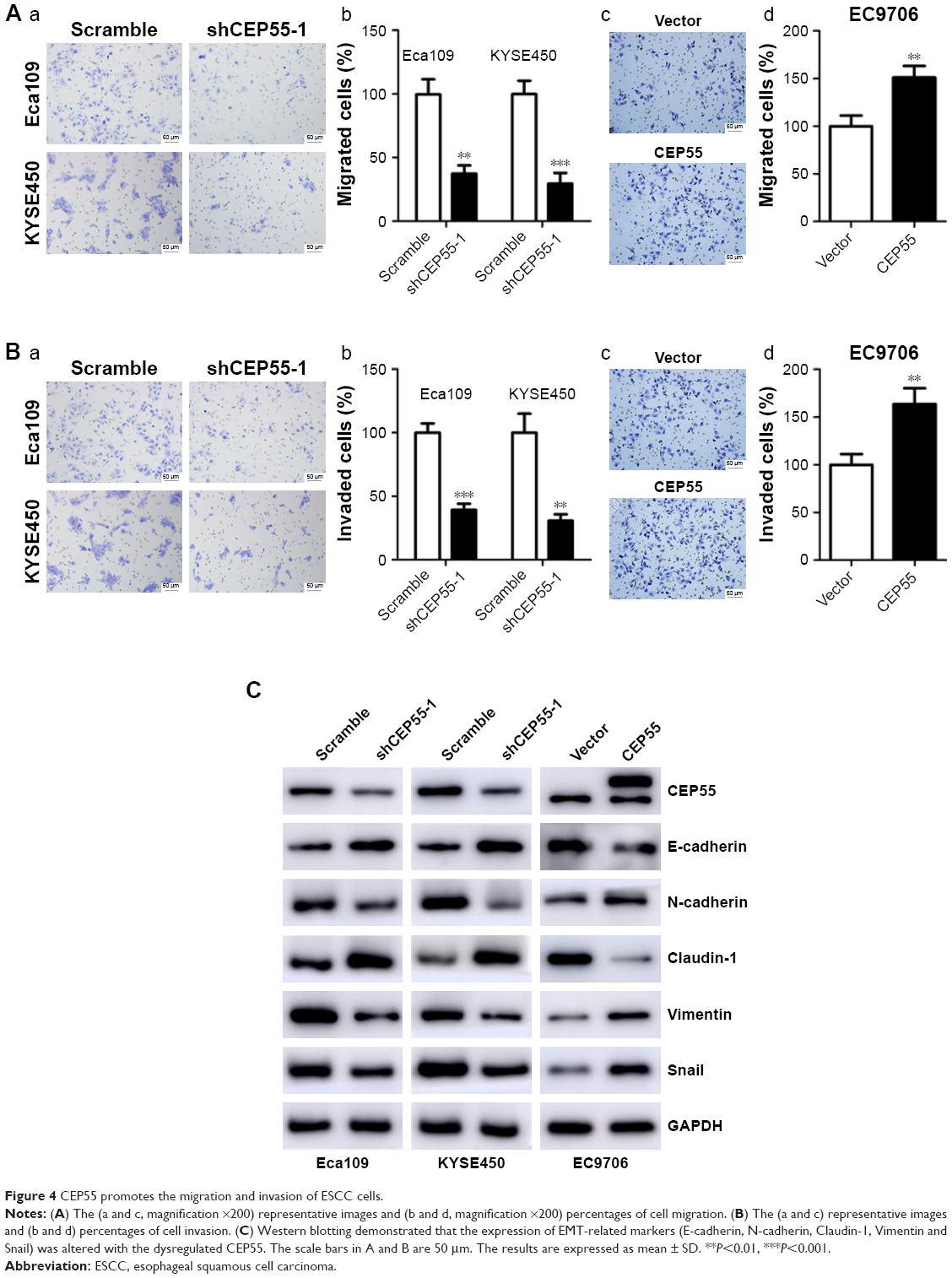 | Figure 4 CEP55 promotes the migration and invasion of ESCC cells. |
Furthermore, we detected the expression of EMT markers (E-cadherin, N-cadherin, Claudin-1, Vimentin and Snail) based on CEP55 expression using Western blotting. As indicated in Figure 4C, downregulation of CEP55 increased the expression of E-cadherin and Claudin-1, epithelial markers, and decreased the expression of N-cadherin, Vimentin and Snail, mesenchymal markers, in Eca109 and KYSE450 cells. By contrast, upregulation of CEP55 increased these mesenchymal markers (N-cadherin, Vimentin and Snail) and decreased the epithelial markers (E-cadherin and Claudin-1) in EC9706 cells.
CEP55 may promote the malignancy of ESCC through activation of the PI3K/Akt pathway
To detect whether CEP55 promoted the malignancy of ESCC via the PI3K/Akt pathway like other cancers, we investigated the effects of CEP55 knockdown or upregulation on the expression of phosphorylated AktS473 (pAktS473) and phosphorylated AktT308 (pAktT308). Western blotting confirmed that the expression of pAktS473 and pAktT308 was decreased in CEP55-deficient cells (Figure 5A). Correspondingly, both pAktS473 and pAktT308 were significantly upregulated in CEP55-overexpressed cells (Figure 5A).
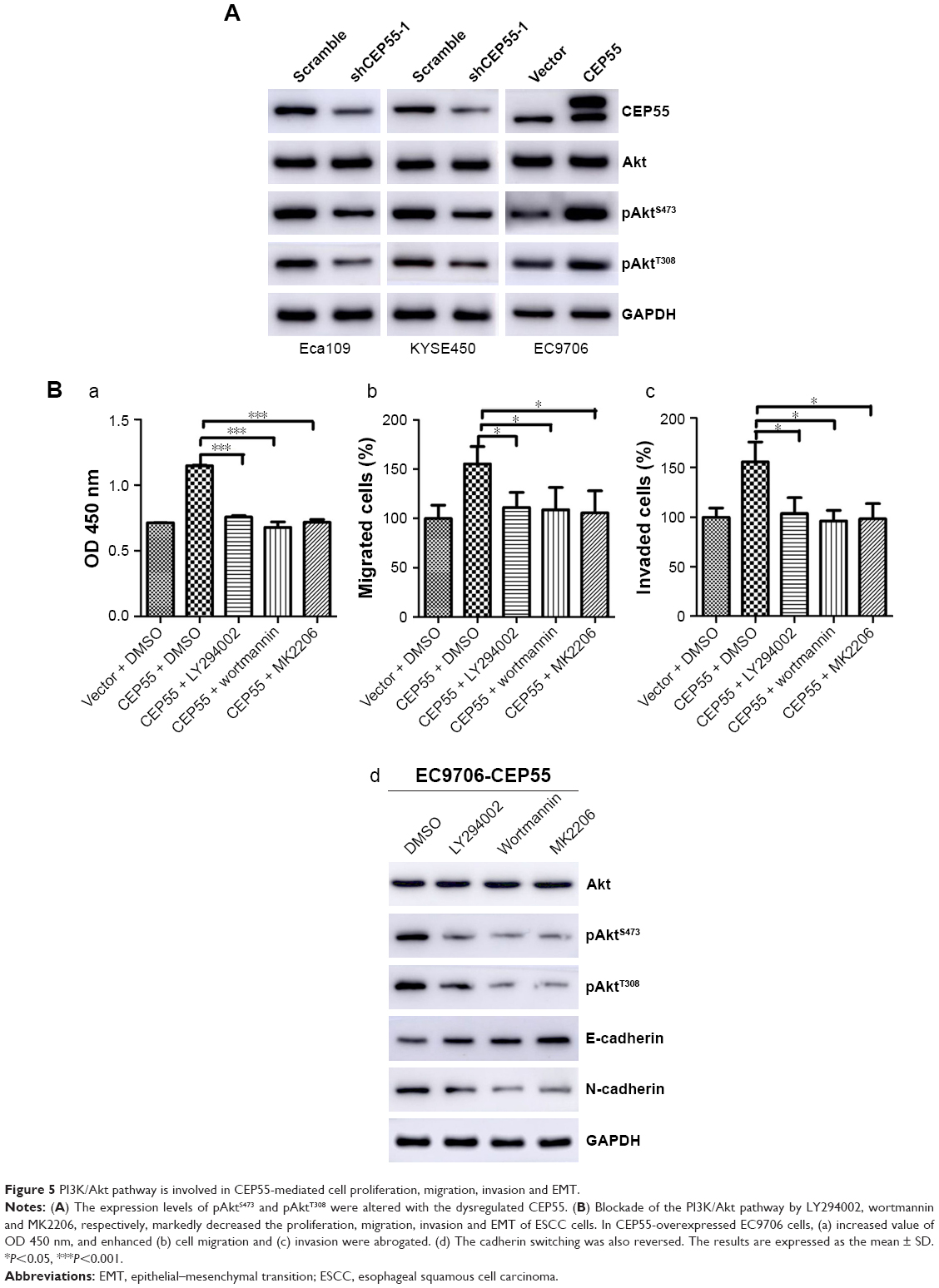 | Figure 5 PI3K/Akt pathway is involved in CEP55-mediated cell proliferation, migration, invasion and EMT. |
Furthermore, we used LY294002 and wortmannin, PI3K inhibitors and MK2206, an Akt inhibitor, to verify the role of the PI3K/Akt pathway in CEP55-modulated cell proliferation, migration and invasion. Administration of LY294002 (20 μM), wortmannin (5 μM) and MK2206 (1 μM) markedly decreased the proliferation of ESCC cells. The increased OD value of 450 nm was abrogated in CEP55-overexpressed EC9706 cells (P<0.001; Figure 5Ba). Similarly, the potential of migration (P<0.05; Figure 5Bb) and invasion (P<0.05; Figure 5Bc) was also prevented in Transwell assays. In addition, to confirm that CEP55 regulated the EMT process via the PI3K/Akt pathway, we detected whether cadherin switching (downregulation of E-cadherin and upregulation of N-cadherin) can be reversed by the blockade of the PI3K/Akt pathway. As shown in Figure 5Bd, cadherin switching was reversed by inhibition of the PI3K/Akt pathway, showing E-cadherin upregulation and N-cadherin downregulation. Taken together, CEP55 promoted proliferation, and enhanced migration and invasion by modulating EMT, at least partly, through activation of the PI3K/Akt pathway in ESCC cells.
Discussion
Despite the significant improvements in therapeutic regimens, surgical resection is considered the first-line treatment for early and localized ESCC.30 Ivor-Lewis esophagectomy with thoracic–abdominal two-field lymph node dissection has been widely accepted and commonly used due to its relatively lower surgical trauma and postoperative complications.31,32 pN0 ESCC patients in China are a particular entity; patients in this stage tend to accept primary surgery if the tumor can be completely resected. However, the prognosis of patients in this stage is not promising, and their prognostic tendencies vary greatly. Thus, there is a need to stratify individuals with pN0 ESCC based on their different prognoses and provide personalized treatment.
CEP55 was reported to be overexpressed in multiple cancers, such as head and neck squamous cell carcinoma, oral cavity squamous cell carcinoma, gastric cancer, breast cancer, lung cancer and pancreatic cancer.7–11,13–16 Additionally, CEP55 was elucidated to act as a promising prognostic predictor of malignant diseases. Chen et al11 reported that CEP55 overexpression may lead to a poor prognosis in patients with oral cavity squamous cell carcinoma. Additionally, the prognosis of nasopharyngeal squamous cell carcinoma was identified to be correlated with the CEP55/Akt pathway.33 Interestingly, in this study, we also found CEP55 overexpression in pN0 ESCC and identified similar significance of CEP55, showing that its overexpression can predict a lower 5-year OS and PFS of patients.
Increasing evidence has shown that CEP55 may promote the malignancy of multiple tumor cells.7–9,15,16 Coincidentally, we identified that CEP55 was overexpressed in ESCC cell lines and individuals with ESCC in this research. The proliferative, migratory and invasive abilities of cells may be altered with the changes in CEP55 expression.8,12,15 Additionally, in this research, we found that the downregulation of CEP55 attenuated the malignant abilities of ESCC cells, and upregulation of CEP55 promoted these malignant behaviors, with an increased OD value of 450 nm and more migrated and invaded cells. EMT is an important process of cancer metastasis, with 90% of tumors exhibiting different degrees of EMT during tumor development.34–36 During this process, the epithelial cells attenuate the expression of epithelial characteristics and gain mesenchymal phenotype properties.37,38 In this study, we surprisingly found that the downregulation of CEP55 suppressed EMT in ESCC cells, including the upregulation of epithelial markers (E-cadherin and Claudin-1) and downregulation of mesenchymal markers (N-cadherin, Vimentin and Snail). Additionally, the upregulation of CEP55 had the reverse effect on the expression levels of the EMT markers. Thus, CEP55 modulated the migration and invasion of ESCC cells and was correlated with the metastasis of ESCC. Zhang et al once reported that CEP55 may modulate EMT in epithelial ovarian cancer, a finding that was consistent with the result in this research.16
The PI3K/Akt pathway is one of the major pro-survival pathways.39 It plays an important role in regulating cell survival, cell cycle, angiogenesis, metastasis and metabolism.40–42 Several studies have demonstrated that the PI3K/Akt signaling pathway is activated in the progression of ESCC.43,44 Additionally, CEP55 was reported to bind and interact with PI3KCA (p110), and this complex may increase the activation of AktS473.13,45 CEP55 overexpression was elucidated to promote cell migration and invasion via PI3K/Akt in lung adenocarcinoma and hepatocellular carcinoma.13,15 In addition, the PI3K/Akt pathway was also demonstrated to play an important role in the process of the CEP55-mediated cell cycle in gastric cancer.9 In this study, we found that the activation of AktS473 and AktT308 was modulated by CEP55. Thus, CEP55 knockdown decreased the expression of pAktS473 and pAktT308 and CEP55 overexpression increased the activation of AktS473 and AktT308. We hypothesized that PI3K/Akt may participate in the CEP55-modulated biological behavior of ESCC cells. Next, LY294002, wortmannin and MK2206 were used to block the PI3K/Akt pathway. Consistent with our assumption, these inhibitors partly abolished the pro-proliferation, pro-migration and pro-invasion effects and reversed the EMT of CEP55 in ESCC. Taken together, CEP55 promoted proliferation, enhanced migration and invasion by modulating EMT, at least partly, through the PI3K/Akt pathway activation.
Previous studies have demonstrated that patients may benefit from postoperative adjuvant radiotherapy after Ivor-Lewis esophagectomy and two-field lymph node dissection.46,47 Additionally, in this study, we found that CEP55 may predict the poor prognosis of pN0 ESCC patients after surgery. Thus, we believe that postoperative adjuvant radiotherapy may be imposed on patients with CEP55 overexpression to act as a compensation mechanism to reduce locoregional lymphatic recurrence and, in return, improve the overall survival of patients. In addition, we identified that CEP55 promoted proliferation, migration and invasion via the PI3K/Akt pathway in ESCC cells. Thus, some novel drugs targeting CEP55 or the PI3K/Akt pathway may be promising treatment modalities.
This study was retrospective and had a limited sample size. Although we are the first to demonstrate the prognostic value of CEP55 in pN0 ESCC patients, these studies should be repeated with different parameters and prospectively. Multicenter randomized studies are also needed to confirm the prognostic value of CEP55.
Conclusion
CEP55 may serve as a promising predictive biomarker of pN0 ESCC patients after surgery; it can promote proliferation and enhance migration and invasion by modulating the EMT of ESCC cells via the PI3K/Akt pathway. Thus, some regimens, including postoperative adjuvant radiotherapy and target drugs, may serve as complementary treatments to improve the survival of patients with an otherwise poor prognosis.
Acknowledgment
This work was supported by the Natural Science Foundation of Shandong Province (ZR2017MH089).
Disclosure
The authors report no other conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. | ||
Napier KJ, Scheerer M, Misra S. Esophageal cancer: A Review of epidemiology, pathogenesis, staging workup and treatment modalities. World J Gastrointest Oncol. 2014;6(5):112–120. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol. 2006;24(14):2137–2150. | ||
Eloubeidi MA, Desmond R, Arguedas MR, Reed CE, Wilcox CM. Prognostic factors for the survival of patients with esophageal carcinoma in the U.S. Cancer. 2002;95(7):1434–1443. | ||
Jeffery J, Sinha D, Srihari S, Kalimutho M, Khanna KK. Beyond cytokinesis: the emerging roles of CEP55 in tumorigenesis. Oncogene. 2016;35(6):683–690. | ||
Peng T, Zhou W, Guo F, et al. Centrosomal protein 55 activates NF-κB signalling and promotes pancreatic cancer cells aggressiveness. Sci Rep. 2017;7(1):5925. | ||
Wang G, Liu M, Wang H, et al. Centrosomal Protein of 55 Regulates Glucose Metabolism, Proliferation and Apoptosis of Glioma Cells via the Akt/mTOR Signaling Pathway. J Cancer. 2016;7(11):1431–1440. | ||
Tao J, Zhi X, Tian Y, et al. CEP55 contributes to human gastric carcinoma by regulating cell proliferation. Tumour Biol. 2014;35(5):4389–4399. | ||
Waseem A, Ali M, Odell EW, Fortune F, Teh MT. Downstream targets of FOXM1: CEP55 and HELLS are cancer progression markers of head and neck squamous cell carcinoma. Oral Oncol. 2010;46(7):536–542. | ||
Chen CH, Chien CY, Huang CC, et al. Expression of FLJ10540 is correlated with aggressiveness of oral cavity squamous cell carcinoma by stimulating cell migration and invasion through increased FOXM1 and MMP-2 activity. Oncogene. 2009;28(30):2723–2737. | ||
Chen CH, Shiu LY, Su LJ, et al. FLJ10540 is associated with tumor progression in nasopharyngeal carcinomas and contributes to nasopharyngeal cell proliferation, and metastasis via osteopontin/CD44 pathway. J Transl Med. 2012;10:93. | ||
Chen CH, Lu PJ, Chen YC, et al. FLJ10540-elicited cell transformation is through the activation of PI3-kinase/AKT pathway. Oncogene. 2007;26(29):4272–4283. | ||
Wang Y, Jin T, Dai X, Xu J. Lentivirus-mediated knockdown of CEP55 suppresses cell proliferation of breast cancer cells. Biosci Trends. 2016;10(1):67–73. | ||
Chen CH, Lai JM, Chou TY, et al. VEGFA upregulates FLJ10540 and modulates migration and invasion of lung cancer via PI3K/AKT pathway. PLoS One. 2009;4(4):e5052. | ||
Zhang W, Niu C, He W, et al. Upregulation of centrosomal protein 55 is associated with unfavorable prognosis and tumor invasion in epithelial ovarian carcinoma. Tumour Biol. 2016;37(5):6239–6254. | ||
Coutant C, Rouzier R, Qi Y, et al. Distinct p53 gene signatures are needed to predict prognosis and response to chemotherapy in ER-positive and ER-negative breast cancers. Clin Cancer Res. 2011;17(8):2591–2601. | ||
Jones J, Otu H, Spentzos D, et al. Gene signatures of progression and metastasis in renal cell cancer. Clin Cancer Res. 2005;11(16):5730–5739. | ||
Hu Y, Wu G, Rusch M, et al. Integrated cross-species transcriptional network analysis of metastatic susceptibility. Proc Natl Acad Sci U S A. 2012;109(8):3184–3189. | ||
Cuzick J, Swanson GP, Fisher G, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol. 2011;12(3):245–255. | ||
Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. | ||
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–644. | ||
Mccubrey JA, Steelman LS, Chappell WH, et al. Advances in targeting signal transduction pathways. Oncotarget. 2012;3(12):1505–1521. | ||
Liu M, Hu Y, Zhang MF, et al. MMP1 promotes tumor growth and metastasis in esophageal squamous cell carcinoma. Cancer Lett. 2016;377(1):97–104. | ||
Nie ZC, Weng WH, Shang YS, et al. MicroRNA-126 is down-regulated in human esophageal squamous cell carcinoma and inhibits the proliferation and migration in EC109 cell via PI3K/AKT signaling pathway. Int J Clin Exp Pathol. 2015;8(5):4745–4754. | ||
Chen J, Lan T, Zhang W, et al. Platelet-activating factor receptor-mediated PI3K/AKT activation contributes to the malignant development of esophageal squamous cell carcinoma. Oncogene. 2015;34(40):5114–5127. | ||
Chen G, Wang Z, Liu XY, Liu FY. Adjuvant radiotherapy after modified Ivor-Lewis esophagectomy: can it prevent lymph node recurrence of the mid-thoracic esophageal carcinoma? Ann Thorac Surg. 2009;87(6):1697–1702. | ||
Jia Y, Zhang M, Jiang W, Zhang Z, Huang S, Wang Z. Overexpression of IFITM3 predicts the high risk of lymphatic metastatic recurrence in pN0 esophageal squamous cell carcinoma after Ivor-Lewis esophagectomy. PeerJ. 2015;3:e1355. | ||
Jiang W, Wang Z, Jia Y. CEP55 overexpression predicts poor prognosis in patients with locally advanced esophageal squamous cell carcinoma. Oncol Lett. 2017;13(1):236–242. | ||
Stahl M, Mariette C, Haustermans K, Cervantes A, Arnold D; ESMO Guidelines Working Group. Oesophageal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(Suppl 6):vi51–vi56. | ||
Nakagawa S, Kanda T, Kosugi S, Ohashi M, Suzuki T, Hatakeyama K. Recurrence pattern of squamous cell carcinoma of the thoracic esophagus after extended radical esophagectomy with three-field lymphadenectomy. J Am Coll Surg. 2004;198(2):205–211. | ||
Law S, Wong J. Two-field dissection is enough for esophageal cancer. Dis Esophagus. 2001;14(2):98–103. | ||
Hwang CF, Shiu LY, Su LJ, et al. Oncogenic fibulin-5 promotes nasopharyngeal carcinoma cell metastasis through the FLJ10540/AKT pathway and correlates with poor prognosis. PLoS One. 2013;8(12):e84218. | ||
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | ||
Nieto MA. Epithelial-Mesenchymal Transitions in development and disease: old views and new perspectives. Int J Dev Biol. 2009;53(8–10):1541–1547. | ||
Kahata K, Dadras MS, Moustakas A. TGF-β Family Signaling in Epithelial Differentiation and Epithelial-Mesenchymal Transition. Cold Spring Harb Perspect Biol. 2018;10(1):a022194. | ||
da Silva SD, Morand GB, Alobaid FA, et al. Epithelial-mesenchymal transition (EMT) markers have prognostic impact in multiple primary oral squamous cell carcinoma. Clin Exp Metastasis. 2015;32(1):55–63. | ||
Garside VC, Chang AC, Karsan A, Hoodless PA. Co-ordinating Notch, BMP, and TGF-β signaling during heart valve development. Cell Mol Life Sci. 2013;70(16):2899–2917. | ||
Hemmings BA, Restuccia DF. The PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2015;7(4):a026609. | ||
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. | ||
Martini M, de Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Ann Med. 2014;46(6):372–383. | ||
de Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16(Suppl 2):S17–S27. | ||
Yoshioka A, Miyata H, Doki Y, et al. The activation of Akt during preoperative chemotherapy for esophageal cancer correlates with poor prognosis. Oncol Rep. 2008;19(5):1099–1107. | ||
Liu M, Hu Y, Zhang MF, et al. MMP1 promotes tumor growth and metastasis in esophageal squamous cell carcinoma. Cancer Lett. 2016;377(1):97–104. | ||
Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Curr Cancer Drug Targets. 2013;13(3):234–244. | ||
Chen G, Wang Z, Liu XY, Zhang MY, Liu FY. Clinical study of modified Ivor-Lewis esophagectomy plus adjuvant radiotherapy for local control of stage IIA squamous cell carcinoma in the mid-thoracic esophagus. Eur J Cardiothorac Surg. 2009;35(1):1–7. | ||
Yu Y, Wang Z, Yang Z, Liu XY. Therapeutic efficacy evaluation of postoperative adjuvant radiotherapy in mid-thoracic esophageal carcinoma patients underwent Ivor Lewis esophagectomy with two-field lymphadenectomy. Med Oncol. 2015;32(2):348. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.