Back to Journals » Drug Design, Development and Therapy » Volume 8
CCL19 and CCL21 modulate the inflammatory milieu in atherosclerotic lesions
Authors Akhavanpoor M, Gleissner CA, Gorbatsch S, Doesch A, Akhavanpoor H, Wangler S, Jahn F, Lasitschka F, Katus HA, Erbel C
Received 7 August 2014
Accepted for publication 10 September 2014
Published 27 November 2014 Volume 2014:8 Pages 2359—2371
DOI https://doi.org/10.2147/DDDT.S72394
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Mohammadreza Akhavanpoor,1,2,* Christian A Gleissner,1,2,* Stephanie Gorbatsch,1,2 Andreas O Doesch,1,2 Hamidreza Akhavanpoor,1,2 Susanne Wangler,1,2 Frederik Jahn,1,2 Felix Lasitschka,3 Hugo A Katus,1,2 Christian Erbel1,2
1Department of Cardiology, University of Heidelberg, 2DZHK (German Centre for Cardiovascular Research), partner site Heidelberg/Mannheim, 3Institute of Pathology, University of Heidelberg, Germany
*These authors contributed equally to this work
Abstract: Despite advances in the pharmacologic and interventional treatment of coronary artery disease, atherosclerosis remains the leading cause of death worldwide. Atherosclerosis is a chronic inflammatory disease, and elevated expression of CCL19 and CCL21 has been observed in ruptured lesions of coronary arteries of patients with myocardial infarction and carotid plaques of patients with ischemic symptoms, as well as in plasma of coronary artery disease patients. However, the exact role of CCL19 and CCL21 in atherosclerosis remains unknown. In order to identify CCL19 and CCL21 as a novel therapeutic target, we performed bone marrow transplantation as an immunomodulatory treatment concept. Bone marrow of plt/plt mice (lacking CCL19 and CCL21-Ser) was transplanted into atherogenic Ldlr-/- mice. The study demonstrated a significantly increased inflammatory cellular infiltration into the lesions of plt/plt/Ldlr-/- mice versus controls. Although the level of chemoattraction was increased, messenger ribonucleic acid and protein levels in thoracic aorta and serum of several proinflammatory cytokines (TNFα, IFNγ, IL-6, IL-12, and IL-17) were significantly reduced in plt/plt/Ldlr-/- versus control mice. Increased influx, accompanied by reduced activation of leukocytes in atherosclerotic lesion, was accompanied by increased plaque stability but unchanged lesion development. In conclusion, modulation of the chemokines CCL19 and CCL21 represents a potent immunoregulatory treatment approach, and thus represents a novel therapeutic target to stabilize atherosclerotic lesions.
Keywords: atherosclerosis, chemokines, immunomodulatory therapy, bone marrow transplantation
Introduction
Atherosclerosis and its complications are still the leading cause of death worldwide.1,2 Acute coronary syndrome, one of the major complications of atherosclerosis, is caused by the rupture of vulnerable lesions, leading to atherothrombosis and vessel occlusion.3 Today, atherosclerosis is believed to be a chronic inflammatory disease, involving innate and adaptive immunity.2,4–8 Despite significant progress in the treatment of acute myocardial infarction, effective prevention strategies against atherosclerosis are still lacking. Therefore, for a better understanding of the pathophysiology in atherogenesis and for the identification of novel therapeutic targets and development of novel therapies, studying lesional biology is of great importance.
The migration and recruitment of leukocytes by different chemokines promote all stages of atherosclerosis, including plaque vulnerability, leading to myocardial infarction or stroke.9,10 CCL19 and CCL21 and their common receptor, CCR7, are involved in different autoimmune diseases, such as rheumatoid arthritis and bowel disease.11,12 Stromal cells in T-cell zones of lymph nodes and spleen mainly express CCL19.13,14 CCL21 is mainly expressed by high endothelial venules in lymph nodes and endothelial cells of lymphoid tissues in several organs.15 Mice possess two different CCL21 molecules: CCL21-Ser and CCL21-Leu. Both isoforms differ in the amino acids serine (CCL21-Ser) and leucine (CCL21-Leu) at position 65.16,17 CCL21-Ser is expressed in secondary lymphoid organs, whereas CCL21-Leu is mainly expressed in the lymphatic endothelium of nonlymphatic tissues.18
Spontaneous mutant mouse strain plt/plt mice lack the expression of CCL19 and CCL21-Ser in secondary lymphoid organs due to a deletion of the genes encoding for CCL19 and CCL21-Ser.19 Interestingly, plt/plt mice still express CCL21-Leu in nonlymphoid organs, allowing the mobilization of leukocytes in the periphery.20 Due to these genetic changes plt/plt mice reveal a dysregulation in leukocytes trafficking to lymphoid tissues.14 In spite of these defects, plt/plt mice are capable of inducing immune responses.18,21
Recent studies suggest a potential role of CCL19 and CCL21 in atherogenesis. In 2007, our group demonstrated expression of CCL19 and CCL21 in human carotid artery and coronary artery plaques.7 The expression of these chemokines was significantly increased in unstable atherosclerotic lesions and in coronary artery lesions leading to myocardial infarction, suggesting a crucial role in plaque progression, instability, and rupture.7 In line with our findings, Damås et al also showed expression of CCL19 and CCL21 in atherosclerotic lesions of Apoe−/− mice, in human carotid lesions, and in plasma of coronary artery disease patients.22 In patients with unstable angina pectoris, the plasma levels of CCL19 and CCL21 were significantly higher than patients with stable angina pectoris and healthy control patients.22 More recently, Halvorsen et al revealed significantly increased expression levels of CCL19 and CCL21 in carotid plaques of symptomatic patients compared to asymptomatic patients.23
To evaluate CCL19 and CCL21 further as a novel therapeutic target in atherogenesis, we performed bone marrow transplantation of plt/plt mice into Ldlr−/− mice, investigating the effects of both chemokines on the inflammatory process in atherosclerotic lesions.
Materials and methods
Animals
Male Ldlr−/− mice on a C57BL/6J background and 6–8 weeks of age were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Male plt/plt mice 6–8 weeks of age on a C57BL/6J background (kindly obtained by R Foerster, Institute of Immunology, Hannover Medical School, Germany and H Nakano, Laboratory of Respiratory Biology, National Institute of Environmental Health Science, NC, USA) were kept within the animal-care facility of the University of Heidelberg, whereas housing and care of animals and all the procedures done in the study were in accordance with the guidelines and regulations of the local Animal Care Committee (Institutional Review Board approval AZ 35-9185.81/G222/12).
To study the effects of the chemokines CCL19 and CCL21, 18 Ldlr−/− mice were irradiated with 9.5 Gy followed by bone marrow transplantation (1×106 cells) within 24 hours, either of plt/plt mice on a C57BL/6J background (plt/plt/Ldlr−/−, therapy group, n=9), or of C57BL/6J mice (C57BL/6/Ldlr−/−, control group, n=9). Four weeks after bone marrow transplantation, animals were fed a Western-type diet with 21% milk fat, 0.2% cholesterol, and metabolic energy of 4,566 kcal/kg (Altromin, Germany) for an additional 14 weeks. Since it is known that irradiation decelerates the development of atherosclerotic lesions, we prolonged the observational period up to 14 weeks. Eighteen weeks after bone marrow transplantation, the aortic root was dissected and embedded in optical coherence tomography for immunohistochemistry, the thoracic aorta snap-frozen for gene-expression analysis, and blood serum stored at −20°C for enzyme-linked immunosorbent assay (ELISA).
Determination of plasma lipid concentration and plasma cell composition
Total serum cholesterol, high-density lipoprotein (HDL) cholesterol, low-density lipoprotein (LDL) cholesterol, triglycerides, and blood count were analyzed at the Department of Clinical Chemistry of the University of Heidelberg with a standard method.
Tissue processing
For ribonucleic acid (RNA) isolation, the aortic arch and thoracic aorta were dissected and snap-frozen as described previously.24 The entire aortic root, a predilection site for lesion development in atherogenic mice,25 was serially cryosectioned at 5 μm for immunohistochemistry as described previously.24
Quantitative real-time polymerase chain reaction
Total RNA was isolated from the thoracic aorta using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and the Fermentas First Strand cDNA [complementary deoxyribonucleic acid] Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturers’ instructions. For quantitative real-time polymerase chain reaction (qPCR), Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA), Fermentas dNTP Mix (Thermo Fisher Scientific), and SYBR green/nucleic acid gel stain (Lonza, Basel, Switzerland) were used. The relative expression method was used for analyzing data as described previously.24 For primer sequences, refer to Table 1.
 | Table 1 Primer sequences, including forward (FW) and reverse (RV) sequences and optimal temperatures used for quantitative real-time polymerase chain reaction |
Immunohistochemistry
Sections were fixed in acetone, air-dried, and incubated with primary antibodies as described previously.24,26 For immunohistochemistry the following primary antibodies were used and titrated to optimum performance: α-smooth muscle (SM) actin for detection of SM cells (Dianova, Hamburg, Germany), B220 for B cells (R&D Systems, Minneapolis, MN, USA), CD3 for T-cells (BD, San Jose, CA, USA), MAC-2 for macrophages (Accurate, Westbury, NY, USA). Isotype- and concentration-matched rat control monoclonal antibodies as well as normal Armenian hamster serum (both Dianova, eBioscience) served as negative controls.
Immunoenzyme stainings were performed on 5 μm cryosections using standard avidin–biotin anti-alkaline phosphatase (for B220; Vector Laboratories, Burlingame, CA, USA) and ZyMax™ horseradish peroxidase–streptavidin conjugate (for α-SM actin, CD3, and MAC2; Invitrogen) according to the manufacturer’s instructions. Goat antirat biotin (R&D Systems; for B220), Biotin goat antirat IgG (H + L) conjugate (Invitrogen; for MAC2), biotin goat antirat IgG (BD; for CD3), polyclonal swine antigoat, mouse, and rabbit Ig/biotinylated (Dako, Glostrup, Denmark; for α-SM actin) were used as a secondary reagent (30 minutes at room temperature).
To quantify the atherosclerotic lesions, all Oil Red O-stained sections were examined by microscopy, and subsequently the lumen, lesion area, and degree of stenosis were analyzed by using imaging software (NIS-Elements AR; Nikon). We calculated the lesion area by taking the mean of the surface of the lesion of each section of each mouse (μm2). Fractional stenosis was calculated by dividing the surface of the lesion by the surface of the vessel and multiplying by 100 (%). Maximum stenosis was analyzed by dividing the vessel area including the lesion area by the vessel area without the lesion area and multiplying by 100 (%). Movat’s pentachrome staining was performed to illustrate plaque composition. Detection of apoptosis was performed using an in situ cell-death detection kit (Roche, Mannheim, Germany) according to the manufacturer’s protocols.
Multiplex ELISA
To perform quantitative analysis of different cytokines and chemokines, a multiplex analysis kit (Quansys Biosciences, USA) was used according to the manufacturer’s protocols.
Flow cytometry
For fluorescence-activated cell sorting (FACS) analysis, cells were isolated from lymph nodes (n=9/group) and spleens of mice (n=5/group) and labeled with specific antibodies for detection of different cells, cell subtypes, and the grade of cellular activation: T-cells (CD3, CD4, CD8, CD44), B cells (CD19, CD25), dendritic cells (DCs; CD11c, CD83, CD86), and monocytes (CD62L, CD11b, Ly6C). Flow cytometry was performed as described previously.24,26
Stimulation of macrophages
Murine macrophages (RAW cells, 264.7; Cell Line Service, Germany) were obtained in Roswell Park Memorial Institute 1640 culture medium. The culture medium was supplemented with 10% fetal bovine serum, penicillin G (100 U/mL), and streptomycin (100 μg/mL). Cells were maintained at 37°C in humidified air containing 5% CO2. RAW cells were stimulated with 100 ng CCL19 or 100 ng CCL21 for 2 hours and 8 hours. Unstimulated RAW cells served as controls. Cells were harvested at the end of the experimental period and subjected to qPCR analysis as described earlier.
Oxidized low-density lipoprotein-induced foam-cell formation
Human peripheral blood was obtained from healthy volunteers with approval from the institutional review board as described previously.27 Peripheral blood mononucleated cells were isolated from human peripheral blood using Histopaque (Sigma, Germany) following negative isolation with magnetic beads (Stem Cell, Canada), yielding >95% CD14+ monocytes. Cells were cultured in macrophage serum-free medium (Life Technologies, USA) supplemented with Nutridoma SP (Roche, Germany) and penicillin/streptomycin (Sigma, Germany) for 6 days in the presence of 100 ng/mL recombinant human macrophage colony-stimulating factor (Peprotech, USA). Macrophages were exposed to 15 μg/mL oxidized LDL (oxLDL) (Hycultec, Germany) in addition to 100 ng/mL CCL19 or 100 ng/mL CCL21 for 24 hours. The experiment was repeated five times. After washing the cells, we performed Oil Red O staining. For Oil Red O-staining analysis, we used the program ImageJ and the plugin colored convolution for ImageJ (National Institutes of Health). We measured the positively stained area in relation to the total area of the cells for each group. Untreated macrophages served as negative controls.
Statistical analysis
The nonparametric Mann–Whitney U-test was used to compare the treatment group to control groups. A P-value <0.05 was considered statistically significant. Data are presented as means ± standard deviation or as box plots displaying median and 25th and 75th percentiles as boxes and whiskers representing 10th and 90th percentiles.
Results
Animal and blood analysis
No animals died during the in vivo studies. No growth retardation was seen during the study period. On the day of tissue harvesting, there were no significant differences in body weight and height of mice, serum concentrations of total cholesterol, HDL and LDL cholesterol, triglycerides, or whole blood counts between plt/plt/Ldlr−/− mice and the control group (Table 2).
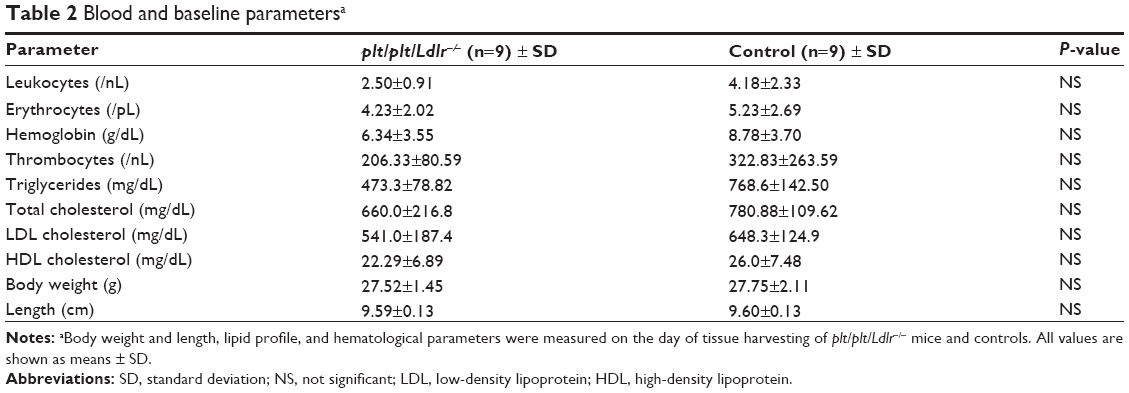 | Table 2 Blood and baseline parametersa |
Gene-expression levels of Ccl19, Ccl21-Ser, Ccl21-Leu, and Ccr7 in thoracic aorta
In order to confirm the success of the bone marrow transplantation, we quantified gene-expression levels of Ccl19, Ccl21-Ser, and Ccl21-Leu in the thoracic aorta by qPCR analysis. Ccl19 was found to be significantly reduced within the thoracic aorta of plt/plt/Ldlr−/− compared to control mice (P=0.0031, Table 3). Interestingly, Ccl21-Ser expression was clearly downregulated accordingly (P=0.0002, Table 3), whereas Ccl21-Leu was upregulated in plt/plt/Ldlr−/− mice compared to control mice (P=0.0002, Table 3). The expression level of Ccr7, the receptor of CCL19 and CCL21, was significantly upregulated in the thoracic aorta of plt/plt/Ldlr−/− mice compared to control mice (P=0.0001, Table 3).
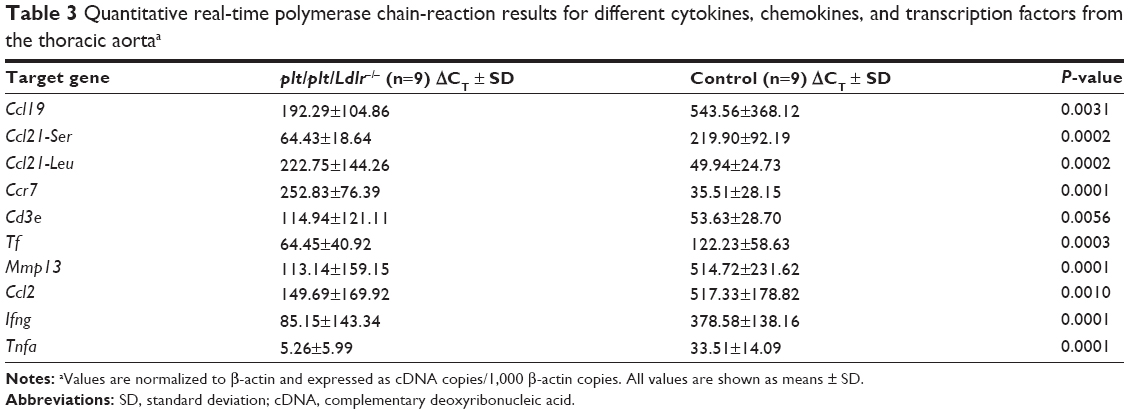 | Table 3 Quantitative real-time polymerase chain-reaction results for different cytokines, chemokines, and transcription factors from the thoracic aortaa |
No changes in plaque burden but increased plaque stability in plt/plt/Ldlr−/− mice
The development of atherosclerotic lesions was analyzed by immunohistochemistry staining with Oil Red O of the aortic root. Representative immunostaining photographs are shown in Figure 1A. There were no significant influences on lesion size, fractional stenosis, or maximum stenosis rate between the therapy and control groups (Figure 1B).
 | Figure 1 No changes on plaque burden in plt/plt/Ldlr−/− mice. |
To illustrate effects of our therapy on plaque stability, α-SM actin staining for identification of vascular SM cells in atherosclerotic lesion as well as Movat’s pentachrome for evaluation of collagen and elastin content in plaque were performed. Representative photomicrographs of the aortic root are shown in Figure 2, A–D. There were no significant differences in collagen and elastin content between both groups, but vascular SM cells were significantly increased in the whole atherosclerotic lesions (P=0.0401) and especially in the region of the fibrous cap (P=0.0387) of plt/plt/Ldlr−/− mice compared to the control group (Figure 2, A, B, E, and F). By qPCR analysis of the thoracic aorta, we also found that the plaque-destabilizing Mmp13 and the prothrombotic Tf were significantly reduced in plt/plt/Ldlr−/− mice (Mmp13, P=0.0001; Tf, P=0.0003; Table 3). In order to investigate apoptosis within atherosclerotic lesions, terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick-end labeling/4′,6-diamidino-2-phenylindole staining was performed. However, the number of apoptotic cells was too low for statistical analysis in both groups (data not shown).
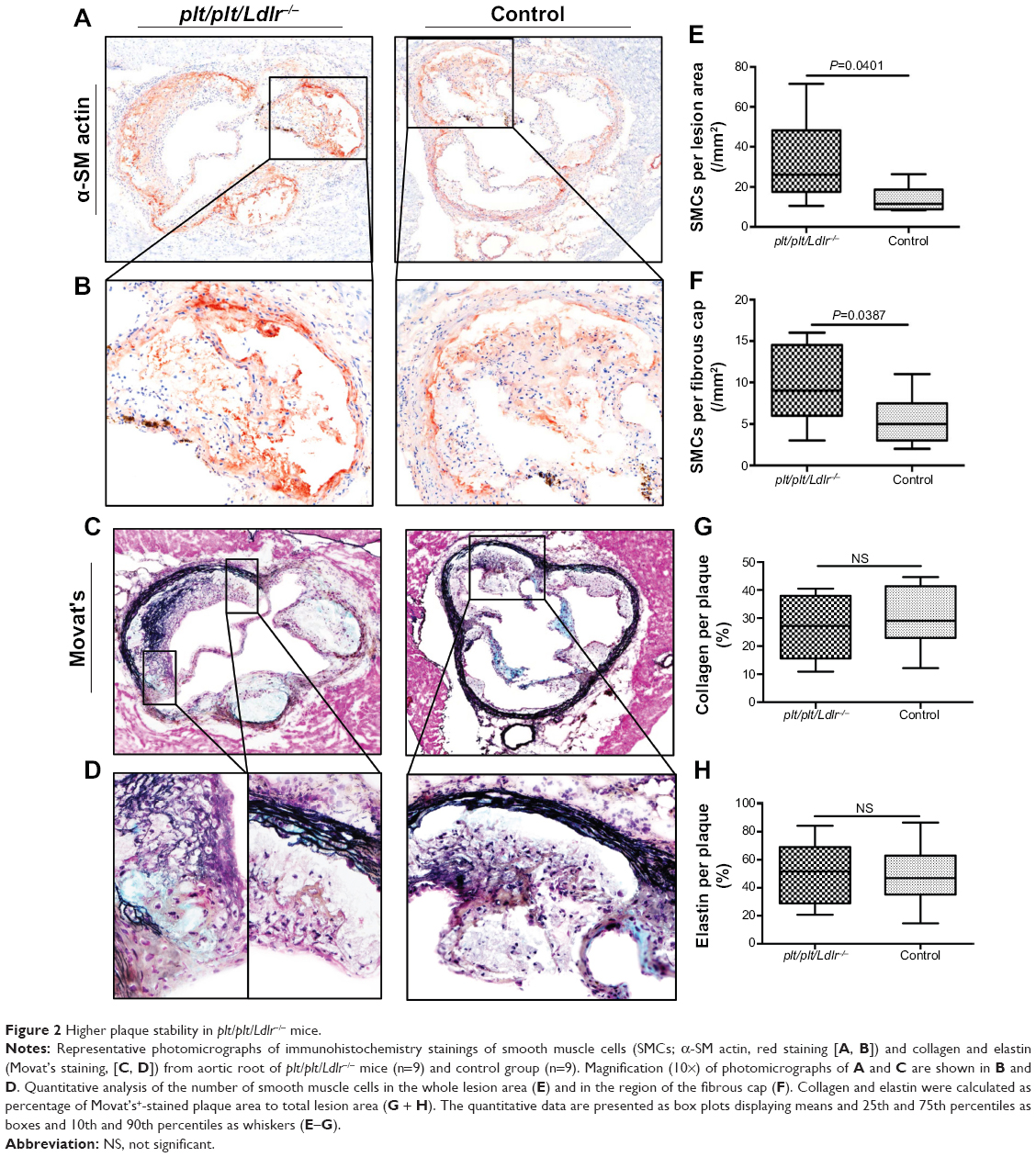 | Figure 2 Higher plaque stability in plt/plt/Ldlr−/− mice. |
Increased leukocyte infiltration into atherosclerotic lesions of plt/plt/Ldlr−/− mice
Next we analyzed the cellular infiltration of leukocytes in atherosclerotic lesion by immunohistochemistry staining for T-cells, B cells, and macrophages. Immunohistochemistry staining for CD3+ T-cells showed a significantly increased proportion of CD3+ T-cells (cell number/plaque area) within atherosclerotic lesions of plt/plt/Ldlr−/− mice compared to the control group (P=0.0066; Figure 3, A, and D). qPCR analysis of the thoracic aortas of mice confirmed the immunohistological results. Tissue levels of cDNA transcripts for Cd3e, part of the CD3 receptor on T-cells, were distinctly higher in the thoracic aorta of plt/plt/Ldlr−/− than in control mice (P=0.0056, Table 3). In order to identify macrophages within the lesion, we stained for MAC2+ macrophages. The proportion of macrophages (cell number/plaque area) was also significantly increased in plt/plt/Ldlr−/− mice (P=0.0043; Figure 3, B, and D). Immunohistochemistry staining for B220+ B cells showed a significantly higher amount of B cells within atherosclerotic lesions of plt/plt/Ldlr−/− mice compared to control mice (P=0.0168; Figure 3, C and D). Taken together, these results show that leukocyte recruitment into atherosclerotic lesions was significantly upregulated in plt/plt/Ldlr−/− mice.
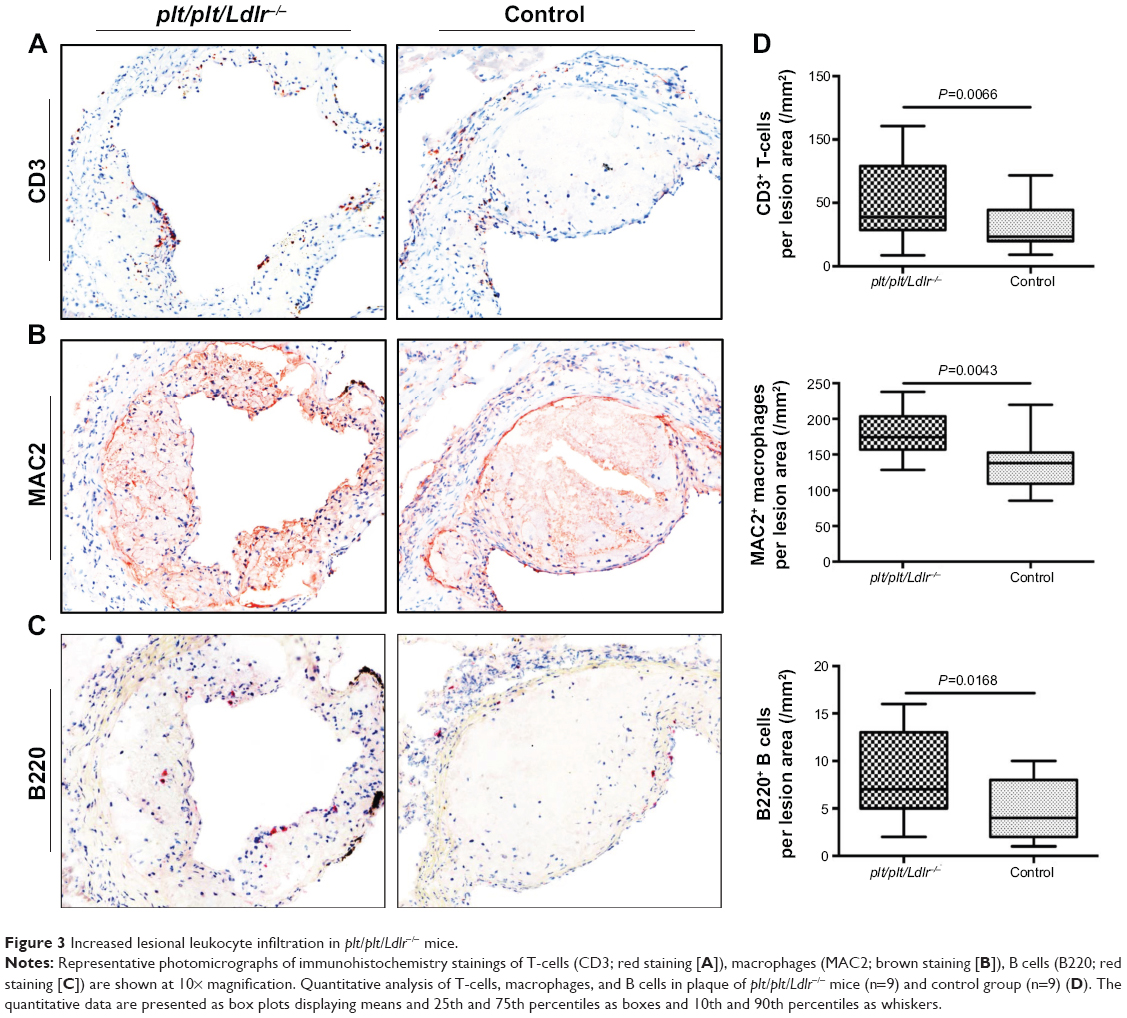 | Figure 3 Increased lesional leukocyte infiltration in plt/plt/Ldlr−/− mice. |
Subsequently, we measured the serum levels of several CC chemokines, which are mainly involved in the recruitment of leukocytes into atherosclerotic lesions. Interestingly, the expression of CCL2 (MCP-1) was significantly decreased in the thoracic aortas of plt/plt/Ldlr−/− mice (P=0.001, Table 3), which corresponded to significantly reduced CCL2 serum levels in plt/plt/Ldlr−/− mice (P=0.0111, Table 4). By contrast, serum levels of CCL3 (P=0.0401) and CCL5 (P=0.0055) were significantly higher in plt/plt/Ldlr−/− mice (Table 4).
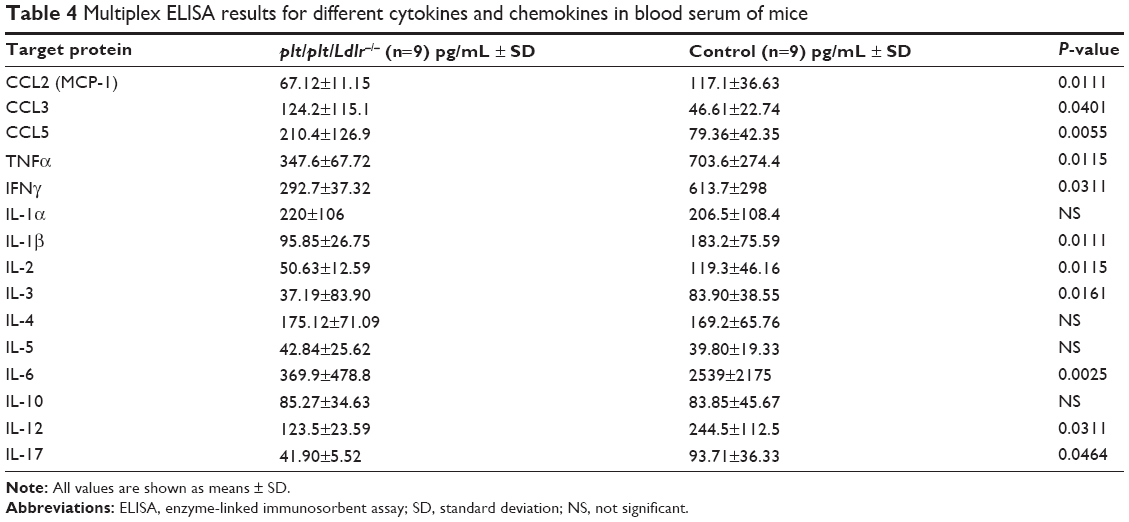 | Table 4 Multiplex ELISA results for different cytokines and chemokines in blood serum of mice |
Activation of leukocytes in the thoracic aorta and blood serum of plt/plt/Ldlr−/− mice
In order to investigate local and systemic effects of CCL19 and CCL21 on proinflammatory activation, we performed qPCR analysis of the thoracic aorta for gene-expression analysis of different proinflammatory cytokines and ELISA analysis for proinflammatory cytokines in the serum of the mice. In the thoracic aorta of the plt/plt/Ldlr−/− mice, there was a significant decrease in the proinflammatory cytokines Tnfa and Ifng compared to the control group (Table 3). In parallel, TNFα and IFNγ levels in the serum of plt/plt/Ldlr−/− mice were also significantly downregulated (Table 4). Interestingly, some other proinflammatory cytokines, such as IL-1β, IL-2, IL-3, IL-6, IL-12, and IL-17, showed significantly reduced serum levels (Table 4).
Since TNFα and IFNγ are the most important proinflammatory cytokines mainly expressed by macrophages in atherosclerotic lesions, we focused on these two cytokines and analyzed the effect of CCL19 and CCL21 on RAW cells, a murine macrophage cell line. CCL19 induced a significant upregulation of the expression of both cytokines TNFα and IFNγ by RAW cells (Figure 4, A and B), whereas CCL21 did not affect expression of either cytokine (Figure 4, C and D). Taken together, our data indicate that the reduced grade of cellular proinflammatory activation may be a consequence of the reduced CCL19 expression in the plt/plt/Ldlr−/− mice.
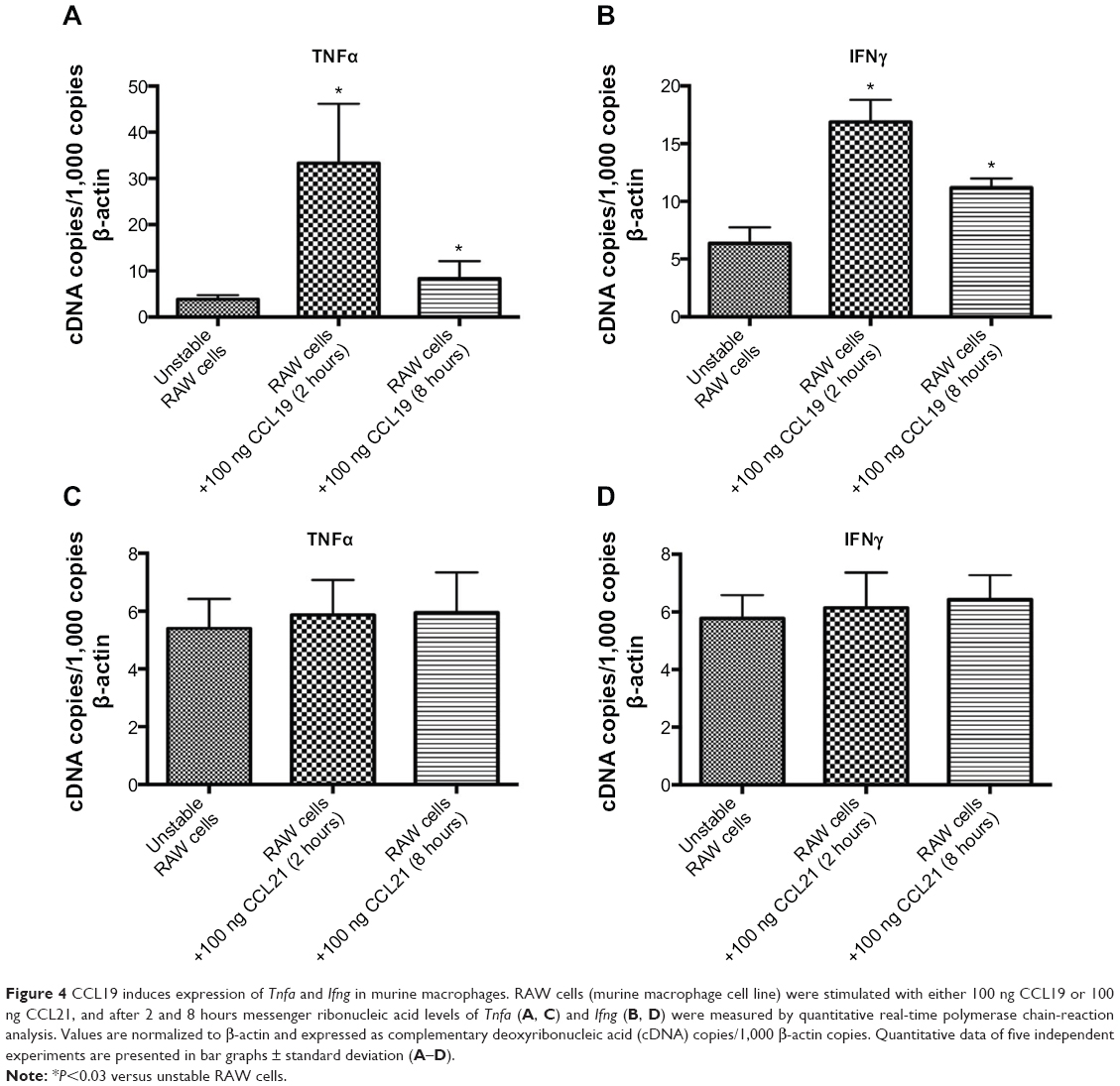 | Figure 4 CCL19 induces expression of Tnfa and Ifng in murine macrophages. RAW cells (murine macrophage cell line) were stimulated with either 100 ng CCL19 or 100 ng CCL21, and after 2 and 8 hours messenger ribonucleic acid levels of Tnfa (A, C) and Ifng (B, D) were measured by quantitative real-time polymerase chain-reaction analysis. Values are normalized to β-actin and expressed as complementary deoxyribonucleic acid (cDNA) copies/1,000 β-actin copies. Quantitative data of five independent experiments are presented in bar graphs ± standard deviation (A–D). |
Changes in the cellular composition of lymph nodes and spleens in plt/plt/Ldlr−/− mice
To understand whether the increased influx of leukocytes into atherosclerotic lesions was due to an upregulated chemoattraction or an altered lymphocyte homing, we analyzed the cellular composition of the lymph nodes and spleens by flow cytometry. The density of different T-cell subsets (CD3+, CD4+, CD8+, and CD4+CD8+), B cells, and DCs in lymph nodes and spleens was not different between the groups (Table 5). However, CD4+CD44+-activated T-cells were significantly reduced in lymph nodes of the plt/plt/Ldlr–/– versus the control group (P=0.0163, Table 5). There were no significant changes in activated B cells or DCs (Table 5). FACS analysis for different monocyte subsets in lymph nodes of mice based on CD11b and Ly-6C expression (CD11b+Ly-6Chi, CD11b+Ly-6Cmid, and CD11b+Ly-6Clow) did not differ between both groups (data not shown). Similar results were seen in spleens of the mice (data not shown).
 | Table 5 Cellular composition of peripheral lymph nodes and spleena |
CCL19 promotes oxLDL uptake and foam-cell formation via CD36 expression on monocyte-derived macrophages
Given the important role of leukocytes, especially macrophages in plaque formation and development, plt/plt/Ldlr−/− mice may result in large plaque volumes due to an increased infiltration of T and B cells and macrophages. However, plaque burden was not different compared to control mice. To further investigate this finding, we focused on macrophages, which are the most prevalent cell type in atherosclerotic lesions,28 and analyzed the average size of macrophages within the atherosclerotic lesions. The average size of macrophages was significantly smaller in plt/plt/Ldlr−/− mice compared to control mice (P<0.0001; Figure 5, A and B). Since the size of macrophages in atherosclerotic lesions is dependent on the grade of oxLDL uptake and foam-cell formation, we analyzed the effect of chemokines CCL19 and CCL21 on human monocyte-derived macrophages. Interestingly, stimulation with CCL19 and not CCL21 (data not shown) led to a significant increase of foam-cell formation in monocyte-derived macrophages (Figure 5, C and D). Moreover, FACS analysis demonstrated a significant increase of CD36 expression on macrophages as the underlying mechanism for the increased oxLDL uptake after stimulation with CCL19 (Figure 5E). Taken together, these results indicate that CCL19 can promote foam-cell formation in macrophages, and thereby contribute to the formation and development of atherosclerotic lesions.
 | Figure 5 Lipid-loaded macrophages in atherosclerotic lesions and effects of CCL19 on oxidized low-density lipoprotein (oxLDL) uptake and foam-cell formation. Representative photomicrographs of immunohistochemistry stainings of macrophages (MAC2; brown staining) from aortic roots of plt/plt/Ldlr−/− mice (n=9) and control group (n=9) are shown at 4× magnification. Magnification (20×) shown below the images (black arrows point at macrophages) (A). Quantitative analysis of the size of lesional macrophages (μm2) is demonstrated. Results are presented as box plots displaying mean and 25th and 75th percentiles as boxes and 10th and 90th percentiles as whiskers (B). Effects of CCL19 on foam-cell formation (C, D). Representative Oil Red O staining of human monocyte-derived macrophages are shown. Groups: control (no oxLDL), oxLDL, and oxLDL plus CCL19 (C). Quantitative analysis of the Oil Red O-positive cell area/cell (oxLDL/cell [%]). Data are demonstrated as bar graphs ± standard deviation of five independent experiments (D). Quantitative analysis of five independent fluorescence-activated cell-sorting experiments for CD36 expression on the surface of human monocyte-derived macrophages after stimulation with 1,1′-dioctadecyl-3,3,3′3′-tetra-methylindocyanide percholorate (Dil)-labeled oxLDL or Dil-labeled oxLDL in addition to CCL19 or unstimulated macrophages (control). Data are presented as bar graphs ± standard deviation (E). |
Discussion
In the past few decades, significant improvements in the therapy of atherosclerosis, including novel pharmacological and interventional treatments,29 have been achieved. However, atherosclerosis remains the leading cause of death worldwide.30–32 Inflammation as the underlying cause and primarily the impact of plaque macrophages in all stages of atherosclerosis are well accepted.24,33–36 The secretion of metalloproteinases and collagenases by lesional macrophages can cause destabilization and rupture of atherosclerotic lesions, followed by a vessel occlusion, resulting in myocardial infarction or stroke.37,38
In the current study, we undertook a novel therapeutic approach by transplanting the bone marrow of plt/plt mice (lacking expression of CCL19 and CCL21-Ser) into Ldlr−/− mice. We were able to downregulate the expression of CCL19 and CCL21-Ser in the thoracic aorta of the plt/plt/Ldlr−/− mice; there was a compensatory upregulation of CCL21-Leu and its receptor – CCR7. This imbalance of the chemokines together with CCR7 led to significantly increased inflammatory cellular infiltration into the lesions of plt/plt/Ldlr−/− mice. Simultaneously, messenger RNA and protein levels in thoracic aorta and serum of several proinflammatory cytokines were significantly reduced in plt/plt/Ldlr−/− due to CCL19 downregulation. The reduced activation of leukocytes led to increased plaque stability, despite increased influx of leukocytes in atherosclerotic lesions.
CCL21 plays a crucial part in the recruitment of cells expressing its receptor, CCR7, to periphery lymphoid tissues,39 as well as in the recruitment of leukocytes in different peripheral nonlymphoid tissue, such as pancreatic islets,40,41 and in chronically inflamed synovial tissue.42 The chemotactic capacity of CCL21 seems to be more effective than CCL19,40,43,44 whereas CCL19 is a strong inducer of cellular activation and proliferation for DCs and T-cells.45,20 Plt/plt mice lack the expression of CCL19 and CCL21-Ser in secondary lymphoid organs due to a deletion of the genes encoding for CCL19 and CCL21-Ser, but still express CCL21-Leu in nonlymphoid organs.19,41 Both forms of CCL21 (CCL21-Ser and CCL21-Leu) show biologically equivalent effects.41 CCL19 and CCL21 bind to its receptor – CCR7. The receptor is expressed by various subsets of immune cells like B cells, T-cells, and DCs.20 CCR7 and its ligands are essentially involved in the regulation of immune-cell trafficking between peripheral tissues and lymphoid organs.39,46 In our study, the bone marrow transplantation resulted in an increase of CCL21-Leu and CCR7, as well as a decrease of CCL19- and CCL21-Ser-expression levels in the thoracic aortas of the treated mice. We believe that the upregulation of CCL21-Leu and CCR7 is a compensatory effect for the downregulation of CCL19 and CCL21-Ser. These changes resulted in an increased cellular influx in atherosclerotic lesions in plt/plt/Ldlr−/− versus control mice. In contrast, the grade of activation of leukocytes, such as T-cells, B cells, and macrophages, in the lesions was markedly downregulated in plt/plt/Ldlr−/− mice versus controls. Analysis of the cellular composition of lymph nodes and spleens of mice did not show significant difference between the mouse groups. Therefore, the increased leukocyte infiltration into atherosclerotic lesions was due to an upregulated chemoattraction, partly by CCL21-Leu and CCR7 upregulation, and not an altered lymphocyte homing.
CCL19 can induce the activation of T cells and DCs.45,20 Since macrophages represent the most prevalent cell type in atherosclerotic lesions and given their important role in atherogenesis, we investigated the influence of CCL19 and CCL21 on macrophages. Murine macrophages were stimulated with both chemokines. CCL19 was able to activate murine macrophages by inducing the expression of TNFα and IFNγ, whereas CCL21 showed no effects on these cells. Therefore, CCL19 may have a significant role in cellular activation, especially of macrophages in atherosclerotic lesions, whereas CCL21 may have strong chemotactic but no relevant effects on activation of leukocytes.
Our study found no difference in plaque development between the groups, but increased plaque stability in plt/plt/Ldlr−/− mice. Owing to the increased infiltration of leukocytes into the plaques of plt/plt/Ldlr−/− mice, we expected increased plaque development and instability. One possible reason for the lack of reduced plaque burden and at the same time increased lesion stability may have been the reduced proinflammatory activation of leukocytes, especially macrophages, due to the downregulation of CCL19 in plt/plt/Ldlr−/− mice. Another possible reason could be associated with lesional macrophages. We found a significant reduction in macrophage size in atherosclerotic lesions of plt/plt/Ldlr−/− mice compared to the control group. Our in vitro data extended the finding by showing an important role of CCL19 on oxLDL uptake and foam-cell formation in macrophages. The increased oxLDL uptake was due to an upregulation of the CD36 receptor on the surface of macrophages. In vivo, the downregulation of CCL19 resulted in a decreased uptake of oxLDL in the lesional macrophages of plt/plt/Ldlr−/− mice, resulting in smaller lesional macrophages. Although there were more macrophages in the lesions, plaque burden remained unchanged because of the reduced size of the macrophages. Therefore, the inhibitory effects on macrophages were compensated by an increase cellular influx in the lesion and led to an unchanged plaque volume, but upregulated plaque stability between the groups. This upregulated plaque stability in plt/plt/Ldlr−/− mice can be explained by the reduced proinflammatory activation of lesional macrophages.
In conclusion, by this immunomodulatory approach we revealed that both chemokines CCL19 and CCL21 and their receptor, CCR7, modulate the inflammatory processes in atherosclerotic lesions. CCL21-Leu coordinates the chemoattraction of leukocytes into atherosclerotic lesions, whereas CCL19 influences the activation of leukocytes, lipid uptake of macrophages, and foam-cell formation. Immunomodulatory modification of both chemokines represents a potential new therapeutic target in the treatment of atherosclerosis.
Acknowledgments
We greatly appreciate the expert technical assistance of Nadine Wambsganss and Jutta Scheuerer. This work was supported by the German Research Foundation (DFG) to CE (ER 682/2-1) and FL (SFB 938/TP Z2), by the German Heart Foundation/German Foundation of Heart Research to CE (F/23/12), the German Centre for Cardiovascular Research (DZHK) to HAK, and by the German Ministry of Education and Research (BMBF) to HAK.
Disclosure
The authors report no conflicts of interest in this work. The abstract of this paper was presented at the 120. DGIM Conference 2014 as a poster presentation with interim findings. The poster’s abstract was published in “Poster abstracts” in the journal Der Internist (2014;55[1 Suppl]). The actual paper, however, has never been published.
References
Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–241. | ||
Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6(7):508–519. | ||
Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20(5):1262–1275. | ||
Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340(2):115–126. | ||
Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362(6423):801–809. | ||
Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. | ||
Erbel C, Sato K, Meyer FB, et al. Functional profile of activated dendritic cells in unstable atherosclerotic plaque. Basic Res Cardiol. 2007;102(2):123–132. | ||
Erbel C, Dengler TJ, Wangler S, et al. Expression of IL-17A in human atherosclerotic lesions is associated with increased inflammation and plaque vulnerability. Basic Res Cardiol. 2011;106(1):125–134. | ||
Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394(6696):894–897. | ||
Aukrust P, Berge RK, Ueland T, et al. Interaction between chemokines and oxidative stress: possible pathogenic role in acute coronary syndromes. J Am Coll Cardiol. 2001;37(2):485–491. | ||
Burman A, Haworth O, Hardie DL, et al. A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J Immunol. 2005;174(3):1693–1700. | ||
Middel P, Raddatz D, Gunawan B, Haller F, Radzun HJ. Increased number of mature dendritic cells in Crohn’s disease: evidence for a chemokine mediated retention mechanism. Gut. 2006;55(2):220–227. | ||
Ngo VN, Tang HL, Cyster JG. Epstein-Barr virus-induced molecule 1 ligand chemokine is expressed by dendritic cells in lymphoid tissues and strongly attracts naive T cells and activated B cells. J Exp Med. 1998;188(1):181–191. | ||
Luther SA, Tang HL, Hyman PL, Farr AG, Cyster JG. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc Natl Acad Sci U S A. 2000;97(23):12694–12699. | ||
Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci U S A. 1998;95(1):258–263. | ||
Vassileva G, Soto H, Zlotnik A, et al. The reduced expression of 6Ckine in the plt mouse results from the deletion of one of two 6Ckine genes. J Exp Med. 1999;190(8):1183–1188. | ||
Nakano H, Gunn MD. Gene duplications at the chemokine locus on mouse chromosome 4: multiple strain-specific haplotypes and the deletion of secondary lymphoid-organ chemokine and EBI-1 ligand chemokine genes in the plt mutation. J Immunol. 2001;166(1):361–369. | ||
Mori S, Nakano H, Aritomi K, Wang CR, Gunn MD, Kakiuchi T. Mice lacking expression of the chemokines CCL21-ser and CCL19 (plt mice) demonstrate delayed but enhanced T cell immune responses. J Exp Med. 2001;193(2):207–218. | ||
Gunn MD, Kyuwa S, Tam C, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. 1999;189(3):451–460. | ||
Förster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol. 2008;8(5):362–371. | ||
Xu B, Aoyama K, Kusumoto M, et al. Lack of lymphoid chemokines CCL19 and CCL21 enhances allergic airway inflammation in mice. Int Immunol. 2007;19(6):775–784. | ||
Damås JK, Smith C, Øie E, et al. Enhanced expression of the homeostatic chemokines CCL19 and CCL21 in clinical and experimental atherosclerosis: possible pathogenic role in plaque destabilization. Arterioscler Thromb Vasc Biol. 2007;27(3):614–620. | ||
Halvorsen B, Dahl TB, Smedbakken LM, et al. Increased levels of CCR7 ligands in carotid atherosclerosis: different effects in macrophages and smooth muscle cells. Cardiovasc Res. 2014;102(1):148–156. | ||
Erbel C, Chen L, Bea F, et al. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J Immunol. 2009;183(12):8167–8175. | ||
Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14(1):133–140. | ||
Klingenberg R, Nofer JR, Rudling M, et al. Sphingosine-1-phosphate analogue FTY720 causes lymphocyte redistribution and hypercholesterolemia in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27(11):2392–2399. | ||
Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol. 2010;184(9):4810–4818. | ||
Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–721. | ||
Go AS, Mozaffarian D, Roger VL, et al. Executive summary: heart disease and stroke statistics – 2014 update: a report from the American Heart Association. Circulation. 2014;129(3):399–410. | ||
Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet. 1997;349(9063):1436–1442. | ||
Myerburg RJ, Interian A Jr, Mitrani RM, Kessler KM, Castellanos A. Frequency of sudden cardiac death and profiles of risk. Am J Cardiol. 1997;80(5B):10F–19F. | ||
Naghavi M, Libby P, Falk E, et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part I. Circulation. 2003;108(14):1664–1672. | ||
Swirski FK, Pittet MJ, Kircher MF, et al. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci U S A. 2006;103(27):10340–10345. | ||
Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–1143. | ||
Tomey MI, Narula J, Kovacic JC. Advances in the understanding of plaque composition and treatment options: year in review. J Am Coll Cardiol. 2014;63(16):1604–1616. | ||
Gleissner CA, Shaked I, Erbel C, Bockler D, Katus HA, Ley K. CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ Res. 2010;106(1):203–211. | ||
Newby AC. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc Med. 2007;17(8):253–258. | ||
Sakakura K, Nakano M, Otsuka F, Ladich E, Kolodgie FD, Virmani R. Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ. 2013;22(6):399–411. | ||
Forster R, Schubel A, Breitfeld D, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99(1):23–33. | ||
Weninger W, Carlsen HS, Goodarzi M, et al. Naive T cell recruitment to nonlymphoid tissues: a role for endothelium-expressed CC chemokine ligand 21 in autoimmune disease and lymphoid neogenesis. J Immunol. 2003;170(9):4638–4648. | ||
Chen SC, Vassileva G, Kinsley D, et al. Ectopic expression of the murine chemokines CCL21a and CCL21b induces the formation of lymph node-like structures in pancreas, but not skin, of transgenic mice. J Immunol. 2002;168(3):1001–1008. | ||
Wengner AM, Hopken UE, Petrow PK, et al. CXCR5- and CCR7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum. 2007;56(10):3271–3283. | ||
Luther SA, Bidgol A, Hargreaves DC, et al. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol. 2002;169(1):424–433. | ||
Link A, Vogt TK, Favre S, et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. 2007;8(11):1255–1265. | ||
Marsland BJ, Battig P, Bauer M, et al. CCL19 and CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity. 2005;22(4):493–505. | ||
Debes GF, Arnold CN, Young AJ, et al. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat Immunol. 2005;6(9):889–894. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.