Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 8
Causes and consequences of obesity: epigenetics or hypokinesis?
Authors Graham M, Baker J ![]() , Davies B
, Davies B
Received 11 February 2015
Accepted for publication 11 February 2015
Published 16 September 2015 Volume 2015:8 Pages 455—460
DOI https://doi.org/10.2147/DMSO.S82629
Checked for plagiarism Yes
Editor who approved publication: Professor Ming-Hui Zou
Michael R Graham,1 Julien S Baker,2 Bruce Davies3
1Llantarnam Research Academy, Cwmbran, Torfaen, UK; 2Exercise Science Research Laboratory, Institute of Clinical Exercise and Health Science, School of Science, University of the West of Scotland, Hamilton, UK; 3Science Department, University of South Wales, Newport, UK
Epigenetics can be defined as the study of heritable changes that affect gene function without modification of the deoxyribonucleic acid (DNA) sequence.1 The transfer of epigenetic marks through generations is not well understood, and their transmission is in dispute.2 Epigenetic marks are tissue-specific and include DNA methylation and histone modifications that mediate biological processes, such as imprinting (Figure 1). Many imprinted genes are regulators of gene expression controlling growth. Imprinting disorders often feature obesity as one of their characteristics.3
Dear editor
Epigenetics can be defined as the study of heritable changes that affect gene function without modification of the deoxyribonucleic acid (DNA) sequence.1 The transfer of epigenetic marks through generations is not well understood, and their transmission is in dispute.2 Epigenetic marks are tissue-specific and include DNA methylation and histone modifications that mediate biological processes, such as imprinting (Figure 1). Many imprinted genes are regulators of gene expression controlling growth. Imprinting disorders often feature obesity as one of their characteristics.3
 | Figure 1 Epigenetic mechanisms. |
Genomic imprinting determines expression of alleles, creating an equilibrium between the expression of the parental alleles influencing growth and metabolism.4
The identification of susceptible loci, and finding the causal genes and variants in each locus for obesity may allow molecular and physiological investigations. However, the knowledge of the mechanisms through which these contribute to susceptibility to obesity will not necessarily provide the requisite management strategies.
Geneticists have long since identified the obesity gene (FTO)5 and a low-fat gene (APOA5),6 and that variations in the adiponectin gene (single nucleotide polymorphism [SNP] 276 G allele) can lead to hypoadiponectinemia, which then results in insulin resistance, the metabolic syndrome, increased atherosclerosis, morbidity, and ultimately, premature mortality.7 Using exome sequencing, a low-frequency coding variant has been identified in the SYPL2 gene associated with morbid obesity.8 This gene may be involved in the development of excess body fat.
Hypokinesis, in conjunction with excess caloric intake over expenditure, leads to obesity, which can then result in the metabolic syndrome, type 2 diabetes mellitus, cardiovascular disease (CVD), coronary heart disease, and cerebrovascular accident, or “stroke”.9–12
Obesity is also believed to predispose to certain types of cancer,13 such as breast14 and colon cancer.15
However, the assertion that the complete absence of fat cells (adipocytes) is metabolically advantageous is a fallacy. Adipocytes play an important role in health by their hormonal role in the regulation of energy intake and fat storage, and function as an endocrine gland (Figure 2).16,17 Adipocytes secrete approximately 50 adipokines with diverse functional roles.18
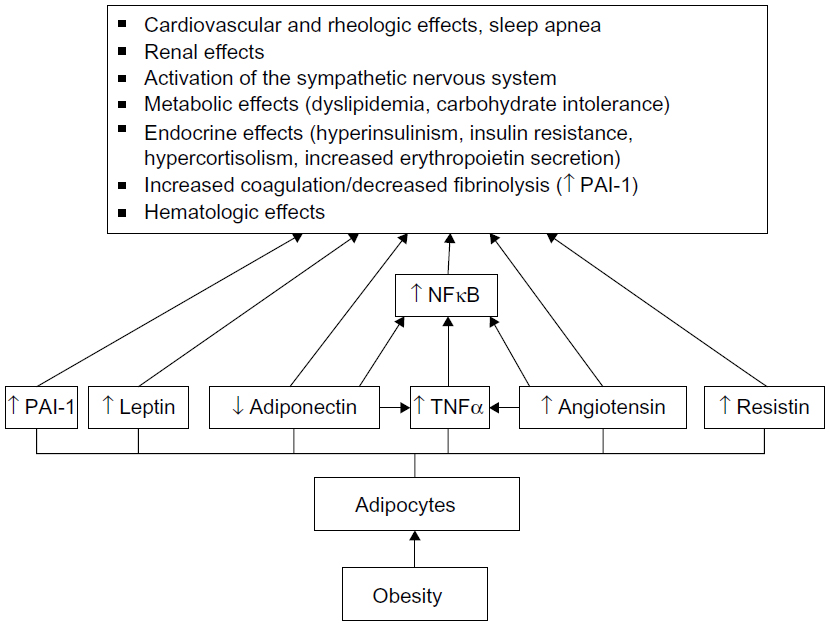 | Figure 2 The endocrine function of an adipocyte. |
The average human phenotype contains 30 billion fat cells, weighing approximately 13.5 kg.19 To establish the dynamics within the stable population of adipocytes in adults, adipocyte turnover can be measured by analyzing the integration of 14C derived from nuclear bomb tests in genomic DNA. If excess weight is gained as an adult, fat cells increase approximately fourfold in size before dividing and increasing the absolute number of fat cells present. However, even after marked weight loss, the number of fat cells stays constant in adulthood in lean and obese individuals, indicating that the number of adipocytes is set during childhood and adolescence.19 Approximately 10% of fat cells die and are replaced annually. The expansion of the fat cell population and the final number of adipocytes in the adult body appears to occur at an earlier age in obese people. Fatter people experience a period of rapid adipoctye production around the age of 2 years and reach their adult number of fat cells when they are approximately 16.5 years old.19 Lean people, however, recruit fat cells most rapidly at approximately 6 years old, with their fat cell population reaching its adult size at approximately 18.5 years. Obese people have as many as twice the number of adipocytes as lean people.19
Scientific evidence supports the concept that gene expression in our physically active ancestors reduced adverse physiological effects that may occur with a sedentary lifestyle. To prevent the development of these adverse physiological effects, children who experience appropriate nutrition and regular exercise during their growing years display healthy patterns of physical maturation consistent with their genetic potential.20 However, a genetic explanation cannot explain how humans have become so fat over such a short time frame.21 Heredity can only explain 67% of the population variance in obesity.22
Genetics are unlikely to account fully for the prevalence of obesity – it must be due to the declining rates of physical activity and an increase in the consumption of energy-dense foods.23,24
The differences in phenotypes and physiology between two geographically separate but genetically similar Pima Indian tribes suggests the importance of the environmental effect, especially physical activity, on the genome.25 The Mexican Pima Indians were found to expend 2,100–2,520 kJ day-1 more in physical activity than did their obese Arizonian Pima equivalents, to have had a diet lower in fat and higher in fiber content, and to weigh 26 kg less.25 As a consequence, the Mexican Pima Indians did not have the diabetes prevalence of the Arizonian Pima Indians.26
Children of pregnant women exposed to the Dutch famine of 1944 were more susceptible to diabetes, obesity, CVD, renal disease, and other health problems.27 The children of the women who were pregnant during the famine were smaller, and when these children grew up and had children, those children were also smaller than average.28 This suggested that the famine experienced by the mothers caused epigenetic changes that were passed down to the next generation.
Genetic predisposition is the primary cause of most of the rare diseases that affect one in 17 people.29,30 Every human possesses approximately three million sequence variants of which approximately 400 can predispose to pathology, in adverse environmental circumstances.31
Could lifestyle assessments and avoidance of potential risks have beneficial effects on a person’s health despite genetic variants? This question can only truly be answered if we are able to sequence individual genomes on demand and then, only if it is cost effective.
The general public should be concerned with prevention rather than the management of obesity and its sequelae. Despite considerable financial investment, the pharmaceutical industry has provided only palliation of obesity rather than cure.
Physical activity should be the main agenda of the Department of Health and the responsibilities for its effective prescription and assessments placed in the hands of appropriate health professionals. More consideration should be given to effective exercise prescription rather than the premature introduction of prescription medicines, such as the latest insulin analog or glucagon-like peptide-1 agonist (GLP-1 agonist), to type 2 diabetics, which appears to be only palliating the disease state and elevating the public purse’s annual budget.32
In the UK in 1998 the anti-obesity drugs’ bill was £20,000.00 and in 2005 it was £38 million, with increased prescribing of drugs such as orlistat and sibutramine.33 In 2010 it was £50 million.34
The annual drugs bill in the UK is currently around £12 billion, which equates to just approximately 10% of National Health Service (NHS) expenditure, having risen around 3.5% a year between 2007 and 2013.
When the NHS was launched in 1948 it had a budget of £437 million (circa £9 billion at today’s value). For 2015 it is predicted to be £115.4 billion.35
The obesity epidemic that we are experiencing is a global phenomenon. In 2005 more than 1.6 billion adults over the age of 15 years, were overweight and 400 million were obese. By 2015 it has been predicted that more than 2.3 billion adults over the age of 15 years, will be overweight and at least 700 million will be obese.36 With global projections, more than 2.16 billion will be overweight and 1.12 billion will be obese individuals by 2030.36
Watson and Crick discovered the structure of DNA in 1953,37 and Sanger identified the amino acid sequence of insulin in 1955, and nucleic acid sequencing becoming a major target of early molecular scientists.38
However, we are no nearer to eradicating the most basic of diseases, associated with obesity.
Have we really had a single or series of genetic mutations to account for a 21.4% and a 24% increase in obesity in males and females respectively, in a mere third of a century?
Sequencing the first human genome took 13 years and was completed in 1986 at a cost of £2 billion.39 Today we can sequence a complete human genome in two days at a cost of approximately £4,000.00.39
Bulk sequencing all 20–25,000 human protein coding genes, the exome, now costs less than £500.29
Targeting the screening of a specific panel of genes known to be associated with a particular phenotype, as opposed to screening mutations, would appear necessary in the belief that eating five pieces of fruit and vegetables a day will keep us slim, healthy, and disease free.40 Scientists are required to identify variants in genes, known to be pathogenic in an effort to decide on preventative, curative or palliative action.
The investment continues as the government’s decision to gene sequence 100,000 whole genomes at a cost to the public purse of £100 million hoping to identify the reason for the current obesity pandemic, pursuant to the work of the Wellcome Trust’s Sanger Institute initiating the Human Genome Project.29,41 However, will such an investment stimulate the general population to cease eating refined processed foods and commence exercising?
The fact there is so much money available for such a resource is surprising. Should not that money be available to employ more exercise scientists, who can advise these obese phenotypes that eating the correct diet and performing even limited amounts of exercise can bring about positive transgenerational epigenetic changes to the major CVD risk factors.42,43
Currently the vast sums of money spent in researching pharmacotherapy alone appear to be having very little effect. A greater investment should be applied to promoting exercise in conjunction with exome sequencing and epigenetic research.
Human growth hormone (hGH) which controls the body’s ability to metabolize carbohydrate, fat, and protein and deposit any excess fat in the appropriate site, commences a downward spiral, after full adult growth has been attained.44 It decreases by 14% per decade from the age of 20 years.44 Exercising for more than 10–20 minutes at an oxygen uptake (VO2max) of 70%, stimulates pituitary release of hGH by five- to tenfold.45 Protein consumption stimulates pituitary release of hGH.46 Carbohydrate and fat ingestion inhibit pituitary release of hGH.47 Also obesity causes a blunted response to hGH compounding the adverse situation.48
Physical activity can ultimately prevent many problems associated with obesity.43 There is a lower cancer incidence rates among the adult Amish population, compared with non-Amish population, due to increased physical activity.49 Physical activity differences can be identified in childhood in the Amish compared with non-Amish population and accounts for Amish children being approximately 3.3 times less likely to be overweight compared to non-Amish children.50 The average American would aspire to walk approximately 10,000 steps per day to maintain a healthy body mass. In one study the average number of steps walked per day was 18,425 for Amish men versus 14,196 for Amish women, who have a very low incidence of obesity. The Old Order Amish refrain from driving cars, using electrical appliances, and employing other modern conveniences. Labor-intensive farming is still the preferred occupation.51
Conclusion
Obesity is a consequence of a high saturated fat, a refined carbohydrate diet, and an increased input over expenditure, combined with reduced exercise. Proactive research scientists are working to identify explanations, at the molecular level, relating exercise to health improving epigenetic changes.
Prevention must always be better than cure. When a large segment of the population adopts modest improvements in health behaviors, the overall disease burden in the population is likely to be reduced more dramatically than if a modest segment of the population adopts large changes.52
Disclosure
The authors report no conflicts of interest in this communication.
References
Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–398. | |
Chong S, Youngson NA, Whitelaw E. Heritable germline epimutation is not the same as transgenerational epigenetic inheritance. Nat Genet. 2007;39(5):574–575; author reply 575. | |
Herrera BM, Keildson S, Lindgren CM. Genetics and epigenetics of obesity. Maturitas. 2011;69(1):41–49. | |
Smith FM, Garfield AS, Ward A. Regulation of growth and metabolism by imprinted genes. Cytogenet Genome Res. 2006;113(1–4):279–291. | |
Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889–894. | |
Corella D, Lai CQ, Demissie S, et al. APOA5 gene variation modulates the effects of dietary fat intake on body mass index and obesity risk in the Framingham Heart Study. J Mol Med (Berl). 2007;85(2):119–128. | |
González-Sánchez JL, Zabena CA, Martínez-Larrad MT, et al. An SNP in the adiponectin gene is associated with decreased serum adiponectin levels and risk for impaired glucose tolerance. Obes Res. 2005; 13(5):807–812. | |
Jiao H, Arner P, Gerdhem P, et al. Exome sequencing followed by genotyping suggests SYPL2 as a susceptibility gene for morbid obesity. Eur J Hum Genet. Epub 2014 Nov 19. | |
Rathmann W, Giani G. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(10):2568–2569; author reply 2569. | |
Li Z, Bowerman S, Heber D. Health ramifications of the obesity epidemic. Surg Clin North Am. 2005;85(4):681–701, v. | |
Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444(7121):875–880. | |
Kahn HS. Obesity and risk of myocardial infarction: the INTERHEART study. Lancet. 2006;367(9516):1053–1054. | |
Anderson AS, Caswell S. Obesity management – an opportunity for cancer prevention. Surgeon. 2009;7(5):282–285. | |
Morimoto LM, White E, Chen Z, et al. Obesity, body size, and risk of postmenopausal breast cancer: the Women’s Health Initiative (United States). Cancer Causes Control. 2002;13(8):741–751. | |
Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86(3):556–565. | |
Frayn KN, Karpe F, Fielding BA, Macdonald IA, Coppack SW. Integrative physiology of human adipose tissue. Int J Obes. 2003;27(8):875–888. | |
Li Y, Fromme T, Schweizer S, Schöttl T, Klingenspor M. Taking control over intracellular fatty acid levels is essential for the analysis of thermogenic function in cultured primary brown and brite/beige adipocytes. EMBO Rep. 2014;15(10):1069–1076. | |
Greenberg AS, Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr. 2006;83(2):461S–465S. | |
Spalding KL, Arner E, Westermark PO, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453(7196):783–787. | |
Booth FW, Chakravarthy MV, Spangenburg EE. Exercise and gene expression: physiological regulation of the human genome through physical activity. J Physiol. 2002;543(Pt 2):399–411. | |
Toye AA, Dumas ME, Blancher C, et al. Subtle metabolic and liver gene transcriptional changes underlie diet-induced fatty liver susceptibility in insulin-resistant mice. Diabetologia. 2007;50(9):1867–1879. | |
Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27(4):325–351. | |
Brownson RC, Boehmer TK, Luke DA. Declining rates of physical activity in the United States: what are the contributors? Annu Rev Public Health. 2005;26:421–443. | |
Ledikwe JH, Blanck HM, Kettel Khan L, et al. Dietary energy density is associated with energy intake and weight status in US adults. Am J Clin Nutr. 2006;83(6):1362–1368. | |
Esparza J, Fox C, Harper IT, et al. Daily energy expenditure in Mexican and USA Pima indians: low physical activity as a possible cause of obesity. Int J Obes Relat Metab Disord. 2000;24(1):55–59. | |
Pratley RE. Gene-environment interactions in the pathogenesis of type 2 diabetes mellitus: lessons learned from the Pima Indians. Proc Nutr Soc. 1998;57(2):175–181. | |
Roseboom T, de Rooij S, Painter R. The Dutch famine and its long-term consequences for adult health. Early Hum Dev. 2006;82(8):485–491. | |
Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DI, Roseboom TJ. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG. 2008;115(10):1243–1249. | |
Burn J. Should we sequence everyone’s genome? Yes. BMJ. 2013;346:f3133. | |
Department of Health. The 2009 Annual Report of the Chief Medical Officer. The 2009 Annual Report of the Chief Medical Officer. London: Department of Health; 2010. | |
Xue Y, Chen Y, Ayub Q, et al; 1000 Genomes Project Consortium. Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet. 2012;91(6):1022–1032. | |
Scholz GH, Fleischmann H. Basal insulin combined incretin mimetic therapy with glucagon-like protein 1 receptor agonists as an upcoming option in the treatment of type 2 diabetes: a practical guide to decision making. Ther Adv Endocrinol Metab. 2014;5(5):95–123. | |
Padwal R, Li SK, Lau DC. Long-term pharmacotherapy for obesity and overweight. Cochrane Database Syst Rev. 2004;3:CD004094. | |
Dramatic rise in slimming drugs prescriptions leaves NHS with £50 m bill. The Daily Mail. 2010 Sept 20. Available from: http://www.dailymail.co.uk/health/article-1313362/Slimming-drugs-cost-NHS-50m.html. Accessed January 3, 2015. | |
nhs.uk [homepage on the Internet]. The NHS in England: About the National Health Service (NHS). National Health Service; 2015 [updated January 7, 2015; cited January 3, 2015]. http://www.nhs.uk/NHSEngland/thenhs/about/Pages/overview.aspx. Accessed February 11, 2015. | |
Fontvieille AM, Ravussin E. Metabolic rate and body composition of Pima Indian and Caucasian children. Crit Rev Food Sci Nutr. 1993;33(4–5):363–368. | |
Watson JD, Crick FH. A structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737–738. | |
Stretton AOW. The first sequence. Fred Sanger and insulin. Genetics. 2002;162(2):527–532. | |
Flinter F. Should we sequence everyone’s genome? No. BMJ. 2013;346:f3132. | |
Keys A, Anderson JT, Aresu M, et al. Physical activity and the diet in populations differing in serum cholesterol. J Clin Invest. 1956;35(10):1173–1181. | |
Wright CF, Middleton A, Burton H, et al. Policy challenges of clinical genome sequencing. BMJ. 2013;347:f6845. | |
Buchan DS, Young JD, Boddy LM, Baker JS. Independent associations between cardiorespiratory fitness, waist circumference, BMI, and clustered cardiometabolic risk in adolescents. Am J Hum Biol. 2014;26(1):29–35. | |
Denham J, Marques FZ, O’Brien BJ, Charchar FJ. Exercise: putting action into our epigenome. Sports Med. 2014;44(2):189–209. | |
Iranmanesh A, Lizarralde G, Veldhuis JD. Age and relative adiposity are specific negative determinants of the frequency and amplitude of growth hormone (GH) secretory bursts and the half-life of endogenous GH in healthy men. J Clin Endocrinol Metab. 1991;73(5):1081–1088. | |
Felsing NE, Brasel JA, Cooper DM. Effect of low and high intensity exercise on circulating growth hormone in men. J Clin Endocrinol Metab. 1992;75(1):157–162. | |
Root AW, Root MJ. Clinical pharmacology of human growth hormone and its secretagogues. Curr Drug Targets Immune Endocr Metabol Disord. 2002;2(1):27–52. | |
Masuda A, Shibasaki T, Nakahara M, et al. The effect of glucose on growth hormone (GH)-releasing hormone-mediated GH secretion in man. J Clin Endocrinol Metab. 1985;60(3):523–526. | |
Scacchi M, Pincelli AI, Cavagnini F. Growth hormone in obesity. Int J Obes Relat Metab Disord. 1999;23(3):260–271. | |
Katz ML, Ferketich AK, Broder-Oldach B, et al. Physical activity among Amish and non-Amish adults living in Ohio Appalachia. J Community Health. 2012;37(2):434–440. | |
Hairston KG, Ducharme JL, Treuth MS, et al. Comparison of BMI and physical activity between old order Amish children and non-Amish children. Diabetes Care. 2013;36(4):873–878. | |
Bassett DR, Schneider PL, Huntington GE. Physical activity in an Old Order Amish community. Med Sci Sports Exerc. 2004;36(1):79–85. | |
Rose G. Environmental factors and disease: the man made environment. Br Med J (Clin Res Ed). 1987;294(6577):963–965. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.