Back to Journals » ImmunoTargets and Therapy » Volume 14
Causal Association Between Circulating Inflammatory Proteins and Autoimmune Liver Disease: a Bidirectional Two-Sample Mendelian Randomization Study
Authors Leng L ![]() , Li Y, Xu T
, Li Y, Xu T ![]() , Shen J
, Shen J ![]() , Li L, Li X
, Li L, Li X ![]()
Received 9 December 2024
Accepted for publication 13 March 2025
Published 26 March 2025 Volume 2025:14 Pages 279—289
DOI https://doi.org/10.2147/ITT.S508140
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sarah Wheeler
Lina Leng,1,* Ying Li,2,* Tao Xu,3 Jingfang Shen,1 Lianju Li,1 Xiaoli Li1
1Department of Rheumatology, Xingtai People’s Hospital, Xingtai, 054001, Hebei Province, People’s Republic of China; 2Department of Oncology, 82 Group Hospital of Chinese People’s Liberation Army, Baoding, Hebei Province, 071000, People’s Republic of China; 3Department of Internal Medicine, Graduate School of Hebei North University, Zhangjiakou, Hebei Province, 075000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoli Li, Email [email protected]
Introduction: To investigate whether there is a direct causal relationship between circulating inflammatory proteins and autoimmune liver disease (AILD).
Materials and Methods: We collected genetic data for various AILD from the Genome Wide Association Studies (GWAS) dataset. The latest research provides GWAS data for 91 proteins associated with inflammation. Perform bidirectional two sample Mendelian randomization (MR) analysis using inverse variance weighted (IVW) to determine the causal relationship between inflammatory proteins and AILD, and use Mendelian randomization Egger method (MR Egger), weighted median (WM), and weighted mode as supplementary evaluations. In addition, we conducted sensitivity analysis.
Results: Positive MR analysis showed that CDCP1 (OR=1.363, p=0.0465) and IL-18 (OR=1.416, p=0.0477) were associated with higher including autoimmune hepatitis (AIH) risk. Higher CXCL11 (OR=1.574, p=9.23× 10-5) were associated with an increased risk of primary biliary cholangitis (PBC). Lower levels of three inflammatory proteins were associated with increased risk of PBC. TNFSF12 (OR=1.827, p=0.0001, p_adj_fdr=0.0063), CD6 isoform (OR=1.126, p=0.0389), CCL20 (OR=1.880, p=0.0395) are associated with increased risk of primary sclerosing cholangitis (PSC). Reverse MR imaging showed that PBC may promote the expression levels of CCL4 (OR=1.023, p=0.0201) and OSM (OR=1.022, p=0.0236). PSC may promote the expression of five inflammatory proteins. Sensitivity analysis further excluded the effects of heterogeneity and horizontal pleiotropy.
Conclusion: This study indicates a potential association between circulating inflammatory proteins and AILD, which may become a new diagnostic indicator or drug target for clinical application in the prevention and treatment of AILD. However, further investigation is needed.
Keywords: autoimmune liver disease, Mendelian randomization, circulating inflammatory protein, autoimmune hepatitis, primary biliary cholangitis, primary sclerosing cholangitis
Introduction
Autoimmune liver diseases (AILD) are a spectrum of liver pathologies triggered by autoimmune dysregulation, characterized by lymphocytic infiltration of the liver, elevated levels of circulating immunoglobulins, increased transaminases, and the production of specific autoantibodies.1 Based on distinct histopathological features and the expression of autoantibodies, AILD can be categorized into autoimmune hepatitis (AIH), primary biliary cholangitis (PBC), and primary sclerosing cholangitis (PSC).2,3 Initially, AILD may be asymptomatic. However, as the disease progresses, it can lead to liver fibrosis, cirrhosis, and even hepatocellular carcinoma.4,5 Currently, the incidence of AILD is on the rise, with global rates of AIH ranging from 0.4 to 2.39 per 100,000 people, PBC from 0.84 to 2.75, and PSC from 0.1 to 4.39.1 Moreover, there is currently no cure for AILD. Despite aggressive treatment, the response rate is suboptimal,6,7 and long-term pharmacological therapy often leads to systemic adverse effects such as infections, osteoporosis, and diabetes mellitus.8 Consequently, AILD has become a severe chronic liver condition, imposing a significant economic burden on society.
The etiology of AILD is multifactorial, involving environmental triggers and genetic anomalies that result in chronic inflammation and T-cell mediated liver damage.9,10 Recent research has revealed the significant role of inflammatory proteins in the development of AILD.11,12 Therefore, focusing on these inflammatory elements could be beneficial. Inflammatory proteins in circulation may provide valuable prognostic information to improve the outcomes of AILD, representing a substantial potential strategy for its prevention and treatment. However, comprehensive preclinical and clinical studies are still lacking, and the clinical significance of these inflammatory proteins remains unclear. Observational studies may be confounded by potential biases and reverse causality. The relationship between inflammatory proteins and AILD observed in previous observational studies requires further investigation.
Mendelian randomization (MR) is an innovative method in genetic epidemiology that employs genetic variants (typically single nucleotide polymorphisms) as instrumental variables (IVs) to establish causal relationships between risk factors (exposures) and diseases (outcomes), essentially mimicking randomized controlled trials (RCTs).13 By leveraging genetic diversity, the MR method aims to elucidate the causality between exposures and outcomes, effectively addressing inherent biases such as confounding, reverse causality, and biased sampling in epidemiological studies, while circumventing the practical limitations of randomized trials.14 Although MR has broad applications, it has not yet been explored as a tool to investigate the potential causal relationship between AILD and circulating inflammatory proteins. Therefore, we conducted a bidirectional MR analysis to assess the causal relationship between changes in inflammatory protein levels and the likelihood of developing AILD.
Materials and Methods
Study Design
We employed a two-sample MR study to investigate the causal relationship between inflammatory proteins and AILD. Obtaining valid results from MR analysis hinges on fulfilling three key assumptions. Firstly, the genetic instrumental variable (IV) must have a strong association with the exposure, which is the risk factor of interest. Secondly, the genetic variant must be independent of unmeasured confounders that could affect the exposure-outcome association. Lastly, it is presumed that the IV affects the outcome solely through its association with the exposure, thereby minimizing the potential for pleiotropy effects. This study has been proofread based on the STrengthening the Reporting of OBservational studies in Epidemiology - Mendelian Randomization (STROBE-MR) checklist to ensure the transparency and reproducibility of our findings.15 Figure 1 depicts the flowchart of the MR study process.
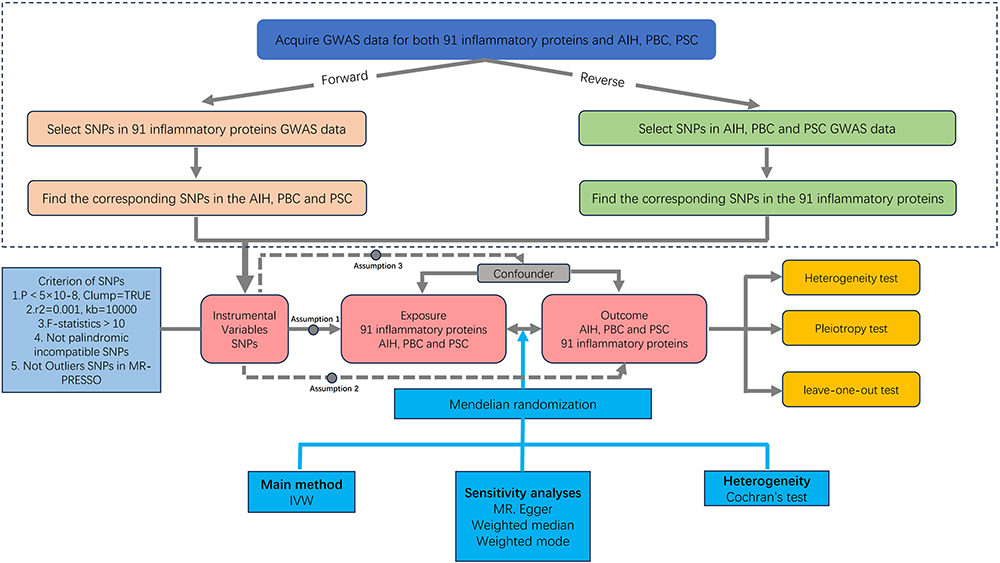 |
Figure 1 Overview of the assumptions of the Mendelian randomization design and the study design. Abbreviation: GWAS, genome-wide association study; IVs, instrumental variables; AIH, autoimmune hepatitis; PBC, primary biliary cholangitis; PSC, primary sclerosing cholangitis; SNPs, single-nucleotide polymorphisms. |
Data Source
The GWAS summary data for AIH included 484,413 European controls and 821 European patients (GWAS ID: ebi-a-GCST90018785). The GWAS summary data for PBC included 16,489 European controls and 8,021 European patients (GWAS ID: ebi-a-GCST90061440), and the genetic data for PSC included 12,019 European controls and 2,871 European patients (GWAS ID: ieu-a-1112). The data for the 91 circulating inflammatory proteins were obtained from a recent GWAS conducted by Zhao et al.16 This study provided summary statistics for the genetic associations with these proteins, which we used as the exposure data in our Mendelian randomization analysis. The GWAS included a large cohort of European ancestry participants, ensuring robust genetic associations. At the same time, it ensures that the samples between the exposure and outcome did not overlap.
IV Selection
In adherence to the three assumptions underlying MR analysis, we initially selected single nucleotide polymorphisms (SNPs) associated with the exposure and ensured that the chosen SNPs were significantly associated with the target exposure at the genome-wide significance threshold (P < 5×10^-8) to meet the first assumption. Secondly, we set the linkage disequilibrium (LD) window to kb = 10000kb, r2 < 0.001 to ensure the independence of the selected genetic variants. The third assumption is the unidirectional effect: the IV should only affect the outcome through the exposure and not directly influence the outcome itself. Adhering to these principles ensures the logical consistency of causal analysis. However, in the case of reverse causality, where AIH is considered the exposure and inflammatory proteins the outcome, the number of independent SNPs with a P-value below the 5×10^−8 threshold was insufficient for MR analysis. Therefore, a more lenient genome-wide significance threshold of P < 5×10^−6 was established to select SNPs closely associated with inflammatory proteins. Before each MR analysis, a Steiger test was conducted to avoid reverse causality. Only SNPs with “TREU” results were included. The MR pleiotropy residual and outlier (MR-PRESSO) test was employed to identify and remove any outliers in the data.17 Subsequently, for each SNP, the F-statistic was determined by performing beta2 /se2 (BETA2/SE2). An F-statistic greater than 10 was considered to have sufficient strength for the selected IV.18 In this study, all F-statistics met the criterion of F > 10. Finally, to enhance data quality, we also removed palindromic SNPs that are genomically mirror-symmetric. Detailed information about all IVs for exposures is provided in the supplementary materials.
MR analysis and sensitivity analysis.
The primary analysis utilized the multiplicative random effects inverse variance weighted (IVW) method, which combines the cumulative causal estimates from the Wald ratios derived from each IV,19 and it offers the greatest statistical power in the absence of pleiotropy. Supplementary analyses were conducted using the Mendelian Randomization Egger (MR-Egger),20 Weighted Median (WM),21 and Weighted Mode methods22 to ensure the validity and robustness of the study results. These MR methods are explained in detail in the corresponding references. False Discovery Rate (FDR) adjustment was applied to correct the results of the IVW method.23 Cochran Q test was used to assess the heterogeneity of MR results. A p-value less than 0.05 was considered indicative of heterogeneity, leading to the use of a random effects model for analysis.24 Additionally, we assessed pleiotropy by employing the MR-Egger intercept test and the MR-PRESSO global test. P-values exceeding 0.05 for both the MR-Egger regression and the MR-PRESSO global test indicate the absence of horizontal pleiotropy.25 Ultimately, to ascertain whether individual SNPs were the sole contributors to the causal effect, we conducted leave-one-out analyses.26 Statistical analyses were performed using R software (version 4.0.2) along with the TwoSampleMR (version 0.5.6) and MR-PRESSO packages, which can be accessed at the following website: https://cloud.r-project.org.
Results
Detailed Information of Included SNPs
Specific information regarding the 91 circulating inflammatory proteins can be found in Supplementary Table 1. Detailed SNP information for the MR analysis and reverse MR analysis of AIH, PBC, PSC, and the 91 circulating inflammatory proteins is presented in Supplementary Tables 2–7. All SNPs demonstrated “TRUE” results in the Steiger test, indicating the absence of reverse causality (Supplementary Tables 2–7). This comprehensive collection includes a variety of detailed information such as genetic loci, effect alleles (EA), and effect allele frequencies (EAF). Notably, the F-statistics for the selected IVs exceed 10. By systematically selecting SNPs, consistency in association with the target exposure is ensured. Additionally, efforts have been made to effectively manage the relationship between the outcome variables and potential confounders within acceptable parameter ranges. Consequently, all selected SNPs can be considered reliable and robust instrumental variables. Meanwhile, the baseline levels of 91 inflammatory proteins can be found at this website: https://www.nature.com/articles/s41590-023-01588-w/figures/8.
Exploring the Causal Links Between Inflammatory Proteins and AIH, PBC, PSC Through MR
The MR analysis results examining the causal relationships between the 91 circulating inflammatory proteins and AIH, PBC, and PSC are depicted in Figure 2 and Supplementary Tables 8–10. Utilizing the IVW method, we identified two outcomes with p-values less than 0.05. The IVW method revealed that elevated levels of CUB domain-containing protein 1 (CDCP1) (OR = 1.363, 95% CI = 1.005–1.849, p = 0.0465) and Interleukin-18 (IL-18) (OR = 1.416, 95% CI = 1.004–1.999, p = 0.0477) are associated with an increased risk of AIH. According to the IVW results, higher levels of C-X-C motif chemokine 11 (CXCL11) (OR = 1.574, 95% CI = 1.254–1.976, p = 9.23×10^-5) were associated with an increased risk of PBC. After adjustment for the FDR, the p-value remained statistically significant with p_adj_fdr = 0.0027. Conversely, elevated levels of Interleukin-12 subunit beta (IL-12β) (OR = 0.872, 95% CI = 0.799–0.952, p = 0.0022), TNF-related activation-induced cytokine (TRANCE) (OR = 0.686, 95% CI = 0.518–0.908, p = 0.0085), and C-X-C motif chemokine 10 (CXCL10) (OR = 0.762, 95% CI = 0.586–0.992, p = 0.0437) were associated with a decreased risk of PBC. Notably, after FDR adjustment, the elevated levels of IL-12β (p_adj_fdr = 0.0022) still showed a significant association with a reduced risk of PBC. The findings revealed causal associations between PSC and three inflammatory proteins: Tumor necrosis factor ligand superfamily member 12 (TNFSF12) (OR = 1.827, 95% CI = 1.338–2.495, p = 0.0001, p_adj_fdr = 0.0063), T-cell surface glycoprotein CD6 isoform (CD6 isoform) (OR = 1.126, 95% CI = 1.006–1.261, p = 0.0389), and C-C motif chemokine 20 (CCL20) (OR = 1.880, 95% CI = 1.031–3.430, p = 0.0395), with elevated levels associated with an increased risk of PSC. In addition, we have summarized the positive results of MR analysis of circulating inflammatory proteins and AIH, PBC, PSC, as shown in Supplementary Table 11.
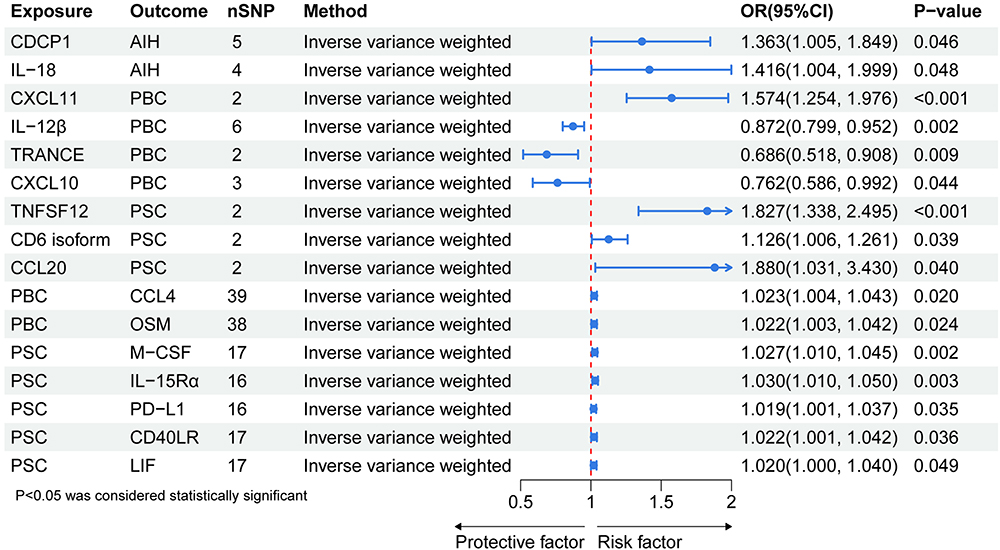 |
Figure 2 MR results between 91 circulating inflammatory proteins and AIH, PBC, PSC. Abbreviation: AIH, Autoimmune hepatitis; PBC, Primary biliary cholangitis; PSC, Primary sclerosing cholangitis; CDCP1, CUB domain-containing protein 1; IL-18, Interleukin-18; CXCL11, C-X-C motif chemokine 11; IL-12β, Interleukin-12 subunit beta; TRANCE, TNF-related activation-induced cytokine; CXCL10, C-X-C motif chemokine 10; TNFSF12, Tumor necrosis factor ligand superfamily member 12; CD6 isoform, T-cell surface glycoprotein CD6 isoform; CCL20, C-C motif chemokine 20; CCL4, C-C motif chemokine 4; OSM, Oncostatin-M levels (CD6); M-CSF, Macrophage colony-stimulating factor 1; IL-15Rα, Interleukin-15 receptor subunit alpha; PD-L1, Programmed cell death 1 ligand 1; CD40LR, CD40L receptor; LIF, Leukemia inhibitory factor. |
Supplementary scatter plots (Supplementary Figure 1) and forest plots (Figure 2) display the causal effects of each SNP in genes encoding the circulating inflammatory proteins that showed positive results on the risk of AIH, PBC, and PSC.
Exploring the Causal Links Between AIH, PBC, PSC and Inflammatory Proteins Through MR
The results of the reverse MR analysis are presented in Supplementary Tables 12–14. In the inverse MR analysis, no association was found between AIH and circulating inflammatory proteins. Regarding the impact of PBC on inflammatory proteins, two positive results were identified. PBC may promote the expression levels of C-C motif chemokine 4 (CCL4) (OR = 1.023, 95% CI = 1.004–1.043, p = 0.0201) and Oncostatin-M levels (CD6) (OSM) (OR = 1.022, 95% CI = 1.003–1.042, p = 0.0236). Furthermore, a positive causal relationship was observed between PSC and five circulating inflammatory proteins: Macrophage colony-stimulating factor 1 (M-CSF) (OR = 1.027, 95% CI = 1.009–1.045, p = 0.0024), Interleukin-15 receptor subunit alpha (IL-15Rα) (OR = 1.030, 95% CI = 1.010–1.050, p = 0.003), Programmed cell death 1 ligand 1 (PD-L1) (OR = 1.019, 95% CI = 1.001–1.037, p = 0.0346), CD40L receptor (CD40LR) (OR = 1.022, 95% CI = 1.001–1.042, p = 0.0364), and Leukemia inhibitory factor (LIF) (OR = 1.019, 95% CI = 1.000–1.040, p = 0.0491). In addition, the positive results of reverse MR analysis of circulating inflammatory proteins and AIH, PBC, PSC are shown in Supplementary Table 15.
Sensitivity Analysis
We observed that the IVW and MR-Egger heterogeneity tests confirmed the absence of statistically significant heterogeneity among the genetic instruments studied (all p-values > 0.05) (Table 1). Furthermore, the lack of statistically significant difference between the MR-Egger Egger intercept and 0 (all p-values > 0.05) indicated no evidence of horizontal pleiotropy (Table 1). In the leave-one-out sensitivity analysis (Supplementary Figure 2), no single SNP significantly disrupted the overall effect of inflammatory proteins on AIH, PBC, and PSC. Similarly, no single SNP significantly disrupted the overall effect of AIH, PBC, and PSC on inflammatory proteins. Due to the limited number of IVs available for positive proteins that meet the criteria, they do not meet the conditions for leave-one-out analysis. Therefore, only four figures are provided. The symmetry and funnel plot absence of bias in the results added credibility to the study’s findings (Supplementary Figure 3). While our funnel plots and sensitivity analyses suggest that the data are unbiased, we acknowledge the importance of considering potential biases in the interpretation of our results. Future studies with larger sample sizes and more comprehensive data sets could provide further validation of our findings and help to minimize the impact of any potential biases.
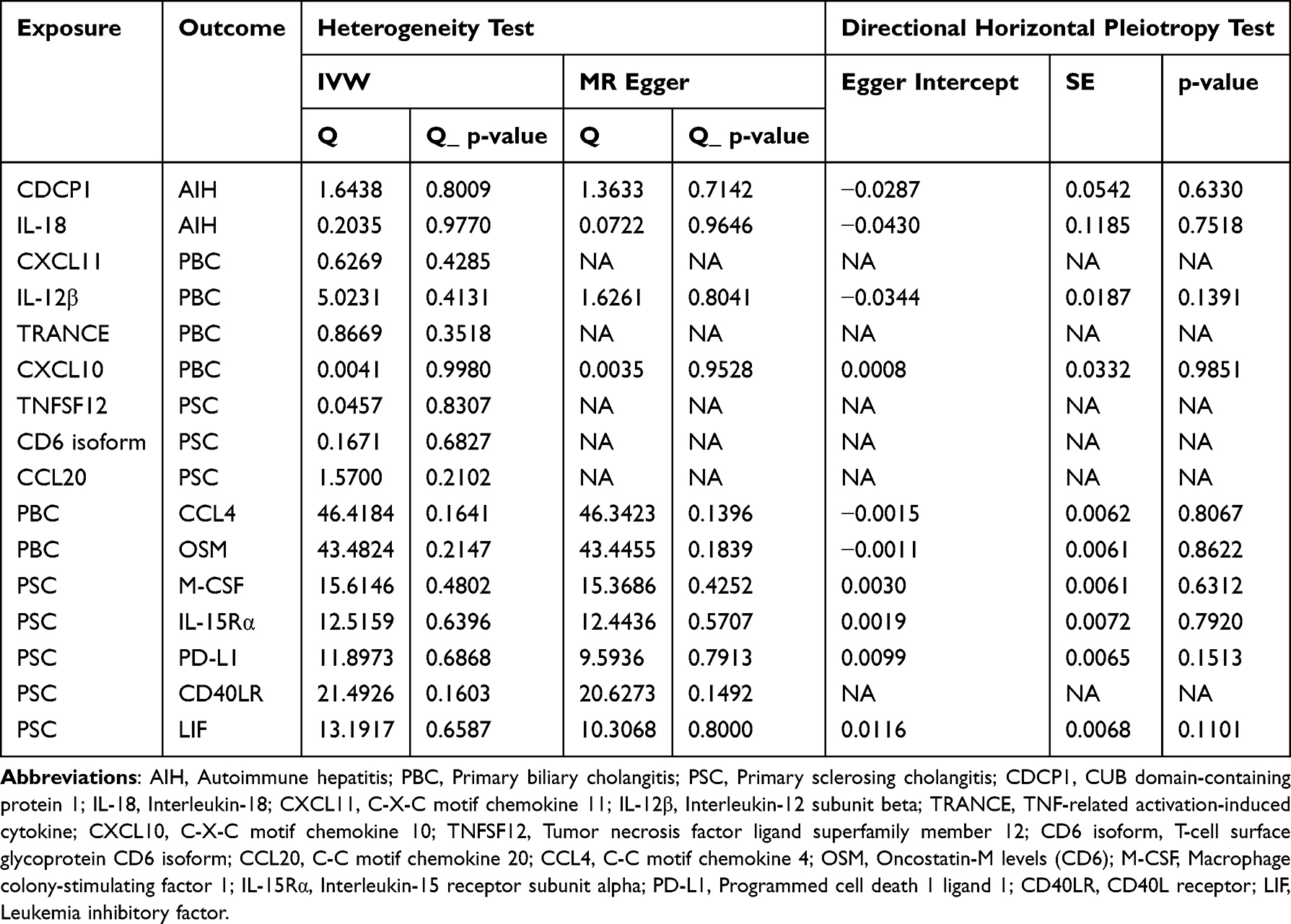 |
Table 1 Heterogeneity and Directional Horizontal Pleiotropy Tests of Circulating Inflammatory Cytokines on AIH, PBC and PSC |
Discussion
In this study, we employed a bidirectional MR approach to elucidate the causal relationships between circulating inflammatory proteins and AILD (including AIH, PBC, and PSC). The results suggest that certain cytokines appear to regulate disease susceptibility in various ways, with complex interactions at play. However, further research is needed to establish the causal effects.
CDCP1, also known as CD318, is a type I transmembrane glycoprotein that is widely upregulated in various malignancies, including liver and pancreatic tumors.27 Most previous studies on CDCP1 have been limited to its role in oncogenesis, suggesting that CDCP1 is essential for cancer cell survival and metastasis through intrinsic mechanisms within tumor cells.28,29 To date, changes in CDCP1 expression in the context of AIH have not been reported. However, CDCP1 has been implicated in related immune diseases. It is identified as a novel ligand for CD6, a surface marker and key regulatory molecule of T cells.30 Polymorphisms in CD6 are associated with multiple sclerosis (MS)31 and autoimmune uveitis,32 and CDCP1 has been shown to promote T cell infiltration, thereby contributing to the development of autoimmune uveitis.33 Both diseases involve a significant role of autoreactive T cells in their pathogenesis. Concurrently, mounting evidence suggests that abnormal T cell (eg, CD4+ T cell) infiltration and activation play a crucial role in the pathogenesis of AIH.34,35 This may partially explain why CDCP1 is a risk factor for AIH in our study. Future preclinical and clinical research is needed to confirm the role of CDCP1 in the development of AIH. IL-18, a cytokine closely related to immune responses and a member of the IL-1 family,36 was found in our forward analysis to be associated with an increased risk of AIH when elevated. IL-18 may play a role in AIH by promoting Th1 cell-mediated reactions, potentially related to the secretion of interferon-gamma (IFN-γ).37 In a mouse model of AIH, dendritic cells in the spleen and liver produce large amounts of IL-18, leading to increased levels of IL-18 in the serum. Administration of anti-IL-18R in vivo can suppress the increase of C-X-C motif chemokine receptor 3 positive (CXCR3+) T cells in the spleen and the progression to fatal AIH.38 Given the significant role of IL-18 in AIH, monoclonal antibodies, inhibitors, and drugs targeting IL-18 may represent a potential therapeutic approach for AIH.
CXCL11 is a chemokine that binds to the CXCR3 receptor. Studies have indicated that the expression levels of CXCL11 in patients with PBC correlate with disease activity. After treatment with ursodeoxycholic acid (UDCA) in PBC patients, the levels of markers such as CXCL11 remain elevated in those who have an incomplete response to UDCA, suggesting that any degree of persistent abnormality may be associated with a poor prognosis.39 In the therapeutic strategy for PBC, modulating related chemokines, including CXCL11, is one of the potential avenues for treatment.40 This is consistent with our research findings. CXCL11 may serve as one of the biomarkers for PBC disease activity, aiding in the assessment of disease severity and therapeutic efficacy. Our study results reveal that IL-12β, TRANCE, and CXCL10 may be protective factors for PBC, which is inconsistent with previous study findings. IL-12β is one of the subunits of IL-12. IL-12 promotes Th1 cell differentiation by activating the STAT4 signaling pathway and induces the production of IFN-γ. In PBC, IFN-γ can activate macrophages and cytotoxic T cells, leading to damage of bile duct epithelial cells.41 Additionally, IL-12β promotes the release of inflammatory factors (such as TNF-α and IL-6) by activating signaling pathways such as NF-κB and STAT4, exacerbating liver inflammation and fibrosis, and driving disease progression in PBC.42 TRANCE, a cytokine belonging to the tumor necrosis factor (TNF) superfamily, plays an important role in immune regulation and bone metabolism by binding to its receptor RANK. TRANCE regulates the interaction between dendritic cells (DCs) and T cells by activating the RANK signaling pathway, promoting autoimmune responses. In PBC, TRANCE may enhance Th1 and Th17 immune responses, leading to damage of bile duct epithelial cells.41 CXCL10, a chemokine produced by various cells (such as hepatocytes, bile duct epithelial cells, and immune cells), recruits Th1-type T cells to the liver and bile duct regions by binding to the receptor CXCR3, promoting inflammatory responses and bile duct injury. Serum and liver tissue levels of CXCL10 are significantly elevated in PBC patients and are associated with disease activity.43 Further research is needed in the future to clarify the causal interactions between the aforementioned factors and PBC.
Studies have shown that under conditions of cholestasis, the expression of TNFRSF12A (the receptor for TNFSF12, also known as TWEAK) is significantly increased in hepatocytes and human cholangitis. The TWEAK/Tnfrsf12a axis plays a crucial role in liver injury induced by cholestasis by promoting hepatocyte pyroptosis through the activation of the NFκB signaling pathway.44 This finding suggests that TNFSF12 may play an important role in the pathogenesis of PSC, which is a special type of cholestatic liver disease. Our research also indicates that TNFRSF12 may be a risk factor for PSC. These findings lay the groundwork for future exploration of the role of TNFSF12 in PSC and may contribute to the development of new therapeutic strategies. CD6 is a glycoprotein expressed on the surface of T cells and belongs to the scavenger receptor family. CD6 regulates the interaction between T cells and antigen-presenting cells (APCs), affecting the activation and differentiation of T cells. The isoforms of CD6 may participate in the pathogenesis of PBC by altering its binding ability with the ligand Activated Leukocyte Cell Adhesion Molecule (ALCAM), affecting the immune response of T cells.45 Similarly, no studies have discussed the role of CCL20 in PSC. CCL20 has a strong chemoattractant effect on lymphocytes and a weaker effect on neutrophils. It acts on target cells by binding and activating the chemokine receptor CCR6, playing an important role in the formation and function of mucosal lymphoid tissues. Through its role in the chemotaxis, activation, and migration of immune cells, it may play a role in the pathogenesis of PSC. However, more research is needed to clarify the specific role and mechanism of CCL20 in PSC.
In reverse MR analysis, we found that PBC may be associated with the expression levels of CCL4 and OSM. CCL4 is an important chemokine that participates in the process of liver inflammation and fibrosis by recruiting monocytes, macrophages, and T cells to the site of inflammation.46 In PBC, the elevation of CCL4 may exacerbate the inflammatory response around the bile duct, leading to damage to bile duct epithelial cells and disease progression.47 In addition, OSM is an IL-6 family cytokine that promotes liver inflammation and fibrosis by activating the STAT3 signaling pathway. The elevation of OSM may further promote the pathological process of PBC, suggesting its potential as a therapeutic target.48 There is a causal relationship between PSC and five circulating inflammatory proteins (M-CSF, IL-15R α, PD-L1, CD40LR, and LIF). These proteins play important roles in immune regulation and inflammatory response: M-CSF can promote monocyte differentiation into macrophages, enhance inflammatory response and tissue damage.49 IL-15R α mainly regulates the activity of NK cells and T cells, and participates in liver immune response.50 PD-L1 participates in the regulation of immune tolerance by inhibiting T cell activity, and its elevation may be related to the immune escape mechanism of PSC.51 CD40LR participates in the interaction between B cells and T cells, promoting the release of inflammatory factors.52 LIF regulates the differentiation and function of immune cells and may be involved in the inflammatory and fibrotic processes of PSC.53 These findings suggest that the pathogenesis of PSC involves abnormal activation of multiple inflammatory pathways, and treatment strategies targeting these proteins may bring new hope to PSC patients.
Nonetheless, it is important to acknowledge that this study has certain limitations. Firstly, the MR method assumes a linear relationship between genetic variants, cytokine levels, and disease risk, which may not always hold true in complex biological systems. Secondly, it is important to note that our study’s findings are based on data from European populations. While this approach reduces the influence of confounding factors such as population stratification, it also limits the extrapolation of our results to other ethnic groups. Different populations may have distinct genetic backgrounds, environmental exposures, and disease susceptibilities, which could influence the observed associations. Therefore, our findings should be interpreted with caution and further validated in diverse populations to ensure their generalizability. Third, due to the complexity of disease progression, MR studies can only reduce confounding factors to a certain extent and cannot completely eliminate them. Fourthly, in this study, we focused our analysis on the relationship between 91 circulating inflammatory proteins and AILD. It is important to note that there are still additional circulating inflammatory proteins associated with the disease that were not included in our analysis. In the future, the connection between circulating inflammatory proteins and AILD should be more comprehensively validated. Lastly, although we have established causal relationships, the exact biological mechanisms behind these associations require further experimental confirmation. Future research should consider conducting relevant basic experimental studies, such as cell experiments and animal model experiments, to further explore the biological basis of the causal relationships identified in this study.
Conclusion
Our findings highlight a significant causal association between inflammatory proteins and the pathogenesis of AILD (AIH, PBC, and PSC). However, while these results provide valuable insights into the potential roles of these proteins, they do not fully explain the complex and multifaceted effects of these mediators in disease progression. Its potential mechanisms and broader impacts require further research.
Publisher’s Note
All claims expressed in this article are the sole responsibility of the authors and do not necessarily reflect the views of their affiliated organizations, or of the publisher, the editors, and the reviewers. Any product that is assessed in this article, or any claims made by its manufacturer, are not guaranteed or endorsed by the publisher. The article has been translated into English and polished.
Data Sharing Statement
The original contributions presented in the study are included in the article/supplementary material. For further inquiries, please contact the corresponding author.
Ethics Statement
The data in this study were obtained from published studies, of which all data had been approved by the institutional review committee. The ethical application for this study was approved by the Ethics Committee of Xingtai People’s Hospital [Approval number: 2025【014】].
Acknowledgments
We extend our sincere gratitude to all authors for their significant contributions to this research. Their invaluable efforts have played a crucial role in the success of the study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors declare that the research, authorship, and/or publication of this article did not receive any financial support.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Trivedi PJ, Hirschfield GM. Recent advances in clinical practice: epidemiology of autoimmune liver diseases. Gut. 2021;70(10):1989–2003. doi:10.1136/gutjnl-2020-322362
2. Horst AK, Kumashie KG, Neumann K, Diehl L, Tiegs G. Antigen presentation, autoantibody production, and therapeutic targets in autoimmune liver disease. Cell mol Immunol. 2021;18(1):92–111. [PMC free article] [PubMed]. doi:10.1038/s41423-020-00568-6
3. Webb GJ, Ryan RP, Marshall TP, Hirschfield GM. The epidemiology of UK autoimmune liver disease varies with geographic latitude. Clin Gastroenterol Hepatol. 2021;19(12):2587–2596. doi:10.1016/j.cgh.2021.01.029
4. Czaja AJ. Hepatocellular carcinoma and other malignancies in autoimmune hepatitis. Dig Dis Sci. 2013;58(6):1459–1476. doi:10.1007/s10620-012-2525-5
5. Jiang X, Lian M, Li Y, et al. The immunobiology of mucosal-associated invariant T cell (MAIT) function in primary biliary cholangitis: regulation by cholic acid-induced interleukin-7. J Autoimmun. 2018;90:64–75. doi:10.1016/j.jaut.2018.01.007
6. Choi J, Choi GH, Lee D, et al. Long-term clinical outcomes in patients with autoimmune hepatitis according to treatment response in Asian country. Liver Int. 2019;39(5):985–994. doi:10.1111/liv.14082
7. Chang C, Tanaka A, Bowlus C, Gershwin ME. The use of biologics in the treatment of autoimmune liver disease. Expert Opin Investig Drugs. 2020;29(4):385–398. doi:10.1080/13543784.2020.1733527
8. Lohse AW, Mieli-Vergani G. Autoimmune hepatitis. J Hepatol. 2011;55(1):171–182. doi:10.1016/j.jhep.2010.12.012
9. Sucher E, Sucher R, Gradistanac T, Brandacher G, Schneeberger S, Berg T. Autoimmune hepatitis‐immunologically triggered liver pathogenesis‐diagnostic and therapeutic strategies. J Immunol Res. 2019;2019:1–19. doi:10.1155/2019/9437043
10. Trivella J, John BV, Levy C. Primary biliary cholangitis: epidemiology, prognosis, and treatment. Hepatol Commun. 2023;7(6). doi:10.1097/.HC9.0000000000000179
11. Engel B, Taubert R, Jaeckel E, Manns MP. The future of autoimmune liver diseases - Understanding pathogenesis and improving morbidity and mortality. Liver Int. 2020;40(Suppl 1):149–153. doi:10.1111/liv.14378
12. He Y, Hwang S, Ahmed YA, et al. Immunopathobiology and therapeutic targets related to cytokines in liver diseases. Cell mol Immunol. 2021;18(1):18–37. doi:10.1038/s41423-020-00580-w
13. Swerdlow DI, Kuchenbaecker KB, Shah S, et al. Selecting instruments for Mendelian randomization in the wake of genome-wide association studies. Int J Epidemiol. 2016;45(5):1600–1616. doi:10.1093/ije/dyw088
14. W. SV, C. RR, R. WBA, et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: the STROBE-MR statement. JAMA. 2021;326(16):1614–1621. doi:10.1001/jama.2021.18236
15. Little J, Higgins JP, Ioannidis JP, et al. STrengthening the reporting of genetic association studies (STREGA): an extension of the STROBE statement. Ann Intern Med. 2009;150(3):206–215. PMID: 19189911. doi:10.7326/0003-4819-150-3-200902030-00011
16. Zhao JH, Stacey D, Eriksson N, et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat Immunol. 2023;24(9):1540–1551. doi:10.1038/s41590-023-01588-w
17. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–698. doi:10.1038/s41588-018-0099-7
18. Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011;40(3):740–752. doi:10.1093/ije/dyq151
19. Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30(7):543–552. doi:10.1007/s10654-015-0011-z
20. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
21. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–314. doi:10.1002/gepi.21965
22. Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985–1998. doi:10.1093/ije/dyx102
23. Shen C, Chen Z, Zhang W, Chen X, Zheng B, Shi C. Preliminary study of the effect of gut microbiota on the development of prostatitis. BMC Med Genomics. 2024;17(1):35. doi:10.1186/s12920-024-01812-y
24. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–665. doi:10.1002/gepi.21758
25. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377–389. doi:10.1007/s10654-017-0255-x
26. Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology. 2017;28(1):30–42. doi:10.1097/EDE.0000000000000559
27. Khan T, Kryza T, Lyons NJ, He Y, Hooper JD. The cdcp1 signaling hub: a target for cancer detection and therapeutic intervention. Cancer Res. 2021;81(9):2259–2269. doi:10.1158/0008-5472.CAN-20-2978
28. Dong Y, He Y, De Boer L, et al. The cell surface glycoprotein cub domain-containing protein 1 (Cdcp1) contributes to epidermal growth factor receptor-mediated cell migration. J Biol Chem. 2012;287(13):9792–9803. doi:10.1074/jbc.M111.335448
29. He Y, Harrington BS, Hooper JD. New crossroads for potential therapeutic intervention in cancer - intersections between cdcp1, egfr family members and downstream signaling pathways. Oncoscience. 2016;3(1):5–8. doi:10.18632/oncoscience.v3i1
30. Enyindah-Asonye G, Li Y, Ruth JH, et al. CD318 is a ligand for CD6. Proc Natl Acad Sci USA. 2017;114(33):E6912–E6921. doi:10.1073/pnas.1704008114
31. Swaminathan B, Cuapio A, Alloza I, et al. Fine mapping and functional analysis of the multiple sclerosis risk gene CD6. PLoS One. 2013;8(4):e62376. doi:10.1371/journal.pone.0062376
32. Zheng M, Zhang L, Yu H, et al. Genetic polymorphisms of cell adhesion molecules in Behcet’s disease in a Chinese Han population. Sci Rep. 2016;6(1):24974. doi:10.1038/srep24974
33. Zhang L, Borjini N, Lun Y, et al. CDCP1 regulates retinal pigmented epithelial barrier integrity for the development of experimental autoimmune uveitis. JCI Insight. 2022;7(18):e157038. doi:10.1172/jci.insight.157038
34. Fan X, Men R, Huang C, et al. Critical roles of conventional dendritic cells in autoimmune hepatitis via autophagy regulation. Cell Death Dis. 2020;11(1):23. doi:10.1038/s41419-019-2217-6
35. Herkel J, Carambia A, Lohse AW. Autoimmune hepatitis: possible triggers, potential treatments. J Hepatol. 2020;73(2):446–448. doi:10.1016/j.jhep.2020.03.015
36. Landy E, Carol H, Ring A, Canna S. Biological and clinical roles of IL-18 in inflammatory diseases. Nat Rev Rheumatol. 2024;20(1):33–47. doi:10.1038/s41584-023-01053-w
37. Ihim SA, Abubakar SD, Zian Z, et al. Interleukin-18 cytokine in immunity, inflammation, and autoimmunity: biological role in induction, regulation, and treatment. Front Immunol. 2022;11(13):919973. doi:10.3389/fimmu.2022.919973
38. Ikeda A, Aoki N, Kido M, et al. Progression of autoimmune hepatitis is mediated by IL-18-producing dendritic cells and hepatic CXCL9 expression in mice. Hepatology. 2014;60(1):224–236. doi:10.1002/hep.27087
39. Jones DEJ, Wetten A, Barron-Millar B, et al. The relationship between disease activity and UDCA response criteria in primary biliary cholangitis: a cohort study. EBioMedicine. 2022;80:104068. doi:10.1016/j.ebiom.2022.104068
40. Yang Y, He X, Rojas M, Leung PSC, Gao L. Mechanism-based target therapy in primary biliary cholangitis: opportunities before liver cirrhosis? Front Immunol. 2023;30(14):1184252. doi:10.3389/fimmu.2023.1184252
41. Lleo A, Invernizzi P, Mackay IR, et al. Etiopathogenesis of primary biliary cirrhosis. World J Gastroenterol. 2008;14(21):3328–3337. doi:10.3748/wjg.14.3328
42. Abe M, Hiasa Y, Onji M. T helper 17 cells in autoimmune liver diseases. Clin Dev Immunol. 2013;2013:607073. doi:10.1155/2013/607073
43. Gulamhusein AF, Hirschfield GM. Primary biliary cholangitis: pathogenesis and therapeutic opportunities. Nat Rev Gastroenterol Hepatol. 2020;17(2):93–110. doi:10.1038/s41575-019-0226-7
44. Li Z, Wang H, Zhu J, et al. Inhibition of TWEAK/Tnfrsf12a axis protects against acute liver failure by suppressing RIPK1-dependent apoptosis. Cell Death Discov. 2022;8(1):328. doi:10.1038/s41420-022-01123-0
45. Pinto M, Carmo AM. CD6 as a therapeutic target in autoimmune diseases: successes and challenges. BioDrugs. 2013;27(3):191–202. PMID: 23568178. doi:10.1007/s40259-013-0027-4
46. Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology. 2014;147(3):577–594.e1. doi:10.1053/j.gastro.2014.06.043
47. Oo YH, Weston CJ, Lalor PF, et al. Distinct roles for CCR4 and CXCR3 in the recruitment and positioning of regulatory T cells in the inflamed human liver. J Immunol. 2010;184(6):2886–2898. doi:10.4049/jimmunol.0901216
48. Matsuda M, Tsurusaki S, Miyata N, et al. Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatology. 2018;67(1):296–312. doi:10.1002/hep.29421
49. Cuevas VD, Simón-Fuentes M, Orta-Zavalza E, et al. The gene signature of activated M-CSF-primed human monocyte-derived macrophages is IL-10-dependent. J Innate Immun. 2022;14(3):243–256. doi:10.1159/000519305
50. Di Scala M, Gil-Fariña I, Olagüe C, et al. Identification of IFN-γ-producing T cells as the main mediators of the side effects associated to mouse interleukin-15 sustained exposure. Oncotarget. 2016;7(31):49008–49026. doi:10.18632/oncotarget.10264
51. Kocheise L, Piseddu I, Vonderlin J, et al. PD-1/PD-L1 immune checkpoint therapy demonstrates favorable safety profile in patients with autoimmune and cholestatic liver disease. Front Immunol. 2024;14:1326078. doi:10.3389/fimmu.2023.1326078
52. Müller MR, Wiesmüller KH, Jung G, et al. Lipopeptide adjuvants: monitoring and comparison of P3CSK4- and LPS-induced gene transcription. Int Immunopharmacol. 2002;2(8):1065–1077. doi:10.1016/s1567-5769(02)00030-9
53. Wu D, Guo J, Qi B, et al. TGF-β1 induced proliferation, migration, and ECM accumulation through the SNHG11/miR-34b/LIF pathway in human pancreatic stellate cells. Endocr J. 2021;68(11):1347–1357. doi:10.1507/endocrj.EJ21-0176
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Causal Association of Primary Biliary Cholangitis with Adverse Pregnancy and Neonatal Outcomes: A Two-Sample Mendelian Randomization Study
Li R, Tan J, Yang X, Ning Z
International Journal of Women's Health 2025, 17:407-415
Published Date: 17 February 2025