Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Cattle encephalon glycoside and ignotin injection improves cognitive impairment in APPswe/PS1dE9 mice used as multitarget anti-Alzheimer’s drug candidates
Authors Gao Y, Hu Y, Li R, Han Z, Geng Y, Xia Z, Du W, Liu L, Zhang H, Wang L
Received 24 November 2014
Accepted for publication 21 January 2015
Published 27 February 2015 Volume 2015:11 Pages 537—548
DOI https://doi.org/10.2147/NDT.S78025
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Ya Gao,1,* Ya-zhuo Hu,1,* Rui-sheng Li,2 Zhi-tao Han,1 Yan Geng,1 Zheng Xia,1 Wen-jin Du,3 Li-xin Liu,4 Hong-hong Zhang,1 Lu-ning Wang5
1Institute of Geriatrics, Chinese PLA General Hospital, Beijing Key Lab of Normal Aging and Geriatrics, 2Research and Technology Service Center, PLA 302 Hospital, 3Department of Neurology, Air Force General Hospital, 4Department of Neurology, Beijing Geriatric Hospital, 5Department of Geriatric Neurology, Chinese PLA General Hospital, Beijing, People’s Republic of China
*These authors contributed equally to this work and are joint first authors
Background: Cattle encephalon glycoside and ignotin injection (CEGI), a multitargeted neurotrophic drug, has been widely used in the treatment of central and peripheral nerve injuries, such as stroke, hypoxic ischemic encephalopathy, and diabetic neuropathy in the People’s Republic of China. However, data regarding the effect of CEGI on Alzheimer’s disease (AD) remain scarce. The present study aimed to investigate the effect of CEGI on learning and memory in an APPswe/PS1dE9 double-transgenic mouse model, a suitable animal model of AD, and elucidate its possible mechanisms.
Materials and methods: Five-month-old APP/PS1 mice were intraperitoneally administered 6.6 mL/kg or 13.2 mL/kg of CEGI for 1 month. After 1 month of administration, all mice received Morris water maze training and a probe test. Mouse brain sections were detected by standard biochemical and immunohistochemical measures.
Results: CEGI treatment significantly improved the spatial learning and memory deficits and decreased cerebral amyloid-β42 levels in brain homogenates of APP/PS1 mice. CEGI treatment elevated the activities of superoxide dismutase, and reduced the levels of malondialdehyde. CEGI attenuated neuronal damage in the hippocampus of APP/PS1 mice and upregulated protein and gene expression of Bcl-2 and the ratio of Bcl-2/Bax. CEGI treatment decreased the number of Iba1+ activated microglia in the cortex of the APP/PS1 mice.
Conclusion: Our results showed that CEGI prevents memory impairment, possibly by decreasing the amyloid-β42 levels in APP/PS1 mice and inhibiting oxidative stress, apoptosis, and inflammation, making CEGI a promising therapeutic agent for AD.
Keywords: Alzheimer’s disease, cognitive impairment, amyloid-β, oxidative stress, apoptosis, inflammation
Introduction
Alzheimer’s disease (AD) is the most common form of dementia worldwide, and the mechanisms have not been clearly elucidated so far. According to the “amyloid cascade” hypothesis, the aggregation of amyloid-β (Aβ) initiates a sequence of events leading to the formation of neurofibrillary tangles, neurodegeneration, and on to the main symptom of dementia. Besides, many studies have found that increased oxidative stress is a key feature involved in the pathogenesis of AD.1–3 In addition, research has shown that cholinergic dysfunction is a characteristic pathological hallmark of AD: it is significantly involved in memory deficits, resulting in the formulation of the “cholinergic hypothesis” of AD.4 Furthermore, some studies suggest that neuroinflammation might contribute to the progression of AD, and anti-inflammatory therapy would be protective against the development of AD in humans.5–7 Therefore, multitarget treatment is expected to improve the prognosis of patients with AD.
Although acetylcholinesterase inhibitors, such as donepezil and galantamine, have been reported to partially delay the progression of AD, they lead to undesirable side effects in AD patients, including nausea, vomiting, diarrhea, and cardiovascular events.8,9 Therefore, it is necessary to find a safe and complementary therapy for AD.
Cattle encephalon glycoside and ignotin injection (CEGI) is a combination of mixed gangliosides (such as GM1), polypeptides (such as carnosine), free amino acids, nucleic acids, and total nitrogen. One milliliter of CEGI contains 50 μg gangliosides, 3.2 mg polypeptides, 1.65 mg free amino acids, 0.3 mg nucleic acids, and 0.925 mg total nitrogen. CEGI (drug approval H22025046; Sihuan Pharmaceutical Holdings Group Ltd, Jilin, People’s Republic of China) was approved by the China Food and Drug Administration in 2010, and is widely used in the treatment of central and peripheral nerve injuries, such as stroke, neonatal hypoxic ischemic encephalopathy, and diabetic peripheral neuropathy, in the People’s Republic of China (PRC), with high safety and few side effects.10 However, the role of CEGI in AD has not been reported.
Certain active constituents of CEGI have been proven to have therapeutic efficacy in AD. For example, continuous intraventricular infusion of GM1 has been shown to have a modest beneficial effect in early onset of AD.11,12 Also, Matsuoka et al reported that peripheral administration of GM1 may reduce or prevent brain amyloidosis in APP/PS1 double-transgenic mice.13 Moreover, GM1 improved spatial learning and memory deficits in a rat model of AD through the inhibition of neuronal oxidative stress and lipid peroxidation.14 Carnosine, a dipeptide consisting of β-alanine and histidine, has been reported to be partially effective in protecting neuronal cultures against Aβ-induced damage in vitro.15 Evidence also suggests that dietary carnosine supplementation (10 mM in drinking water) reduces the Aβ load in triple-transgenic (3× Tg) AD mice.16 Moreover, carnosine prevented the cognitive impairments observed in an APP/PS1 mouse model on a high-fat diet.17 These results suggest that CEGI may be beneficial in the prevention and treatment of AD.
In this study, we used CEGI administration and behavioral testing in an APP/PS1 mouse model of AD, as well as standard biochemical and immunohistochemical measures in the brain tissue, in order to study the effect of CEGI in alleviating memory deficits, and attempted to elucidate the underlying mechanisms.
Materials and methods
Animals
A total of 48 male homozygous APPswe/PS1dE9 double-transgenic mice (5 months of age) and 12 age- and sex-matched wild-type littermates were obtained from Beijing HFK Bio-technology, Institute of Laboratory Animal Science, Chinese Academy of Medical Science (Beijing, PRC). These transgenic mice were maintained on a C57BL/6J background and coexpressed a mouse/human APP Swedish double mutation (K595N/M596L) and human PS1-dE9 mutation under the control of the mouse prion protein promoter, and have previously been reported to exhibit age-related increase in concentrations of Aβ and cognitive deficits, and develop Aβ plaques at 4–5 months of age.18–20 The mice were housed in separate home cages under temperature- (23°C±1°C) and humidity-controlled (55%) conditions with a 12-hour light/dark cycle (lights switched on at 7 am) and free access to food and tap water. All experiments were approved by the Institutional Animal Experiment Committee of the PLA 302 Hospital.
Groups and drug administration
CEGI and donepezil were obtained from Jilin Sihuan Pharmaceutical Holdings Group Ltd and Eisai China, respectively. APPswe/PS1dE9 mice were randomly divided into four groups of 12 mice each, namely a Tg group (intraperitoneal injection of equal volume of 0.9% saline), a low-dose CEGI group (CEGI-L; intraperitoneal injection of 6.6 mL/kg/day CEGI), a high-dose CEGI group (CEGI-H; intraperitoneal injection of 13.2 mL/kg/day CEGI), and a positive control group (intragastric administration of 2 mg/kg/day donepezil). Twelve non-transgenic (nTg) wild-type littermates were used as normal control (intraperitoneal injection of equal volume of 0.9% saline). The mice were weighed every 3 days. After 1 month of drug administration, the Morris water maze (MWM) test was performed.
Behavioral testing
The MWM test included a training session for acquisition of the hidden platform and a probe trial session. The mice were subjected to four trials per day over 5 consecutive days. A mouse was given 60 seconds to find the submerged platform. On reaching the platform, the mouse was allowed to stay on it for 10 seconds. If the mouse failed to locate the platform within 60 seconds, it was guided to the hidden platform by the experimenter and allowed to rest on it for 10 seconds. The time spent by the mice in finding the platform during each session was recorded as the escape latency.
On the day following the last training trial sessions, a probe test was conducted. The mouse was released into the water and allowed to swim freely for 60 seconds. The swimming time spent in the target quadrant was recorded and analyzed.
Brain-tissue preparation
After behavioral testing, all mice were deeply anesthetized with sodium pentobarbital (100 mg/kg intraperitoneally). The mice in each group were transcardially perfused with cold 0.9% saline. Their brains were removed and dissected through the skull. The left hemisphere was immersion-fixed in 4% paraformaldehyde for 48 hours, followed by dehydrate treatment and embedding in paraffin for immunohistochemical and histological analyses. The right hemisphere was stored at −80°C and used for biochemical measurements.
Quantification of total Aβ42 levels in the brains by ELISA
The brain tissues were weighed and homogenized in 10 vol/wt cold guanidine buffer (5 M guanidine HCl/50 mM Tris-HCl) and mixed for 4 hours at room temperature. Dilutions of the extracts were prepared in Dulbecco’s phosphate-buffered saline containing 5% bovine serum albumin and 0.03% Tween 20 (pH 7.4) supplemented with 1× protease-inhibitor cocktail (P8340; Sigma, St Louis, MO, USA). The homogenates were aliquoted and centrifuged at 16,000 g for 20 minutes at 4°C. The supernatants were collected and analyzed with an Aβ42 enzyme-linked immunosorbent assay (ELISA) kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Data obtained from the brain homogenates are expressed as ng/mg total protein.
Assay of oxidative damage markers and cholinergic markers
Briefly, the brain tissues were weighed and homogenized on ice with nine volumes of ice-cold saline and centrifuged at 2,500 g for 10 minutes to obtain the supernatant. Levels of superoxide dismutase (SOD), malondialdehyde (MDA), choline acetyltransferase (ChAT) and acetylcholinesterase (AChE) were measured with assay kits A001-1, A003-1, A079-1, and A024, respectively, from Nanjing Jiancheng Bioengineering Institute, according to the manufacturer’s instructions.
Immunohistochemical and histological analysis
According to The Mouse Brain in Stereotaxic Coordinates, 4 μm-thick contiguous sections were cut in the coronal plane throughout the region of the dorsal hippocampus (bregma −1.46 to −2.18).21 Two successive sections were collected every 40 μm and mounted onto 3-aminopropyltriethoxysilane-coated slides for the following analysis.
To observe the morphological characteristics, after deparaffinization and rehydration, the sections were stained with hematoxylin for 10 minutes, differentiated with 1% hydrochloric acid alcohol, and stained with eosin for 5 minutes. For immunohistochemical staining, after deparaffinization and rehydration, the sections were subjected to heat-mediated antigen retrieval in 10 mM citrate buffer (pH 6.0) and incubated with 3% H2O2 for 10 minutes to eliminate endogenous peroxidase activity. The sections were then incubated overnight with rabbit monoclonal antibodies anti-Bcl-2 (1:100; Millipore, Billerica, MA, USA), anti-Bax (1:200; Abcam, San Francisco, CA, USA), and anti-Iba1 (1:400; Wako, Chuo-ku, Osaka, Japan) at 4°C. The primary antibody was detected with biotinylated secondary antibody and visualized with 3,3′-diaminobenzidine (Dako, Carpinteria, CA, USA). As negative control, sections from the same group of animals were treated in the same manner, except that the primary antibody was replaced by phosphate-buffered saline.
Immunohistochemistry/fluorescence co-staining of brains for microglia and Aβ plaques was conducted. Sections were incubated with anti-Iba1 antibody for the detection of activated microglia as mentioned earlier, then stained with 1% thioflavin S (Sigma) for 10 minutes in the dark for the detection of Aβ plaques. Finally, the sections were mounted with VectaMount and digitally photographed via bright-field microscopy to detect Iba1+ activated microglia, and via fluorescence to visualize the green thioflavin S-fluorescence staining for Aβ plaques. For morphological subtypes of Iba1+ microglia, ramified and intermediate represented quiescent microglia, and amoeboid and round represented activated microglia. Activated microglia and thioflavin S-stained plaques were assessed manually.
All sections from each group containing eight to nine mice were observed using a light microscope (Olympus BX60; Tokyo, Japan), and photo images were taken with the Image-Pro Plus 5.1 image-analysis system. The mean integrated optical density (MIOD = IOD/total area) was measured to assess the expression levels of Bax and Bcl-2 after immunostaining in CA1 of the hippocampus.
RNA isolation and RT-PCR
Total ribonucleic acid (RNA) was extracted from mice brains using the Animal RNAout kit (Tiandz, Beijing, People’s Republic of China) according to the manufacturer’s instructions. The specific primers were utilized for polymerase chain reaction (PCR) amplification, as described previously.22 The reverse-transcription polymerase chain reaction (RT-PCR) procedure was carried out using a PCR apparatus (Thermo, Waltham, MA, USA) according to the manufacturer’s protocol of the One Step RT-PCR kit (Takara, Tokyo, Japan) under the following conditions: 50°C for 30 minutes, 94°C for 2 minutes (30 cycles), 94°C for 30 seconds, 58°C for 30 seconds, and 72°C for 1 minute. The PCR products were electrophoresed on 2.5% agarose gels, stained with ethidium bromide, and analyzed using a Gel Doc™ XR+ imaging system (Bio-Rad, Hercules, CA, USA).
Statistical analysis
All data were expressed as means ± standard error of the mean. P<0.05 was considered statistically significant. The treatment differences in the escape latency in the MWM task were analyzed using repeated-measures one-way analysis of variance (ANOVA). The other data were analyzed using ANOVA followed by the least significant difference test in order to detect intergroup differences.
Results
The general health of the APP/PS1 mice during CEGI treatment was monitored, and no abnormal changes were observed. Administration of CEGI for 1 month tended to increase body weight, but the increase did not reach statistical significance (before injection, nTg 32.0±1.06 g, Tg 32.2±1.64 g, Tg + CEGI-L 32.6±1.88 g, Tg + CEGI-H 32.5±2.04 g, Tg + donepezil 32.4±2.21 g; after injection nTg 32.6±1.19 g, Tg 33.1±1.39 g, Tg + CEGI-L 33.5±1.46 g, Tg + CEGI-H 33.8±1.69 g, Tg + donepezil 33.3±1.97 g). Together, these data indicate that CEGI had no significant toxicity in mice.
CEGI improved spatial learning and memory deficits in APP/PS1 transgenic mice
Repeated-measures ANOVA revealed a significant treatment effect (Figure 1A, P<0.05) on the escape latency. APP/PS1 mice showed significant cognitive impairment compared to the nTg group. The CEGI-L (6.6 ml/kg) group had significantly reduced escape latency compared to the Tg group (Figure 1A, P<0.05). However, the CEGI-H (13.2 mL/kg) and donepezil (2 mg/kg) groups did not have significantly reduced escape latency compared to the Tg group (Figure 1A).
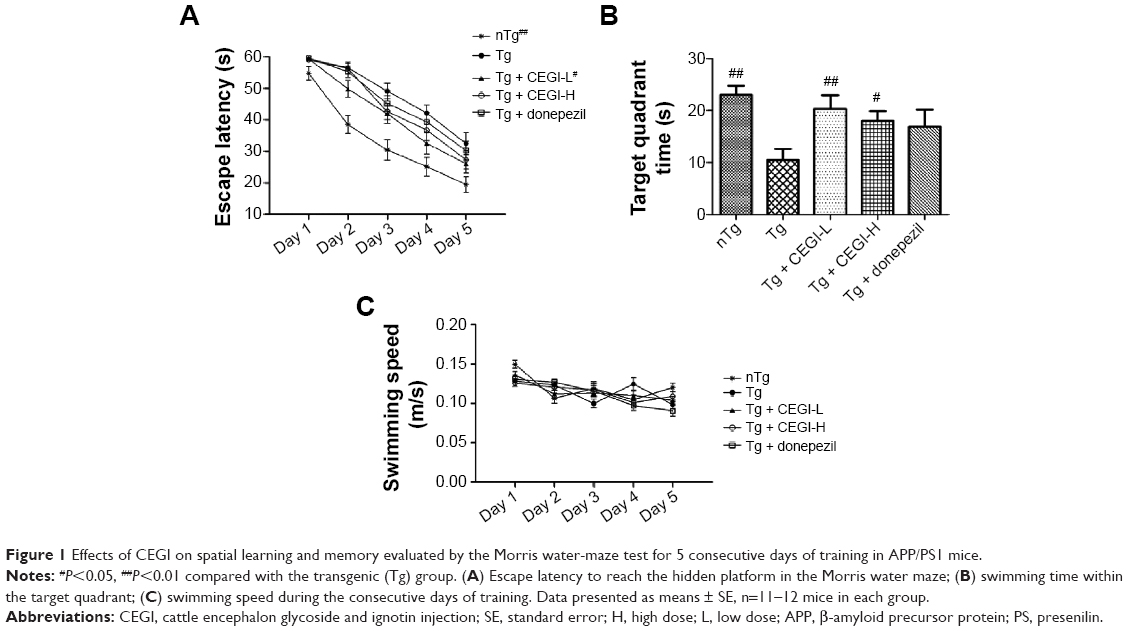 | Figure 1 Effects of CEGI on spatial learning and memory evaluated by the Morris water-maze test for 5 consecutive days of training in APP/PS1 mice. |
The time spent in the target quadrant was significantly reduced among the Tg mice compared to the nTg mice (Figure 1B, P<0.05). The CEGI-L and CEGI-H groups showed significantly increased time spent in the target quadrant compared to the Tg mice (Figure 1B, P<0.01 and P<0.05, respectively). The donepezil (2 mg/kg) group, however, did not significantly increase their time spent in the target quadrant compared to the Tg mice (Figure 1B).
In addition, there was no significant difference in the swimming speed among mice in the five different groups (Figure 1C). This implies that the memory improvement in CEGI mice was not due to motor dysfunctions.
CEGI treatment significantly reduces Aβ42 levels in brains of APP/PS1 transgenic mice
We examined the levels of total Aβ42 in whole brains using specific ELISA tests (Figure 2). The CEGI (6.6 mL/kg or 13.2 mL/kg) group had significantly decreased Aβ42 levels compared to the Tg mice (P<0.01). The donepezil group also had significantly decreased Aβ42 levels compared to the Tg mice (P<0.05).
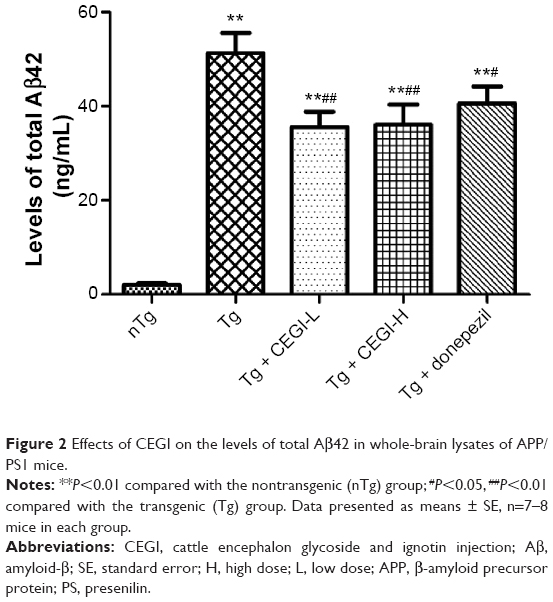 | Figure 2 Effects of CEGI on the levels of total Aβ42 in whole-brain lysates of APP/PS1 mice. |
Activities of oxidative damage markers and cholinergic markers in CEGI-treated APP/PS1 transgenic mice
SOD activity was found to be significantly decreased in Tg group compared to the nTg group (Figure 3A, P<0.01), and a significant elevation was detected in SOD activity in the CEGI-L group compared to the Tg group (Figure 3A, P<0.05). MDA levels were significantly increased in the Tg group compared to the nTg group (Figure 3B, P<0.05), and a significant reduction was detected in MDA levels in the CEGI-L and donepezil groups compared to the Tg group (Figure 3B, P<0.05).
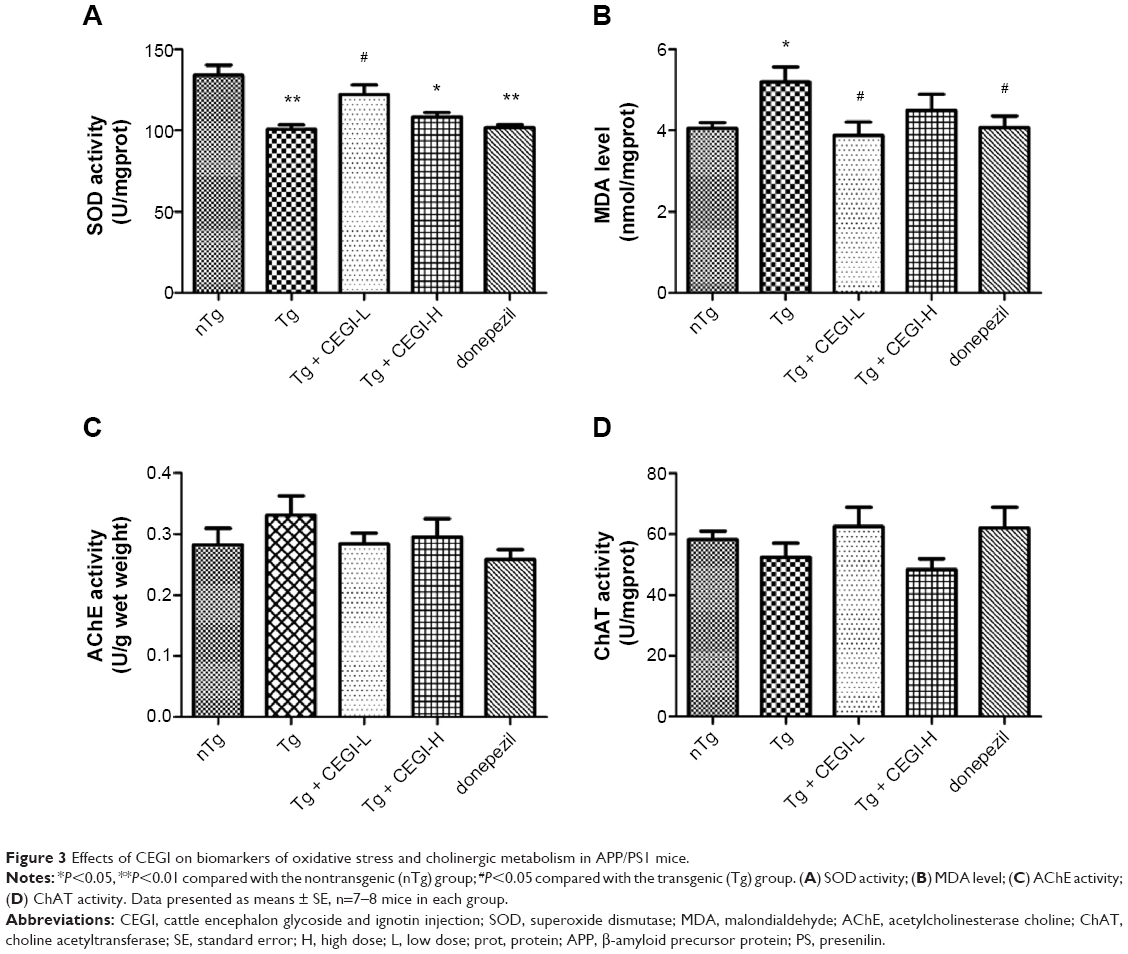 | Figure 3 Effects of CEGI on biomarkers of oxidative stress and cholinergic metabolism in APP/PS1 mice. |
As shown in Figure 4, there were no significant differences in cholinergic measures (AChE and ChAT) among the five different groups. However, mice from the CEGI-L and donepezil groups (especially the donepezil group) showed slightly lower AChE (Figure 3C) and slightly higher ChAT compared to Tg mice (Figure 3D).
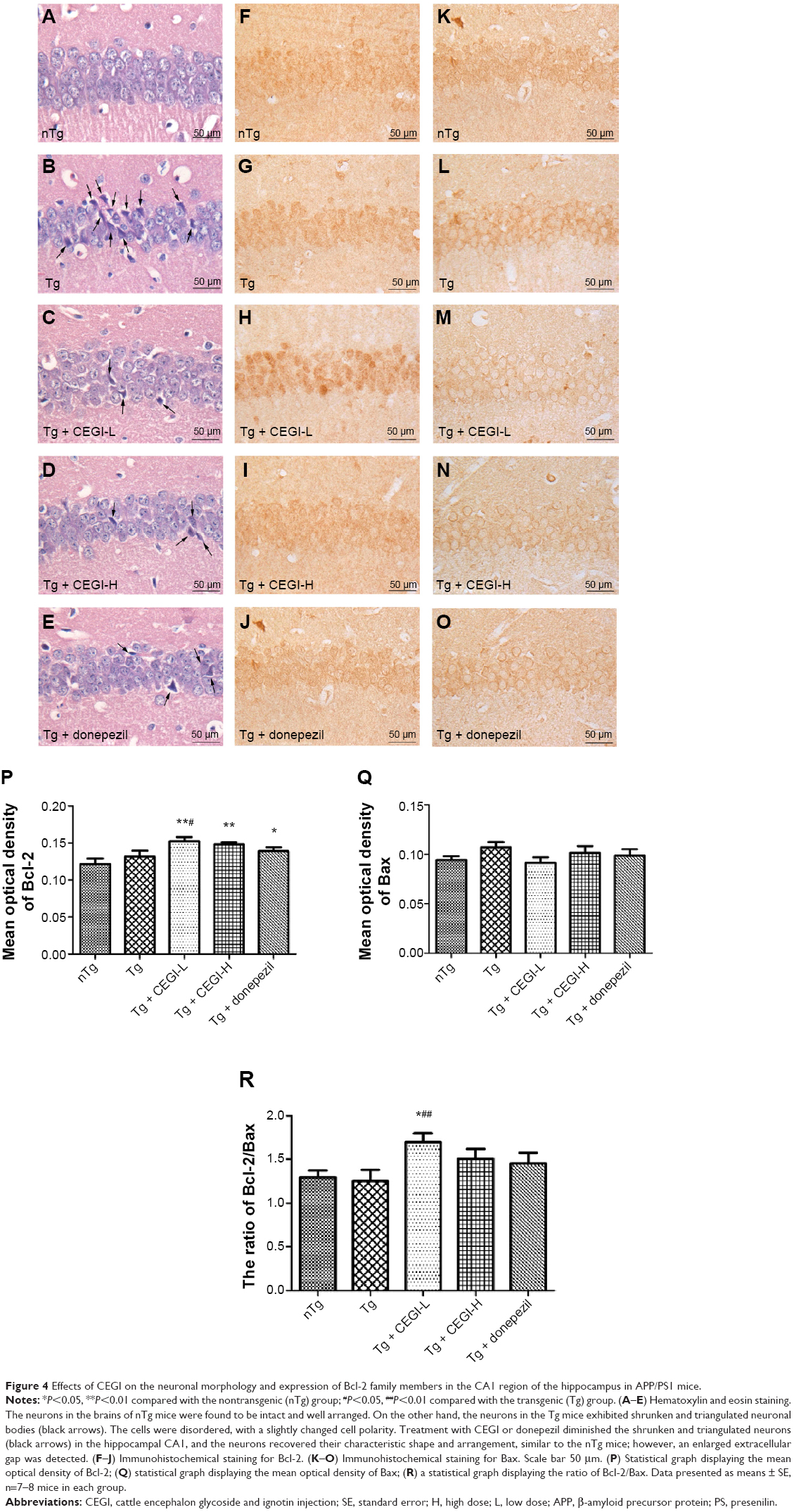 | Figure 4 Effects of CEGI on the neuronal morphology and expression of Bcl-2 family members in the CA1 region of the hippocampus in APP/PS1 mice. |
CEGI treatment changed neuronal morphology and increased protein expression of Bcl-2 and Bcl-2/Bax ratio in the hippocampus of the APP/PS1 transgenic mice
The neurons in the hippocampus CA1 area of Tg mice were arranged in a disordered manner and showed signs of neurodegeneration, such as dark staining and shrunken and triangulated neuronal bodies (Figure 4B). In CEGI- and donepezil-treated mouse brains, the structure of the neurons appeared clear, with abundant cytoplasm and round or oval nuclei, and fewer signs of neurodegeneration (Figure 4C–E).
The expression of Bax and Bcl-2 proteins was detected in the CA1 area of the hippocampus (Figure 4F–O). The Bax staining appeared as specific cytoplasmic granules localized to the cytoplasm, while Bcl-2 proteins were found in the perinuclear regions as well as cytoplasmic regions. The hippocampal CA1 area in mice from the nTg group revealed less Bcl-2+ and Bax+ particles (Figure 4F and K). There were no significant differences in the optical density of Bax+ and Bcl-2+ neurons between nTg mice and Tg mice (Figure 4P and Q). Treatment with CEGI-L showed a significant increase in the optical density of Bcl-2+ neurons (Figure 4H and P; P<0.05), and a decline (but not significant) in the optical density of Bax+ neurons (Figure 4M and Q), resulting in a significant increase in the ratio of Bcl-2/Bax expression relative to Tg mice (Figure 4R, P<0.01).
CEGI treatment increased mRNA expression of Bcl-2 and Bcl-2/Bax ratio in the hippocampus of the APP/PS1 transgenic mice
The CEGI-L group showed significantly elevated messenger RNA (mRNA) levels of Bcl-2 and Bcl-2/Bax ratio compared to the Tg group (Figure 5A, B, and D; P<0.05). However, no significant change was observed in the level of Bax mRNA expression among all treatment groups (Figure 5A and C; P<0.05). The results of protein expression were consistent with the mRNA levels of these genes, which indicated that the protein and gene expression of Bcl-2 and Bcl-2/Bax ratio were affected by the administration of CEGI-L.
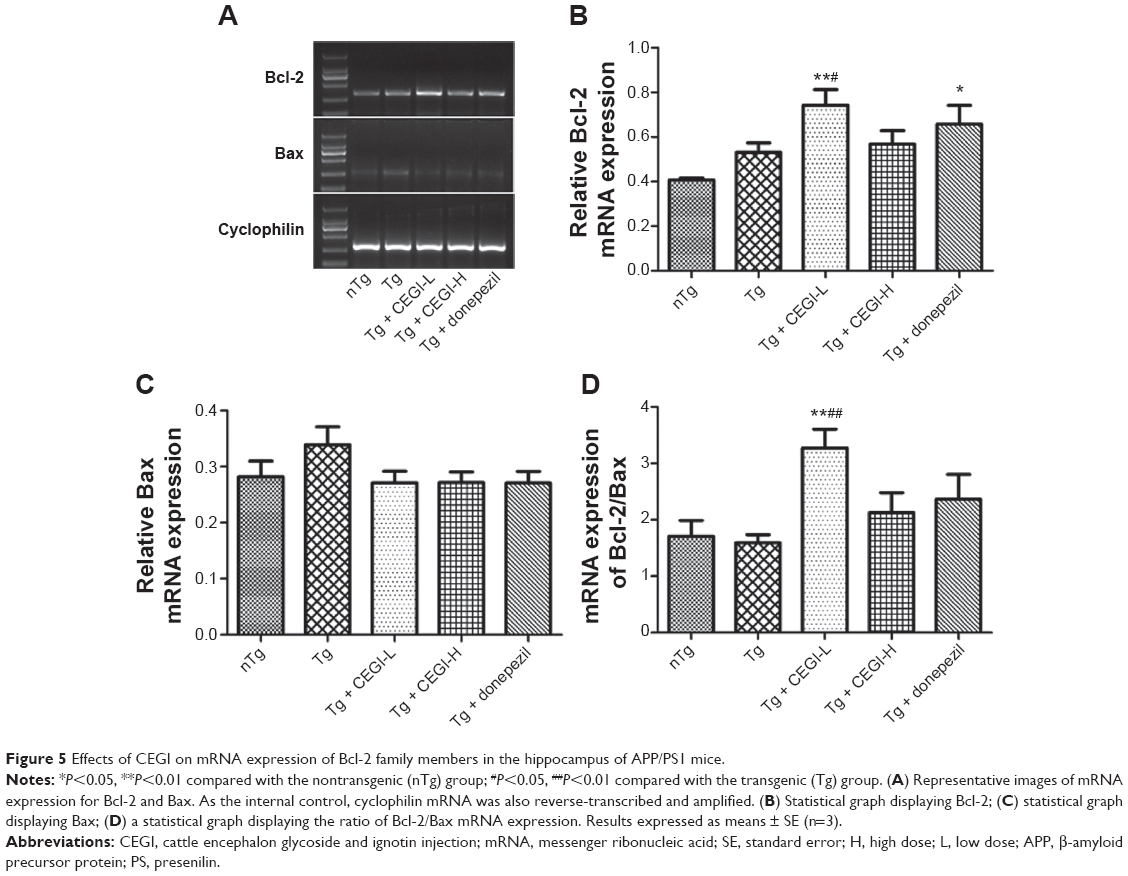 | Figure 5 Effects of CEGI on mRNA expression of Bcl-2 family members in the hippocampus of APP/PS1 mice. |
CEGI treatment decreased the number of Iba1+ activated microglia in the cortex of the APP/PS1 transgenic mice
From the immunohistochemistry/fluorescence co-staining of brains for microglia and Aβ plaques, we observed that Iba1+ activated microglia were present in the vicinity of Aβ deposits (Figure 6A and B). No activated microglia were found in the nTg group (Figure 6C). Treatment of 5-month-old Tg mice with CEGI for 1 month reduced Iba1 immunoreactivity in microglia cells and the numbers of activated microglia in the cortical areas (Figure 6E, F, and H; P<0.05). A decline also occurred in the donepezil-treated group (Figure 6G and H; P<0.05).
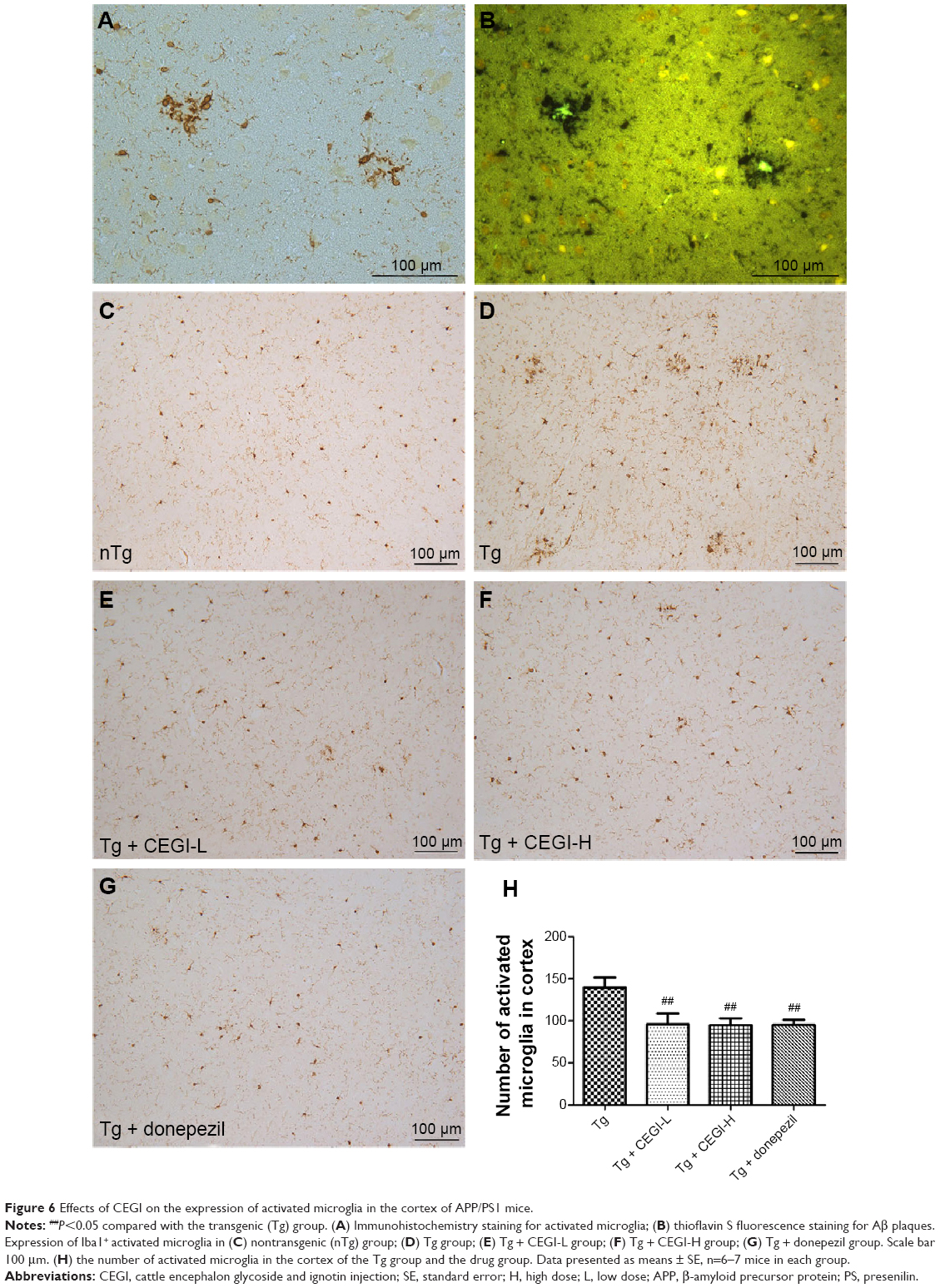 | Figure 6 Effects of CEGI on the expression of activated microglia in the cortex of APP/PS1 mice. |
Discussion
The mechanisms underlying AD include amyloidogenic hypotheses, increased oxidative stress, inflammation and apoptosis, impaired energy metabolism, and cholinergic dysfunction. Given the multifactorial nature of AD, multitarget treatment is expected to improve the prognosis of patients with AD.
Aβ is believed to play an important role in the development of cognitive deficits in AD. Furthermore, it is theorized that increased release of Aβ42 leads to the abnormal deposition of Aβ and the associated neurotoxicity in the brains of affected individuals.23,24 In patients with AD, reduced levels of Aβ42 in cerebrospinal fluid have been described as predictive of AD.25 Administration of CEGI for 1 month significantly improved spatial learning and memory deficits and reduced Aβ42 levels in 6-month-old APP/PS1 mice. Although donepezil treatment also significantly decreased Aβ42 levels in APP/PS1 mice, it did not improve their spatial learning or memory deficits. The beneficial effects were more apparent at the lower dose (6.6 mL/mg). These results suggest that the beneficial effects of CEGI can be better observed within a certain dosage range.
Recent evidence suggests that oxidative stress is an early event in AD and plays a key role in Aβ-induced cell death.26,27 Oxidative stress or damage, such as protein oxidation, lipid oxidation, deoxyribonucleic acid oxidation, and glycoxidation, is closely associated with the development of AD.2 It has also been shown that Aβ-mediated oxidative stress could be attributed to either decreased endogenous antioxidants or increased production of reactive oxygen species.28 SOD is an important antioxidant enzyme against free radicals, and prevents subsequent lipid peroxidation. MDA is an index of lipid peroxidation of biomembranes, and indirectly reflects the extent of injury. In this study, CEGI-treated mice had significantly higher SOD as well as significantly lower MDA compared with the APP/PS1 mice, which suggested that CEGI inhibits oxidative stress in APP/PS1 mice.
It has been well documented that the degree of cognitive impairments in AD patients is significantly correlated with a dysfunction in the cholinergic system.4,29 In terms of cholinergic pathology, mice that overexpress mutant APP and PS1 genes display age-dependent degeneration of cholinergic fibers and alteration of cholinergic metabolic markers.30,31 The activity of ChAT enzymes decreases significantly in the cortex and the hippocampus of middle- or old-aged rather than younger APP/PS1 mice.32,33 Our study showed no significant differences in measures of cholinergic function (AChE and ChAT) between 6-month-old nTg mice and APP/PS1 mice, suggesting that cholinergic function was intact in early stage APP/PS1 mice. However, CEGI- or donepezil-treated APP/PS1 Tg mice (especially the donepezil-treated ones) showed increased ChAT activity and reduced AChE activity in the frontal cortex (difference not significant). Therefore, further confirmation in larger- and older-sample studies is required to prove this finding.
Hippocampal damage is one of the most important causes of amnesia, and neurons in the CA1 area are the principal sites of pathogenesis in AD.34 In support of the histopathology, neurons in the hippocampal CA1 area revealed degenerative changes in the brains of 6-month-old APP/PS1 mice. The improved neuronal status in CEGI-treated mice provided evidence of its neuroprotective effect. Bcl-2 family members, such as Bcl-2 and Bax, are the major regulators of the apoptosis.35 Although our results show Bcl-2 or Bax may not be a player in the cognitive deficits in the APP/PS1 mice, CEGI could markedly upregulate protein and mRNA levels of Bcl-2, playing a major role in the protection of central nervous system cells against a variety of insults.36 Simultaneously, CEGI significantly elevated the ratio of Bcl-2/Bax, which determines survival or death of neuron following Aβ insult.37 We hypothesize that CEGI might play a potential neuroprotective role by upregulating Bcl-2, an antiapoptotic protein. Further studies are necessary to verify the findings involved in the effects of CEGI on apoptosis in neuronal cell cultures.
Increasing evidence has shown that activated microglia as well as their released proinflammatory mediators may enhance the neurotoxicity of Aβ and exacerbate AD-associated pathology.38–40 Also, some investigations have revealed that activated microglia load in the brain was inversely correlated with cognitive functions in AD patients41 and APP/PS1 Tg mice.5,7 In this study, we found that the amount of activated microglia were significantly increased in APP/PS1 mice, suggesting that increased inflammatory responses might contribute to the development of cognitive deficits. Furthermore, CEGI treatment significantly reduced the numbers of activated microglia in the cortex, suggesting that CEGI can decrease inflammatory responses.
Conclusion
In the present study, we demonstrated that administration of CEGI prevents memory impairment in 6-month-old APPswe/PS1dE9 Tg mice, possibly by decreasing the Aβ42 level and inhibiting oxidative stress, apoptosis, and inflammation, which confirmed CEGI as a promising therapeutic agent for AD. However, our experiments were limited to the mouse model of AD, and further studies in human subjects are required to confirm the neuroprotective effects of CEGI.
Acknowledgments
This work was supported by the Institute of Geriatrics of the Chinese PLA General Hospital. We thank Jilin Sihuan Pharmaceutical Holdings Group Ltd for providing CEGI.
Disclosure
The authors report no conflicts of interest in this work.
References
Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic Biol Med. 2007;43(5):658–677. | ||
Feng Y, Wang X. Antioxidant therapies for Alzheimer’s disease. Oxid Med Cell Longev. 2012;2012:472932. | ||
Zhang W, Bai M, Xi Y, et al. Early memory deficits precede plaque deposition in APPswe/PS1dE9 mice: involvement of oxidative stress and cholinergic dysfunction. Free Radic Biol Med. 2012;52(8):1443–1452. | ||
Grothe M, Heinsen H, Teipel SJ. Atrophy of the cholinergic basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biol Psychiatry. 2012;71(9):805–813. | ||
Biscaro B, Lindvall O, Tesco G, Ekdahl CT, Nitsch RM. Inhibition of microglial activation protects hippocampal neurogenesis and improves cognitive deficits in a transgenic mouse model for Alzheimer’s disease. Neurodegener Dis. 2012;9(4):187–198. | ||
Heneka MT, Kummer MP, Weggen S, et al. Molecular mechanisms and therapeutic application of NSAIDs and derived compounds in Alzheimer’s disease. Curr Alzheimer Res. 2011;8(2):115–131. | ||
Zhang W, Bai M, Xi Y, et al. Multiple inflammatory pathways are involved in the development and progression of cognitive deficits in APPswe/PS1dE9 mice. Neurobiol Aging. 2012;33(11):2661–2677. | ||
Inglis F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. Int J Clin Pract Suppl. 2002;(127):45–63. | ||
Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med. 2012;366(10):893–903. | ||
Sha W, Chen L, Lu H, Hu X, Hu Z. Current advances in the clinical application of cattle encephalon glycoside and ignotin injection. Chin Pharm. 2014;23(8):81–84. | ||
Augustinsson LE, Blennow K, Blomstrand C, et al. Intracerebroventricular administration of GM1 ganglioside to presenile Alzheimer patients. Dement Geriatr Cogn Disord. 1997;8(1):26–33. | ||
Svennerholm L, Bråne G, Karlsson I, Lekman A, Ramström I, Wikkelsö C. Alzheimer disease – effect of continuous intracerebroventricular treatment with GM1 ganglioside and a systematic activation programme. Dement Geriatr Cogn Disord. 2002;14(3):128–136. | ||
Matsuoka Y, Saito M, LaFrancois J, et al. Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to beta-amyloid. J Neurosci. 2003;23(1):29–33. | ||
Yang R, Wang Q, Min L, Sui R, Li J, Liu X. Monosialoanglioside improves memory deficits and relieves oxidative stress in the hippocampus of rat model of Alzheimer’s disease. Neurol Sci. 2013;34(8):1447–1451. | ||
Boldyrev A, Koudinov A, Berezov T, Carpenter DO. Amyloid-beta induced cell death is independent of free radicals. J Alzheimers Dis. 2004;6(6):633–638; discussion 673–681. | ||
Corona C, Frazzini V, Silvestri E, et al. Effects of dietary supplementation of carnosine on mitochondrial dysfunction, amyloid pathology, and cognitive deficits in 3xTg-AD mice. PLoS One. 2011;6(3):e17971. | ||
Herculano B, Tamura M, Ohba A, et al. β-Alanyl-L-histidine rescues cognitive deficits caused by feeding a high fat diet in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis. 2013;33(4):983–997. | ||
Minkeviciene R, Ihalainen J, Malm T, et al. Age-related decrease in stimulated glutamate release and vesicular glutamate transporters in APP/PS1 transgenic and wild-type mice. J Neurochem. 2008;105(3):584–594. | ||
Xiong H, Callaghan D, Wodzinska J, et al. Biochemical and behavioral characterization of the double transgenic mouse model (APPswe/PS1dE9) of Alzheimer’s disease. Neurosci Bull. 2011;27(4):221–232. | ||
Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, et al. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24(3):516–524. | ||
Franklin KB, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. 2nd ed. Waltham (MA): Academic Press; 2001. | ||
Jang MH, Jung SB, Lee MH, et al. Melatonin attenuates amyloid β25-35-induced apoptosis in mouse microglial BV2 cells. Neurosci Lett. 2005;380(1–2):26–31. | ||
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. | ||
Barucker C, Harmeier A, Weiske J, et al. Nuclear translocation uncovers the amyloid Peptide Aβ42 as a regulator of gene transcription. J Biol Chem. 2014;289(29):20182–20191. | ||
Jack CR Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):257–262. | ||
Abdul HM, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med. 2008;45(10):1420–1425. | ||
Beal MF. Oxidative damage as an early marker of Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2005;26(5):585–586. | ||
Chauhan V, Chauhan A. Oxidative stress in Alzheimer’s disease. Pathophysiology. 2006;13(3):195–208. | ||
Mukhin VN. [The role of the basal forebrain cholinergic dysfunction in pathogenesis of declarative memory disorder in Alzheimer’s disease]. Ross Fiziol Zh Im I M Sechenova. 2013;99(6):674–681. Russian. | ||
Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283(44):29639–29643. | ||
Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300(5618):486–489. | ||
Perez SE, Dar S, Ikonomovic MD, DeKosky ST, Mufson EJ. Cholinergic forebrain degeneration in the APPswe/PS1DeltaE9 transgenic mouse. Neurobiol Dis. 2007;28(1):3–15. | ||
Savonenko A, Xu GM, Melnikova T, et al. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis. 2005;18(3):602–617. | ||
Stepanichev MY, Zdobnova IM, Zarubenko II, Lazareva NA, Gulyaeva NV. Studies of the effects of central administration of beta-amyloid peptide (25–35): pathomorphological changes in the hippocampus and impairment of spatial memory. Neurosci Behav Physiol. 2006;36(1):101–106. | ||
Jang JH, Surh YJ. Bcl-2 protects against Aβ(25–35)-induced oxidative PC12 cell death by potentiation of antioxidant capacity. Biochem Biophys Res Commun. 2004;320(3):880–886. | ||
Saitoh Y, Ouchida R, Miwa N. Bcl-2 prevents hypoxia/reoxygenation-induced cell death through suppressed generation of reactive oxygen species and upregulation of Bcl-2 proteins. J Cell Biochem. 2003;90(5):914–924. | ||
Li G, Ma R, Huang C, et al. Protective effect of erythropoietin on beta-amyloid-induced PC12 cell death through antioxidant mechanisms. Neurosci Lett. 2008;442(2):143–147. | ||
Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong JS. Microglia enhance beta-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J Neurochem. 2002;83(4):973–983. | ||
Zhang D, Hu X, Qian L, et al. Microglial MAC1 receptor and PI3K are essential in mediating β-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J Neuroinflammation. 2011;8(1):3. | ||
Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25(39):8843–8853. | ||
Parachikova A, Agadjanyan MG, Cribbs DH, et al. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging. 2007;28(12):1821–1833. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.