Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Catching “Early” COPD – The Diagnostic Conundrum
Authors Yip KP ![]() , Stockley RA
, Stockley RA ![]() , Sapey E
, Sapey E ![]()
Received 20 December 2020
Accepted for publication 21 March 2021
Published 13 April 2021 Volume 2021:16 Pages 957—968
DOI https://doi.org/10.2147/COPD.S296842
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Richard Russell
Kay Por Yip,1 Robert A Stockley,2 Elizabeth Sapey1
1Birmingham Acute Care Research Group, Institute of Inflammation and Ageing, University of Birmingham, Edgbaston, Birmingham, UK; 2Department of Respiratory Medicine, University Hospitals Birmingham NHS Foundation Trust, Edgbaston, Birmingham, UK
Correspondence: Kay Por Yip Email [email protected]
Abstract: Chronic obstructive pulmonary disease (COPD) remains a leading cause of morbidity and mortality worldwide. Despite this, there has been little progress so far in terms of disease-modifying therapies over the last few decades and this is in part due to poor understanding of the definition and mechanisms surrounding early disease before it becomes established and increasingly complex. In this review, the nuances and difficulty in defining early disease in COPD are discussed. There are clear benefits in identifying patients early; however, usually diagnosis is made in the presence of significant lung damage. We consider what can be learned of early disease from COPD studies and highlight the lack of inclusion of young smokers (who may be at risk of COPD) or those with mild disease. We discuss promising clinical measures that are being used in an effort to detect early disease. These include symptom assessment, lung physiology measures and computed tomography (CT) imaging modalities. There is emerging evidence for the role of neutrophils and their proteinases in early COPD. This may form an important biomarker to investigate the pathophysiological processes of early COPD. Given the importance of the early disease, it is recommended that future COPD studies focus on capturing the earliest manifestations of disease, to understand the initiating mechanisms and to identify novel treatment targets.
Keywords: COPD, early, neutrophil, lung function, biomarker
Introduction
Chronic obstructive pulmonary disease (COPD) is a major cause of mortality1 and places a high burden on clinical and healthcare resources.2,3 It is widely believed that the disease is precipitated by an abnormal inflammatory response to noxious stimuli, most commonly cigarette smoke. As the disease progresses, multiple pathological and clinical features emerge including but not limited to loss of small airways, chronic bronchitis, mucociliary dysfunction, bacterial colonization and emphysema.
The treatment of COPD is primarily based on bronchodilators and inhaled corticosteroids in varying combinations and these treatments are increased during exacerbations. The only new class of therapeutic agent to emerge over recent decades has been the phosphodiesterase 4 antagonist, roflumilast which is licensed for use in COPD patients with chronic bronchitis. Although this agent improves lung function and reduces exacerbations,4 there is currently no clinical evidence it alters the natural history of the disease.
The search for new therapeutic agents has been hampered by the complexity of patient phenotypes and that several components of the disease can become self-perpetuating, driving inflammation in their own right. These components may or may not influence the initiating pathological processes or physiological progression. For these reasons, studying the earliest stages of the disease has been perceived as critical. This requires identification of individuals at risk exhibiting the earliest changes in symptoms and pathology before the complexities of the clinical phenotypes become manifest. For this review, “early COPD” refers to a prodromal state where people do not yet have spirometrically confirmed airflow obstruction using standard criteria. However, they are on a clear trajectory to meet these criteria, without effective intervention.
Genome-wide associations have proven largely uninformative and the only widely recognized genetic factor to emerge remains alpha-1 antitrypsin deficiency (AATD) which has a clear mechanistic pathway to explain the COPD that emerges and a physiological measurement that precedes the development of COPD (see later). Similarly, biomarker studies have been largely uninformative5 probably reflecting the complex inflammatory processes related to the clinical phenotypic variations of established disease. To overcome these issues and understand the precise mechanisms that instigate the disease process, it is imperative that we appropriately study the initial phases of the disease and establish an understanding of what we mean by “early disease”.
Understanding “Early” vs “Mild” Disease
In other chronic, non-communicable diseases there have been efforts to study early disease to provide biological insight (for example, the BEACON cohort assessing early rheumatoid arthritis).6 Effective intervention strategies at an early stage, potentially before the disease process becomes irreversible, will not only improve long-term clinical outcomes but also mitigate subsequent health economic impact. This concept has also gained momentum in COPD and there is consensus that we need to understand more clearly how COPD develops at the earliest stages of the disease process.
In the past, the terms “mild” and “early” COPD have been used interchangeably. However, these terms refer to different concepts. The term “mild” is used as an established marker of severity whereas the term “early” refers to the initiation of a process in time. For example, an 80-year old with a 60-pack year smoking history and a forced expiratory volume in 1 second (FEV1) of approximately 80% predicted with a decreased FEV1/FVC ratio for the last five years may be classified as having “mild” disease but not necessarily “early” disease as it has likely developed slowly over decades. On the other hand, a 40-year old with the same lung function parameters may have deteriorated from the normal range rapidly and although mild in physiological terms, likely represents a highly active disease process, present for a short period of time.
Longitudinal studies have provided insight into the difficulty in developing a definition for “early disease”. Most studies concentrate on the prevalent FEV1 and its progression as the surrogate for the COPD condition. The rate of change in FEV1 over time is variable in COPD (and indeed decline in FEV1 occurs as part of the normal aging process), with observational studies describing FEV1 decline rates in established disease ranging from 25–79mls/year7–9 compared to 24–32mls/year in non-smokers without COPD.10,11 If these decline rates were consistent throughout the disease course, the time it would take for an individual to reach the diagnostic threshold for COPD would vary (as shown in Figure 1).
 |
Figure 1 Hypothetical trajectories of lung function (adjusted especially for age but also sex, height and race) that may be seen in the general population of smokers. Horizontal colored areas defined by the vertical axis represent COPD severity according to Global Initiative for Chronic Obstructive Lung Disease (GOLD) staging. Trajectory 1 refers to the lung function trajectory of smokers with decline due to age alone. He/she may not experience any respiratory symptoms or develop COPD. Trajectory 2 represents smokers who have mild decline greater than age-related changes. He/she may eventually cross the COPD diagnostic threshold but may only develop mild disease or respiratory symptoms. Trajectory 3 represents a smoker with an even greater lung function decline and will develop more severe COPD in later life with the associated high morbidity burden. The “early disease” process (represented by the shaded rectangle) is rarely identified and yet should contain the initiating clues to development of COPD especially in those with a more active disease process. |
The ideal patient population to study and those who would benefit most from early interventions would be smokers with excess lung function decline (above that seen with ageing), as demonstrated in trajectory 3 of Figure 1. This is suggestive of highly active disease. However, on initial assessment, it can be difficult to differentiate between smokers with a fast decline in lung function and those with a much slower lung function decline (Trajectory 2) or those with a decline consistent only with age-related changes (Trajectory 1). Based purely on conventional spirometry, this differentiation requires years of longitudinal follow-up. Furthermore, some individuals can have super-normal peak lung function reflective of an FEV1 starting above 100% predicted and there is often under-diagnosis of disease in this population as they take longer to reach the “COPD spirometric threshold”. It is imperative that we move on from conventional spirometry for the detection of at-risk populations. While we currently cannot reliably predict which smokers are more likely to develop COPD (other than in those with confirmed AATD), there is mounting evidence providing some guidance to gain further insight into biomarkers of risk.
Knowing Where to Look – Pathological Insights
The classical non-proportional Venn diagram of COPD (Figure 2) was first proposed by Snider12 and was later adopted by the American Thoracic Society.13 The clinical and pathological features described are major components of COPD and there are currently reliable methods to detect these features. (eg, detection of emphysema by CT scans and airflow obstruction using spirometry). However, these features are reflective of late or moderate to severe disease. Recently studies have concentrated on these phenotypes at all stages of the disease and especially in those with spirometry above the COPD threshold.
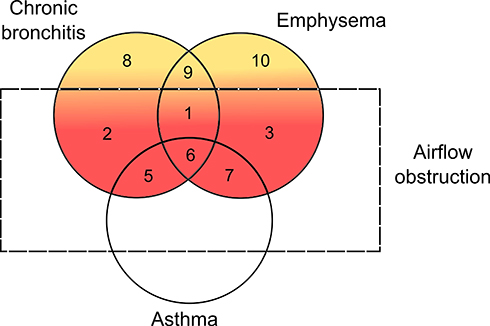 |
Figure 2 Non-proportional Venn diagram of COPD. This diagram illustrates the subsets of patients with chronic bronchitis, emphysema and asthma. The red areas (subsets 1–7) consist of COPD patients with differing clinical and pathological phenotypes of COPD. The majority of patients with COPD will have airflow obstruction together with features of chronic bronchitis and emphysema (subset 1). Some COPD patients may predominantly have symptoms of chronic bronchitis (subset 2) or emphysema (subset 3) or even have features of asthma (subsets 5–7). Those without airflow obstruction (subsets 8–10) are not classified as having COPD but may have pathophysiological features such as chronic bronchitis (subset 8), emphysema (subset 10) or both (subset 9) that if detected and treated early may prevent progression to established COPD. Adapted with permission of the American Thoracic Society. Copyright © 2021 American Thoracic Society. All rights reserved. American Thoracic Society. Definitions, epidemiology, pathophysiology, diagnosis, and staging. Am J Respir Crit Care Med. 1995;152(5pt2):S78–S83. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society. Readers are encouraged to read the entire article for the correct context at https://www.atsjournals.org/doi/10.1164/ajrccm/152.5_Pt_2.S78. The authors, editors, and The American Thoracic Society are not responsible for errors or omissions in adaptations.13 |
In the 1960s, Hogg et al14 using a retrograde catheter technique, demonstrated that small airway resistance is increased up to 40 times in excised emphysematous lungs compared to healthy human lungs and that this is due to narrowing and destruction of the small airways. Since then, further studies have expanded on this concept. Volumetric CT scanning of participants with varying stages of COPD as well as micro-CT analysis of frozen lung specimens from those with and without COPD have shown that small airways are abnormal or lost even in mild COPD and that this precedes the appearance of emphysema.15,16 Small airways disease is thus thought to be an early pathological feature of pending COPD and there have been efforts to employ various clinical and diagnostic measures to quantify this in smokers.
Looking for Early Disease (Using the Right Definition)
There is currently a lack of consensus defining “early disease” in COPD but there has been an attempt to define the “at risk” population operationally for research purposes. Martinez et al17 proposed that early changes leading to COPD should be studied in those younger than 50 years old with ≥10-pack year smoking history with any one of the below:
- early airflow obstruction (post-bronchodilator FEV1/FVC less than the lower limit of normal)
- compatible CT abnormalities such as emphysema, air trapping or bronchial thickening
- an accelerated FEV1 decline (≥60 mls/year) even when in the “normal” range
These criteria need acceptance and validation but represent an important step in moving beyond basic spirometry to identify individuals with high disease activity (and hence a high “inflammatory signal”) at a relatively early age. There are currently several studies ongoing using these criteria to identify and assess early COPD including the Early COPD Development Partnership (ClinicalTrials.gov identifier NCT03480347) and the Determinants of Onset and Progression of COPD in Young Adults. (ClinicalTrials.gov identifier NCT02352220) It is hoped that these studies can help elucidate early key pathophysiological processes to predict future COPD.
Looking for Early Disease (Using Common Symptoms)
The use of symptoms to help identify individuals at risk of COPD has been a hotly debated subject over the last few decades. GOLD released a report in 2001 introducing the GOLD 0 stage defined by the presence of risk factors (smoking) and symptoms (chronic cough and phlegm) in the absence of airflow limitation on spirometry. A retrospective analysis of data from the Copenhagen City Heart cohort later showed that GOLD 0 stage was not a stable feature and not all these characterized individuals eventually progress to establish COPD.18 Because of this study, the GOLD 0 concept was removed from the subsequent 2007 GOLD report.
Although further studies have since supported the dynamic nature of symptoms in smokers, it was suggested that individuals with persistent symptoms have a more pronounced FEV1 decline19 and hence greater risk of developing COPD20 compared to individuals who remain asymptomatic or those whose symptoms resolve. Analysis of data from the National Survey of Health and Development (NSHD) cohort also demonstrated that there was a greater prevalence of chronic bronchitis among smokers between the ages of 36–43 with an associated high risk of incident airflow obstruction in later life.21 Furthermore, it was shown that the longer the symptoms persist, the faster the FEV1 declined.21
There has thus been compelling evidence for the relationship between the presence of persistent symptoms and subsequent development of COPD. However, there also needs to be a refinement of the term “persistent symptoms” and the potential confounding effects of comorbidities before establishing a clear at-risk population among smokers.
Looking for Early Disease (Using the Right Equipment)
Post bronchodilator spirometry is mandatory for the diagnosis and monitoring of COPD.22 However, the forced nature of the test means that results are effort dependent and hence subject to intra-patient variability. Standard spirometry assesses flow in the major airways and largely reflects resistance due to narrowing/loss/early closure of said airways. Hence, they are poor predictors of emphysema23 and are relatively insensitive to the features of early disease. Thus, it is of limited value unless repeated over several years to identify people on the path to develop COPD through assessment of lung function decline. However, various techniques have been explored to quantify small airways dysfunction as a marker of the early manifestation of COPD. These will be discussed below.
Forced oscillometry technique (FOT) is quick, easy to perform and is obtained during tidal breathing, thus requiring no extra effort from the subject.23,24 In the ECLIPSE cohort where over 2000 COPD subjects were observed over 3 years,25 it was shown that peripheral airway resistance (R5-R20), reactance at 5Hz (X5) and area under the reactance curve (Ax) measurements were increased in COPD patients compared to healthy controls and that these measurements were proportionally higher with increasing disease severity.26 However, attempts to evaluate FOT as a marker of “early” disease in smokers has been limited by the differences of oscillation techniques between studies, small sample sizes and lack of focus on younger smokers where the earliest features of pending COPD are likely to be present.27 Thus, conformity and longitudinal studies with FOT will be needed before its utility in detecting early disease can be established.
Assessing the forced expiratory loop provides flow measurements that can reflect dynamic airways collapse due to narrowing or weakness of elastic support in the small airways.23 Maximal mid-expiratory flow (MMEF) quantifies maximal expiratory flow conventionally between 75% and 25% of a forced vital capacity (FVC). It has previously been found that MMEF is significantly lower in GOLD 0 patients and was thus thought to be a marker of early airflow limitation before the diagnostic threshold for COPD is reached by the FEV1/FVC measurement.28 Another study supported this concept in two groups of AATD subjects with a normal FEV1/FVC ratio. Those with a reduced MMEF had noticeably reduced quality of life before emphysema became radiologically apparent and also subsequently showed a faster deterioration in FEV1 than those without.29
Early closure of the small airways has also been assessed by observing gas composition (typically nitrogen) during slow expiration. Such assessments during single-breath washouts are quadriphasic (phase I–IV) and enable the closing volume (CV) of an individual to be quantified.30 CV is known to be increased in COPD due to premature airway closure but observation of the Phase III slope may be a useful indicator of small airway dysfunction before overt COPD develops.31 Multiple-breath washouts, on the other hand, enable the lung clearance index (LCI) to be determined.32 LCI is shown to be a sensitive and repeatable measure of airways disease and can be abnormal before the FEV1 is significantly impaired.33 In fact, it is already widely implemented clinically for the identification of early airway pathological features in patients with cystic fibrosis. A recent study has also demonstrated that it may be useful as an indicator of early disease in AATD before abnormal spirometry parameters emerge.34
Imaging tools have also been proposed as a method to detect the early disease, as well as identifying the different pathological features of COPD (emphysema, bronchial wall thickening and bronchiectasis). In particular, using CT densitometry to quantify emphysema before it becomes macroscopically obvious may enable understanding of early inflammatory mechanisms of this COPD component whilst these subjects retain normal spirometry.35 The small airways themselves are impossible to visualize using CT scanning. However, novel imaging techniques such as parametric response mapping (PRM) have been used to assess air trapping independent of emphysema by comparing lung densities at total inspiration and expiration, which likely acts as a surrogate of small airways dysfunction.36,37 This provides a radiological equivalent of the dynamic airways collapse seen spirometrically.
Apart from the presence of chronic bronchitis and the diagnostic techniques mentioned above, various other concepts have also been explored that may help predict progression to established COPD. Preserved ratio impaired spirometry (PRISm) is defined by a reduced FEV1 in the setting of a preserved FEV1/FVC ratio on spirometry. It was found in recent years that patients with PRISm were at high risk of subsequently developing COPD.38 Due to similar reasons, those with a history of severe childhood asthma39 and those with evidence of lung airway-parenchymal volume mismatch on CT40 have also been of particular interest. However, the evidence base surrounding these concepts are currently limited and the clinical utility of such approaches to identify patients at risk or COPD (or indeed when/if these subjects could be classed as early COPD) is unclear.
As demonstrated, there are both physiological and imaging techniques that have shown promise in detecting small airways dysfunction, which likely reflects damage and loss of the small airways seen pathologically in early disease. However, there is currently a lack of robust prospective studies looking at the use of such techniques to identify smokers with subsequent excessive lung function decline (although these are underway). Thus, it remains to be seen whether any of the above might be a more sensitive marker than FEV1 for the study of the early disease processes in COPD.
The Biomarker Conundrum
Without biological insight, there can be little progress in the development of novel treatments. Furthermore, mapping out early disease mechanisms can help identify molecular biomarkers as well as patient phenotypes that would most likely respond to targeted pharmacological interventions.
In recent years, there has been an extensive array of COPD molecular biomarker study publications. Most of these studies focus on blood biomarkers, instead of lung media, due to ease of access and reproducibility. There are however several issues in the use of such biomarkers in clinical practice and particularly in the field of “early disease”. Firstly, although many studies find statistically significant differences in biomarkers between healthy control subjects and COPD patients, there exists marked overlap between individual groups with similarities seen in other non-COPD lung conditions, rendering them ineffective as a prognostic tool.5 Secondly, there is a significant day-to-day variability of these markers especially in lung secretions which is unexplained but likely reflects sampling issues. However, such variability can be mitigated to a certain degree by taking sequential samples and using a rolling mean as the result.40,41
Thirdly, most biomarker studies focus on COPD populations with the established disease of varying severity and likely varying clinical phenotypes. At this stage, most individuals would already have extensive lung tissue damage with raised levels of inflammatory markers that, at least in part, reflect a physiological response to the damage. It is thus difficult to establish “cause or effect” as the biomarker may simply reflect disease severity rather than disease activity. Thus, biomarker studies in younger smokers with a focus on disease activity before the development of major lung tissue damage coupled with subsequent disease monitoring to assess progression will be needed. This will help identify differences in disease processes in the early stages from those caused by established COPD whilst still reflect future development.
Table 1 illustrates some of the most frequently studied blood biomarkers in patients with COPD. The table does not aim to summarize all studies looking at biomarkers but rather to highlight that such studies tend to focus on the older population (both with and without disease) and patients with a more severe stage of COPD. Therefore, these biomarkers will unlikely be able to capture the key processes in the early disease state and may likely reflect physiological responses to established disease and its severity.
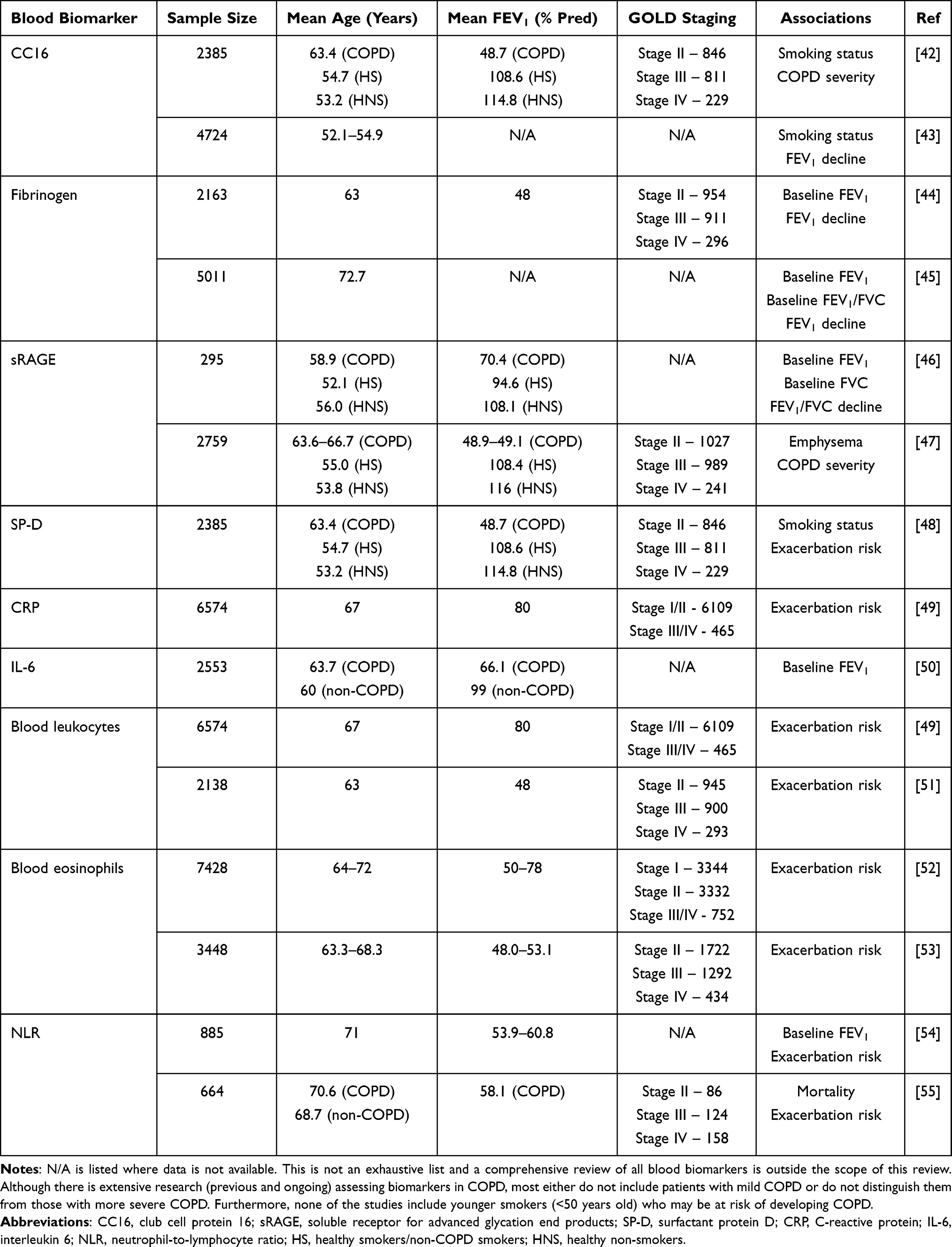 |
Table 1 Studies of Commonly Researched Blood Biomarkers in COPD with Cohort Demographic Features and Associated Clinical/Physiological Status Together with Outcomes Where These Have Been Documented |
Neutrophil Dysfunction: An Early Signal and a Potential Biomarker?
Over the years many potential mechanisms have been implicated in the pathophysiology of COPD. However, the role of neutrophils and proteolytic enzymes has been widely accepted.56 Cell and animal studies have demonstrated the ability of neutrophils to damage lung tissues by the release of serine proteinases, such as neutrophil elastase (NE) and proteinase 3.57 These proteinases degrade all components of the extracellular matrix, leading to the development of emphysema in animal models and (by implication) in humans. These enzymes are also pro-inflammatory in their own right and cause hyperplasia of the submucosal glands and goblet cells, leading to excess mucus secretion and impairment of mucociliary clearance. Subsequently, symptoms of chronic bronchitis and bacterial colonization develop, which further amplifies inflammation.57,58
Assays have been developed recently to quantify footprints of lung NE activity systemically by specific cleavage products of lung elastin59 or the accompanying fibrinogen.60 Aa-Val360 is a NE-specific fibrinogen degradation product reflecting lung elastolytic activity. Plasma levels are raised in stable COPD (especially in AATD) and increase further during exacerbations.61 Whether the activity levels relate to future outcomes requires further study but the elastin-specific cleavage product does reflect long-term mortality62 and the fibrinogen footprint does reflect subsequent FEV1 decline early in subjects with AATD.63
There is also an increased understanding of the potential mechanisms of damage caused by dysfunctional neutrophil responses. In the 1980s, it was found that neutrophils from patients with established emphysema had an increased migratory response to chemoattractants and a more destructive proteinase response than subjects with other neutrophilic lung diseases.64 This defect of peripheral neutrophils has been confirmed demonstrating migration with increased speed in response to chemoattractants but with reduced accuracy. It was also shown that this was a constitutional defect in COPD patients, with no difference in neutrophil migratory function influenced by COPD severity.65 Newer research has shown that peripheral neutrophils from smokers with chronic bronchitis but normal lung function have a similar migratory phenotype to those patients with COPD which may influence early pathophysiological changes leading to excess mucus production.66 However, long-term follow-up is needed to confirm whether this also marks progression to COPD.
These findings taken together could represent an insight into the cause of the early COPD disease process. Neutrophils with dysfunctional migratory dynamics may take a more prolonged and tortuous route towards a chemoattractant source. Thus, as they migrate within the lung architecture, they degrade surrounding lung tissue by creating a trail of obligate neutrophil proteinase activity. This increases bystander tissue damage as the initial high concentration of neutrophil elastase released cannot be inhibited by surrounding protease inhibitors.67 The exact mechanism affecting the neutrophil response has yet to be elucidated although it is not a secondary response to the environment and can be normalized by specific PI3K inhibitors suggesting this pathway is central.65,68
The neutrophil changes discussed above are key mechanisms capable of causing excessive lung tissue damage but individually do not explain susceptibility to COPD. It is becoming clear, however, that a subset of smokers with no spirometric evidence of COPD may already have distinct dysfunctional neutrophil phenotypes.66 These neutrophils respond abnormally to an inflammatory insult with subsequently increased potential for lung damage and hence would increase susceptibility to developing COPD. This could be genetically determined or reflect epigenetic modifications upon exposure to causative environmental factors.69
A prime example of the effect of genetic factors in COPD pathogenesis is AATD. This condition is characterized by low circulating levels of α1-antitrypsin which normally partially protects lung tissue against damage caused by proteolytic enzymes released from activated and migrating neutrophils. However, although a proportion of AATD patients who are never-smokers develop COPD in middle age, many do not.70 This observation suggests that aside from an assumed protease-antiprotease imbalance, other epigenetic factors also play a role, even in what is considered a monogenetic condition.
Despite the emerging interest in neutrophil phenotypes, there have been relatively few studies assessing this in-depth in COPD, and none in early COPD. It has been demonstrated by neutrophil proteomic analysis that there exist neutrophil phenotypes that can be functionally different between patients with COPD even if they are not clinically different in terms of symptoms or lung function.71 Although this study compared neutrophils from healthy subjects and COPD patients, the different stages of COPD severity were not explored. Mapping out these phenotypes in young smokers and assessing whether outliers relate to early disease activity and precede the development of established COPD needs exploration and could provide potential pathways for future therapeutic development.72
Why Look for Early COPD?
Identifying individuals with “early COPD” is also important to determine optimal management strategies for this cohort. There have been studies showing that symptomatic smokers have worse quality of life and clinical outcomes compared to “healthy smokers” irrespective of spirometry measurements.73,74 These individuals are likely to have underlying pathophysiological abnormalities that may require targeted treatment. However, the evidence base surrounding this strategy remains unclear as such patients are not usually included in clinical trials. The RETHINC study is novel as it studies symptomatic current or former smokers with normal lung function on spirometry. This study may provide critical insight into the concept of “early COPD”, assessing the impact of dual bronchodilator therapy in this patient population.75
Discovery of novel therapeutic agents for “early COPD” (or indeed COPD in general) has also been hampered by the lack of robust biomarkers that are closely linked to the underlying disease mechanism/s. Therapeutic trials that depend on FEV1 as the traditional surrogate for COPD progression and outcome are expensive as participants require many years of physiological follow-up to determine the disease-modulating effect. Thus, identifying relevant biomarkers would help facilitate short-term Phase II proof of concept studies that would be key to investment in such large phase III studies in COPD (Figure 3).
 |
Figure 3 Hypothetical timeline of disease progression in susceptible smokers. Subjects who are persistently exposed to risk factors (eg, cigarette smoke, air pollutants, biomass fuel smoke) suffer from low-grade airway inflammation. However, a subset of them (which may be genetically determined or dependent on epigenetic factors) are predisposed to develop COPD in later life over many years. These subjects may initially develop small airway dysfunction and if highly active, will have rapid lung function decline until they cross the diagnostic threshold for COPD. Many COPD patients are diagnosed only when they suffer from established symptoms and impaired health. At this point, the complexities of the pathological and clinical phenotypes are already established and damage is irreversible which increases the challenge of developing disease-modifying therapies for clinical trials. The most logical approach is to identify disease earlier when the pathological inflammation is only influenced by risk factors and gene/environment susceptibility (white to yellow zone) and not by amplification due to tissue damage and progression to variable phenotypes and their combinations (red zone). |
Summary
COPD remains a major non-communicable disease that causes increased morbidity and mortality worldwide. Only 50% of smokers develop COPD, and pathology is likely to progress over many years before the spirometric threshold for diagnosis is passed. To date, the ability to detect patients at risk of progression from early disease to clinically significant disease is limited in clinical practice but remains an important aim in disease prevention and the development of new therapies. This concept remains the most critical target for future COPD research prevention and treatment.
Current therapy (except for smoking cessation) is unable to prevent COPD development or halt its progression once established. Pathological changes are likely irreversible and self-propagating by the time the disease becomes clinically manifest, making it difficult to dissect pathological signals from physiological ones. Neutrophil dysfunction has been studied for many years and is considered central to the development of COPD and its progression. However, it is still unclear if this reflects a response to an abnormal primary factor or whether it is the initiating event in early disease. It is thus important to study younger subjects both at risk of and with the early disease to identify whether this is an early pathophysiological mechanism and hence a novel target for disease modification before it becomes established. Such studies will also help identify biomarkers that reflect causation and predict response to targeted therapies and is crucial for the development of much-needed larger clinical trials.
Disclosure
Professor Robert A Stockley is the investigator lead and reports research funding from Mereobiopharma; reports lecture and travel fees from and chair of Phase 4 advisory board for CSL Behring, member of steering committee for Vertex, chair of Phase 3 Data and Safety Monitoring Board for Kamada, and advisory board for Z factor, during the conduct of the study. Professor Elizabeth Sapey reports grants from HDR-UK, Wellcome Trust, MRC, British Lung Foundation, NIHR, and Alpha 1 Foundation, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Burney PGJ, Patel J, Newson R, Minelli C, Naghavi M. Global and regional trends in COPD mortality, 1990–2010. Eur Respir J. 2015;45(5):1239. doi:10.1183/09031936.00142414
2. Trueman D, Woodcock F, Hancock E. Estimating the Economic Burden of Respiratory Illness in the UK. British Lung Foundation; 2017.
3. Ford ES, Murphy LB, Khavjou O, Giles WH, Holt JB, Croft JB. Total and state-specific medical and absenteeism costs of COPD among adults aged 18 years in the United States for 2010 and projections through 2020. Chest. 2015;147(1):31–45. doi:10.1378/chest.14-0972
4. Martinez FJ, Rabe KF, Calverley PMA, et al. Determinants of response to roflumilast in severe COPD: pooled analysis of two Randomized Trials. Am J Respir Crit Care Med. 2018;198(10):1268–1278. doi:10.1164/rccm.201712-2493OC
5. Stockley RA, Halpin DMG, Celli BR, Singh D. Chronic obstructive pulmonary disease biomarkers and their interpretation. Am J Respir Crit Care Med. 2018;199(10):1195–1204. doi:10.1164/rccm.201810-1860SO
6. Abhishek A, de Pablo P, Cader MZ, Buckley CD, Raza K, Filer A. Diagnostic outcomes associated with ankle synovitis in early inflammatory arthritis: a cohort study. Clin Exp Rheumatol. 2014;32(4):533–538.
7. Lange P, Celli B, Agustí A, et al. Lung-function trajectories leading to chronic obstructive pulmonary disease. N Eng J Med. 2015;373(2):111–122. doi:10.1056/NEJMoa1411532
8. Tantucci C, Modina D. Lung function decline in COPD. Int J Chron Obstruct Pulmon Dis. 2012;7:95–99. doi:10.2147/COPD.S27480
9. Bhatt SP, Soler X, Wang X, et al. Association between functional small airway disease and FEV1 decline in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2016;194(2):178–184. doi:10.1164/rccm.201511-2219OC
10. Leem AY, Park B, Kim YS, Chang J, Won S, Jung JY. Longitudinal decline in lung function: a community-based cohort study in Korea. Sci Rep. 2019;9(1):13614. doi:10.1038/s41598-019-49598-9
11. Chinn S, Jarvis D, Melotti R, et al. Smoking cessation, lung function, and weight gain: a follow-up study. Lancet. 2005;365(9471):1629–1635. doi:10.1016/S0140-6736(05)66511-7
12. Snider GL. Chronic obstructive pulmonary disease: a definition and implications of structural determinants of airflow obstruction for epidemiology. Am Rev Respir Dis. 1989;140(3_pt_2):S3–S8. doi:10.1164/ajrccm/140.3_Pt_2.S3
13. American Thoracic Society. Definitions, epidemiology, pathophysiology, diagnosis, and staging. Am J Respir Crit Care Med. 1995;152(5pt2):S78–S83. doi:10.1164/ajrccm/152.5_Pt_2.S78
14. Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Eng J Med. 1968;278(25):1355–1360. doi:10.1056/NEJM196806202782501
15. McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Eng J Med. 2011;365(17):1567–1575. doi:10.1056/NEJMoa1106955
16. Koo H-K, Vasilescu DM, Booth S, et al. Small airways disease in mild and moderate chronic obstructive pulmonary disease: a cross-sectional study. Lancet Respir Med. 2018;6(8):591–602. doi:10.1016/S2213-2600(18)30196-6
17. Martinez FJ, Han MK, Allinson JP, et al. At the root: defining and halting progression of early chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;197(12):1540–1551. doi:10.1164/rccm.201710-2028PP
18. Vestbo J, Can LP. GOLD stage 0 provide information of prognostic value in chronic obstructive pulmonary disease? Am J Respir Crit Care Med. 2002;166(3):329–332. doi:10.1164/rccm.2112048
19. Brito-Mutunayagam R, Appleton SL, Wilson DH, Ruffin RE, Adams RJ. Global initiative for chronic obstructive lung disease stage 0 is associated with excess FEV1 decline in a representative population sample. CHEST. 2010;138(3):605–613. doi:10.1378/chest.09-2607
20. de Marco R, Accordini S, Cerveri I, et al. Incidence of chronic obstructive pulmonary disease in a cohort of young adults according to the presence of chronic cough and phlegm. Am J Respir Crit Care Med. 2007;175(1):32–39. doi:10.1164/rccm.200603-381OC
21. Allinson JP, Hardy R, Donaldson GC, Shaheen SO, Kuh D, Wedzicha JA. The presence of chronic mucus hypersecretion across adult life in relation to chronic obstructive pulmonary disease development. Am J Respir Crit Care Med. 2016;193(6):662–672. doi:10.1164/rccm.201511-2210OC
22. Vogelmeier C, Martinez FJ, Agusti A, et al. Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease – 2019 Report. Global Initiative for Chronic Obstructive Lung Disease;2018.
23. Stockley JA, Cooper BG, Stockley RA, Sapey E. Small airways disease: time for a revisit? Int J Chron Obstruct Pulmon Dis. 2017;12:2343–2353. doi:10.2147/COPD.S138540
24. Brashier B, Salvi S. Measuring lung function using sound waves: role of the forced oscillation technique and impulse oscillometry system. Breathe. 2015;11(1):57–65. doi:10.1183/20734735.020514
25. Vestbo J, Anderson W, Coxson HO, et al. Evaluation of COPD longitudinally to identify predictive surrogate end-points (ECLIPSE). Eur Respir J. 2008;31(4):869. doi:10.1183/09031936.00111707
26. Lipworth BJ, Jabbal S. What can we learn about COPD from impulse oscillometry? Respir Med. 2018;139:106–109. doi:10.1016/j.rmed.2018.05.004
27. Bhattarai P, Myers S, Chia C, et al. Clinical application of Forced Oscillation Technique (FOT) in early detection of airway changes in smokers. J Clin Med. 2020;9(9):2778. doi:10.3390/jcm9092778
28. Tsushima K, Sone S, Yoshikawa S, et al. Clinical differences in the global initiative for chronic obstructive lung disease stage 0. Respir Med. 2006;100(8):1360–1367. doi:10.1016/j.rmed.2005.11.021
29. Stockley JA, Ismail AM, Hughes SM, Edgar R, Stockley Robert A, Sapey E. Maximal mid-expiratory flow detects early lung disease in α1-antitrypsin deficiency. Eur Respir J. 2017;49(3):1602055. doi:10.1183/13993003.02055-2016
30. Dollfuss RE, Milic-Emili J, Bates DV. Regional ventilation of the lung, studied with boluses of 133xenon. Respir Physiol. 1967;2(2):234–246. doi:10.1016/0034-5687(67)90057-6
31. Gennimata S-A, Palamidas A, Karakontaki F, et al. Pathophysiology of evolution of small airways disease to overt COPD. COPD. 2010;7(4):269–275. doi:10.3109/15412555.2010.497515
32. Becklake MR, New A. Index of the intrapulmonary mixture of inspired air. Thorax. 1952;7(1):111. doi:10.1136/thx.7.1.111
33. Horsley AR, Gustafsson PM, Macleod KA, et al. Lung clearance index is a sensitive, repeatable and practical measure of airways disease in adults with cystic fibrosis. Thorax. 2008;63(2):135. doi:10.1136/thx.2007.082628
34. Fuchs SI, Schwerk N, Pittschieler K, et al. Lung clearance index for monitoring early lung disease in alpha-1-antitrypsin deficiency. Respir Med. 2016;116:93–99. doi:10.1016/j.rmed.2016.04.015
35. Crossley D, Renton M, Khan M, Low EV, Turner AM. CT densitometry in emphysema: a systematic review of its clinical utility. Int J Chron Obstruct Pulmon Dis. 2018;13:547–563. doi:10.2147/COPD.S143066
36. Hoffman EA, Lynch DA, Barr RG, van Beek EJR, Parraga G, Investigators I. Pulmonary CT and MRI phenotypes that help explain chronic pulmonary obstruction disease pathophysiology and outcomes. J Magn Reson Imaging. 2016;43(3):544–557. doi:10.1002/jmri.25010
37. Galbán CJ, Han MK, Boes JL, et al. Computed tomography-based biomarker provides unique signature for diagnosis of COPD phenotypes and disease progression. Nat Med. 2012;18(11):1711–1715. doi:10.1038/nm.2971
38. Wijnant SRA, De Roos E, Kavousi M, et al. Trajectory and mortality of preserved ratio impaired spirometry: the Rotterdam Study. Eur Respir J. 2020;55(1):1901217. doi:10.1183/13993003.01217-2019
39. Tai A, Tran H, Roberts M, Clarke N, Wilson J, Robertson CF. The association between childhood asthma and adult chronic obstructive pulmonary disease. Thorax. 2014;69(9):805. doi:10.1136/thoraxjnl-2013-204815
40. Sapey E, Bayley D, Ahmad A, Newbold P, Snell N, Stockley RA. Inter-relationships between inflammatory markers in patients with stable COPD with bronchitis: intra-patient and inter-patient variability. Thorax. 2008;63(6):493–499. doi:10.1136/thx.2007.086751
41. Stone H, McNab G, Wood AM, Stockley RA, Sapey E. Variability of sputum inflammatory mediators in COPD and α1-antitrypsin deficiency. Eur Respir J. 2012;40(3):561. doi:10.1183/09031936.00162811
42. Lomas DA, Silverman EK, Edwards LD, Miller BE, Coxson HO, Tal-Singer R. Evaluation of serum CC-16 as a biomarker for COPD in the ECLIPSE cohort. Thorax. 2008;63(12):1058. doi:10.1136/thx.2008.102574
43. Park HY, Churg A, Wright JL, et al. Club cell protein 16 and disease progression in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(12):1413–1419. doi:10.1164/rccm.201305-0892OC
44. Vestbo J, Edwards LD, Scanlon PD, et al. Changes in forced expiratory volume in 1 second over time in COPD. N Eng J Med. 2011;365(13):1184–1192. doi:10.1056/NEJMoa1105482
45. Jiang R, Burke GL, Enright PL, et al. Inflammatory markers and longitudinal lung function decline in the elderly. Am J Epidemiol. 2008;168(6):602–610. doi:10.1093/aje/kwn174
46. Iwamoto H, Gao J, Pulkkinen V, Toljamo T, Nieminen P, Mazur W. Soluble receptor for advanced glycation end-products and progression of airway disease. BMC Pulm Med. 2014;14(1):68. doi:10.1186/1471-2466-14-68
47. Cheng DT, Kim DK, Cockayne DA, et al. Systemic soluble receptor for advanced glycation endproducts is a biomarker of emphysema and associated with AGER genetic variants in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(8):948–957. doi:10.1164/rccm.201302-0247OC
48. Lomas DA, Silverman EK, Edwards LD, et al. Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. Eur Respir J. 2009;34(1):95. doi:10.1183/09031936.00156508
49. Thomsen M, Ingebrigtsen TS, Marott JL, et al. Inflammatory biomarkers and exacerbations in chronic obstructive pulmonary disease. JAMA. 2013;309(22):2353–2361. doi:10.1001/jama.2013.5732
50. Walter RE, Wilk JB, Larson MG, et al. Systemic inflammation and COPD: the Framingham Heart Study. CHEST. 2008;133(1):19–25. doi:10.1378/chest.07-0058
51. Müllerova H, Maselli DJ, Locantore N, et al. Hospitalized exacerbations of COPD: risk factors and outcomes in the ECLIPSE Cohort. CHEST. 2015;147(4):999–1007. doi:10.1378/chest.14-0655
52. Vedel-Krogh S, Nielsen SF, Lange P, Vestbo J, Nordestgaard BG. Blood eosinophils and exacerbations in chronic obstructive pulmonary disease. the Copenhagen General Population Study. Am J Respir Crit Care Med. 2015;193(9):965–974. doi:10.1164/rccm.201509-1869OC
53. Yun JH, Lamb A, Chase R, et al. Blood eosinophil count thresholds and exacerbations in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2018;141(6):2037–2047.e2010. doi:10.1016/j.jaci.2018.04.010
54. Lee H, Um S-J, Kim YS, et al. Association of the neutrophil-to-lymphocyte ratio with lung function and exacerbations in patients with chronic obstructive pulmonary disease. PLoS One. 2016;11(6):e0156511. doi:10.1371/journal.pone.0156511
55. Xiong W, Xu M, Zhao Y, Wu X, Pudasaini B, Liu J-M. Can we predict the prognosis of COPD with a routine blood test? Int J Chron Obstruct Pulmon Dis. 2017;12:615–625. doi:10.2147/COPD.S124041
56. Jasper A, McIver W, Sapey E, Walton G. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Research. 2019;8:557. doi:10.12688/f1000research.18411.1
57. Crisford H, Sapey E, Stockley RA. Proteinase 3; a potential target in chronic obstructive pulmonary disease and other chronic inflammatory diseases. Respir Res. 2018;19(1):180. doi:10.1186/s12931-018-0883-z
58. Stockley RA. Role of inflammation in respiratory tract infections. Am J Med. 1995;99(6):8S–13S. doi:10.1016/S0002-9343(99)80304-0
59. Luisetti M, Ma S, Iadarola P, et al. Desmosine as a biomarker of elastin degradation in COPD: current status and future directions. Eur Respir J. 2008;32(5):1146. doi:10.1183/09031936.00174807
60. Carter RI, Mumford RA, Treonze KM, et al. The fibrinogen cleavage product Aα-Val360, a specific marker of neutrophil elastase activity in vivo. Thorax. 2011;66(8):686–691. doi:10.1136/thx.2010.154690
61. Carter RI, Ungurs MJ, Mumford RA, Stockley RA. Aα-Val360: a marker of neutrophil elastase and COPD disease activity. Eur Respir J. 2013;41(1):31–38. doi:10.1183/09031936.00197411
62. Rabinovich RA, Miller BE, Wrobel K, et al. Circulating desmosine levels do not predict emphysema progression but are associated with cardiovascular risk and mortality in COPD. Eur Respir J. 2016;47(5):1365. doi:10.1183/13993003.01824-2015
63. Carter RI, Ungurs MJ, Pillai A, Mumford RA, Stockley RA. The relationship of the fibrinogen cleavage biomarker Aa-Val360 with disease severity and activity in a1-antitrypsin deficiency. CHEST. 2015;148(2):382–388. doi:10.1378/chest.14-0520
64. Burnett D, Hill S, Chamba A, Stockley R. Neutrophils from subjects with chronic obstructive lung disease show enhanced chemotaxis and extracellular proteolysis. Lancet. 1987;330(8567):1043–1046. doi:10.1016/S0140-6736(87)91476-0
65. Sapey E, Stockley JA, Greenwood H, et al. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(9):1176–1186. doi:10.1164/rccm.201008-1285OC
66. Yip K, Hughes M, Stockley R, Sapey E. S70 Inaccurate neutrophil migration in symptomatic smokers without chronic obstructive pulmonary disease. Thorax. 2019;74(Suppl 2):A46–A46.
67. Stockley RA. Neutrophils and protease/antiprotease imbalance. Am J Respir Crit Care Med. 1999;160(supplement1):S49–S52. doi:10.1164/ajrccm.160.supplement_1.13
68. Sapey E, Greenwood H, Walton G, et al. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood. 2014;123(2):239. doi:10.1182/blood-2013-08-519520
69. Ostuni R, Natoli G, Cassatella MA, Tamassia N. Epigenetic regulation of neutrophil development and function. Semin Immunol. 2016;28(2):83–93. doi:10.1016/j.smim.2016.04.002
70. Piitulainen E, Tornling G, Eriksson S. Effect of age and occupational exposure to airway irritants on lung function in non-smoking individuals with alpha 1-antitrypsin deficiency (PiZZ). Thorax. 1997;52(3):244–248. doi:10.1136/thx.52.3.244
71. Loi ALT, Hoonhorst S, van Aalst C, et al. Proteomic profiling of peripheral blood neutrophils identifies two inflammatory phenotypes in stable COPD patients. Respir Res. 2017;18(1):100. doi:10.1186/s12931-017-0586-x
72. Hughes MJ, Sapey E, Stockley R. Neutrophil phenotypes in chronic lung disease. Expert Rev Respir Med. 2019;13(10):951–967. doi:10.1080/17476348.2019.1654377
73. Woodruff PG, Barr RG, Bleecker E, et al. Clinical significance of symptoms in smokers with preserved pulmonary function. N Eng J Med. 2016;374(19):1811–1821. doi:10.1056/NEJMoa1505971
74. Regan EA, Lynch DA, Curran-Everett D, et al. Clinical and radiologic disease in smokers with normal spirometry. JAMA Intern Med. 2015;175(9):1539–1549. doi:10.1001/jamainternmed.2015.2735
75. Han MK, Ye W, Kim D-Y, Woodruff P. Pulmonary trials cooperative I. Design of the redefining therapy in early COPD Study. Chronic Obstr Pulm Dis. 2020;7(4):382–389. doi:10.15326/jcopdf.7.4.2020.0157
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.