Back to Journals » International Journal of Nanomedicine » Volume 16
Carfilzomib and Paclitaxel Co-Loaded Protein Nanoparticles an Effective Therapy Against Pancreatic Adenocarcinomas
Authors Cheng WT, Ho HO ![]() , Lin SY, Liu DZ, Chen LC, Sheu MT
, Lin SY, Liu DZ, Chen LC, Sheu MT ![]()
Received 28 July 2021
Accepted for publication 25 September 2021
Published 5 October 2021 Volume 2021:16 Pages 6825—6841
DOI https://doi.org/10.2147/IJN.S331210
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Farooq A. Shiekh
Wen-Ting Cheng,1,* Hsiu-O Ho,1,* Shyr-Yi Lin,2,3 Der-Zen Liu,4 Ling-Chun Chen,5 Ming-Thau Sheu1
1School of Pharmacy, College of Pharmacy, Taipei Medical University, Taipei, Taiwan, Republic of China; 2Division of Gastroenterology, Department of Internal Medicine, Wan Fang Hospital, Taipei Medical University, Taipei, Taiwan, Republic of China; 3Department of General Medicine, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan, Republic of China; 4Graduate Institute of Biomedical Materials and Engineering, Taipei Medical University, Taipei, Taiwan, Republic of China; 5Department of Biotechnology and Pharmaceutical Technology, Yuanpei University of Medical Technology, Hsinchu, Taiwan, Republic of China
*These authors contributed equally to this work
Correspondence: Ming-Thau Sheu
School of Pharmacy, College of Pharmacy, Taipei Medical University, 250 Wu-Hsing Street, Taipei, 11031, Taiwan, Republic of China
Tel/Fax +886 2 2377 1942
Email [email protected]
Ling-Chun Chen
Department of Biotechnology and Pharmaceutical Technology, Yuanpei University of Medical Technology, No. 306, Yuanpei Street, Hsinchu, 30015, Taiwan, Republic of China
Tel/Fax +886 2 2377 1942
Email [email protected]
Purpose: Therapeutic efficacy of pancreatic adenocarcinomas (PACs) with combined therapy of carfilzomib (CFZ) and paclitaxel (PTX) co-loaded in human serum albumin (HSA) nanoparticles (NPs) was examined.
Methods: CFZ and PTX were encapsulated individually or combined into HSA NPs by a simple reverse self-assembly method developed to achieve an optimal combination ratio for synergistic therapy. CFZ or/and PTX loaded HSA nanoparticles were physically characterized and the evaluation of combination index, drug release, pharmacokinetic, anti-tumor, and biodistribution studies were conducted.
Results: All resultant drug-loaded HSA NPs were spherical with a particle size of < 150 nm and a zeta potential of − 21.1∼− 23.0 mV. Drug loading rates and entrapment efficiencies were 9.1%∼ 10.1% and 90.7%∼ 97.1%, respectively. CFZ and PTX demonstrated synergistic effects in an MIA PaCa-2 cytotoxicity at a 1:2 ratio (CI50 were 0.01∼ 0.25). In vitro dissolution revealed that the CFZ/PTX ratio released from the co-loaded HSA NPs (CFZ/PTX/HSA NPs) was about 1.77∼ 2.08, which conformed to the designated loaded ratio. In vivo evaluation showed that the combined therapy of CFZ and PTX at a 1:2 ratio co-loaded in HSA NPs (CFZ/PTX/HSA NPs) demonstrated optimal synergistic improvement of the growth inhibition of MIA PaCa-2 cells with less systematic toxicity, even though the pharmacokinetic profiles observed did not show obvious beneficial and their biodistributions in tumors were found to be smaller.
Conclusion: The one-pot reverse assembly method developed was environmentally friendly and capable of co-loading an optimal combination ratio of two chemodrugs into HSA NPs for synergistic therapy.
Keywords: human serum albumin, nanoparticles, carfilzomib, paclitaxel, co-loading, synergism
Introduction
Pancreatic adenocarcinomas (PACs) are relatively rare compared to other cancers and represent only 3.2% of all new cancer cases in the US. Nevertheless, the average 5-year survival rate for all stages of PAC is only 10.0% because PACs cannot be detected and treated early. In general, the percent of cases and 5-year relative survival according to various stages of PAC (localized, regional, distant, and unknown) at diagnosis were reported to be 11% and 39.4%, 30% and 13.3%, 52% and 2.9%, and 7% and 6.1%, respectively, showing that the earlier PAC is caught, the better chance a person has of surviving 5 years after being diagnosed.1 Gemcitabine (GEM) is the only approved first-line monotherapy for treating PACs. Unfortunately, it still delivers unsatisfactory therapeutic outcomes in prolonging progression-free survival (PFS) and overall survival (OS) of patients with locally advanced and metastatic PAC.2
Combined therapy has become a major means to combat cancer thanks to its primary advantages of increased efficacy without, or with minimal, addictive toxicities at equal or reduced administered doses. Multiple combination therapies composed of GEM and different cytotoxic and biologic agents have undergone clinical evaluations to examine the therapeutic efficacies for patients at various stages of PAC since 2002 as reported by Lei et al.3 Among them, only the combination regimen of GEM with either erlotinib (Tarceva®) or paclitaxel albumin-bound nanoparticles (NPs) (Abraxane®) demonstrated significant improvements in most clinical outcome parameters compared to GEM alone, leading to the approval of both combination therapies by the US Food and Drug Administration (FDA) as a first-line combination therapy for patients with locally advanced and metastatic PAC in 20154 and 2013,5 respectively. Despite combination therapies having demonstrated improved outcomes in patient survival and quality of life, the overall improvement is still marginal, especially for patients diagnosed with late stages of the disease. There is still an urgent need to generate effective strategies, new single agents, or new combinations, to significantly improve clinical outcomes for treating PACs.
Several studies support the proteasome being an effective therapeutic target against PAC from the perspectives of high heterogeneity and chemoresistance. By unraveling transcriptomic predictive signature data by Fraunhoffer et al,6 a subgroup of PACs sensitive to FDA-approved carfilzomib (CFZ) was identified, and it was ultimately suggested to repurpose CFZ for treating PACs.7 Furthermore, proteasome inhibitors (PIs), such as carfilzomib, can induce apoptosis in PACs by inducing endoplasmic reticular (ER) stress, which facilitates synergistic effects when combined with radiation therapy or chemodrugs like camptothecin and paclitaxel.8 It was further disclosed that a combination of MG-132 (a PI) and camptothecin at a ratio of 5:1 (2.5 µmol/l MG-132: 0.5 µmol/l camptothecin) provided promising results with enhanced cytotoxicity compared to the single compounds in MIA PaCa-2 cells, while that for the combination of MG-132 and paclitaxel at the same 5:1 ratio but with lower concentrations of 0.08 µmol/l MG-132 and 0.016 µmol/l paclitaxel could moderately increase the cytotoxicity to 62% from 46% for paclitaxel alone at the same concentration of 0.016 µmol/l as that in combination.9 This potentially suggests that a combination of PIs, such as CFZ with paclitaxel for treating PACs might be worth pursuing.
The poor biostability and short half-life of CFZ are considered major issues causing CFZ to perform with low efficacy in patients with solid cancers because it is difficult for CFZ to arrive at the proteasome in solid tumors.10,11 Polymer micelles (PMs) composed of biodegradable block copolymers poly(ethylene glycol) (PEG) and poly(caprolactone) (PCL) were reported to improve the metabolic stability of CFZ in vitro. However, despite the in vitro metabolic protection of CFZ, CFZ-loaded PMs or PEG-PCL-deoxycholic acid (CFZ-PMs) did not display superior in vivo anticancer efficacy in mice bearing human lung cancer xenografts (H460) to that of the clinically used cyclodextrin-based CFZ (CFZ-CD) formulation.12 A novel albumin-coated nanocrystal formulation of CFZ (CFZ-alb NC) displayed improved metabolic stability and enhanced cellular interactions, uptake, and cytotoxic effects in breast cancer cells in vitro. Consistently, CFZ-alb NCs showed greater anticancer efficacy in a murine 4T1 orthotopic breast cancer model than the currently used cyclodextrin-based formulation. It was highly suggested that human serum albumin (HSA)-bound NPs could be used as a viable nanocarrier to encapsulate CFZ for cancer therapy.13 As described above, it was paclitaxel (PTX) albumin-bound nanoparticles (Abraxane®), not Taxol® (a traditional dosage form with PTX being dissolved in the mixture of Cremophor EL and ethanol), which was approved for combination with GEM by the FDA as a first-line combination therapy for patients with locally advanced and metastatic PACs. Therefore, HSA-bound NPs could be used as a viable nanocarrier to co-encapsulate PTX and CFZ for combination therapy of PAC.
HSA has garnered considerable interest as a nanocarrier, due to its low toxicity, biocompatibility, and the ability to reduce interactions with phagocytes in the reticuloendothelial system (RES).14–16 Moreover, albumin can interact with cancer cells based on its increased use as an energy source in rapidly proliferating cancer cells.17 It was reported that nanoalbumin-bound (nab)-drugs can aid drug permeation across tumor vessels.18,19 It was also suggested that albumin facilitates the movement of nab-drugs across endothelial cell membranes by binding to the gp60 receptor and sequentially interacting with other albumin-binding proteins such as secreted protein acidic and rich in cysteine (SPARC), which is abundantly expressed in and near cancer cells.20–22 As exemplified, GEM (Gemcitabine)-loaded HSA and PTX-loaded HSA for practical PAC treatment have been reported by Han et al and Yu et al, respectively.23,24 Therefore, it was thought that NPs fabricated with HSA might potentially be an optimal choice for co-delivery of chemotherapeutic drugs with a high drug loading capacity, biodegradability, and good biocompatibility.
Abraxane™ is the first FDA-approved chemotherapeutic formulation based on Nab™ nanotechnology, which relies heavily on the use of organic solvents, namely, chloroform.25 The toxicity introduced by residual chloroform poses a potential risk to patient health. In response to the issue of chronic toxicity, a reversible self-assembling method, which eliminates the dependence on toxic organic solvents during manufacturing, was developed in a preliminary study and demonstrated to be capable of successfully preparing HSA-bound PTX and CFZ nanosuspensions. Furthermore, both NPs formed using this method still retained their suitability for intravenous (IV) administration.25 Therefore, in this study, the preparation of CFZ-loaded, PTX-loaded, and CFZ/PTX co-loaded HSA NPs was developed and optimized. To confirm these advantages, the properties of the three drug-loaded HSA NPs, including the encapsulating efficiency (EE), drug-loading (DL), mean size, polydispersity index (PDI), drug release, and cell growth inhibition against MIA CaPa-2 cells (human pancreatic cancer cell line) were characterized in vitro. Furthermore, the in vivo pharmacokinetic study of the three drug-loaded HSA NPs (CFZ/HSA NPs, PTX/HSA NPs, and CFZ/PTX/HSA NPs) were evaluated in Sprague-Dawley rats and compared to two solvent-based (Sb) drugs of CFZ and PTX (Sb-CFZ and Sb-PTX). The anti-tumor efficacy and systemic toxicity were further evaluated in MIA CaPa-2 tumor-bearing C.B-17 SCID mice.
Materials and Methods
Preparation of Drug Solutions and Drug-Loaded Human Serum Albumin Nanoparticles
Sb-CFZ (solvent-based CFZ) was prepared by dissolving 60 mg of CFZ (Chunghwa Chemical Synthesis & Biotech, New Taipei City, Taiwan), 3000 mg sulfobutylether beta-cyclodextrin (SBE-β-CD), and 57.7 mg citric acid in 29 mL deionized water through sodium hydroxide (NaOH) pH adjustment (pH=3.5). The solution was lyophilized and stored at 4°C until reconstitution for use.26 Sb-PTX (solvent-based PTX) was prepared by solubilizing 6 mg PTX (ScinoPharm, Tainan, Taiwan) in 527 mg of purified Cremophor® EL (polyoxyethylated castor oil; BASF, Ludwigshafen, Germany) in 497 mg (v/v) of dehydrated alcohol.
Drug-loaded HSA NPs were prepared with defatted human serum albumin (HSA) by a self-assembling method developed in our lab. Defatted HSA was produced by adsorption of fatty acids in HSA onto charcoal as previously described.27 Briefly, a marketed 20% HSA solution (Taiwan Blood Services Foundation, Taipei, Taiwan) was diluted with deionized water, and then the pH was adjusted to 2.7 with 1 N HCl. After adding 5 g of activated charcoal, the resulting HSA solution was stirred at 300 rpm and 4°C for 2 h. The mixed solution was centrifuged at 8000 rpm and 4°C for 10 min, and the supernatant was filtered through a 0.45-µm nylon membrane (ChromTech, Bad Camberg, Germany) to remove the charcoal. Finally, the pH of the filtrate was adjusted to 7.0 with 1 N NaOH and lyophilized. The so-obtained lyophilized HSA powder was stored at 4°C.28
The preparation of drug-loaded HSA NPs was divided into three steps. First, the pH of the HSA solution was adjusted to 2.7 with 1.0 N HCl to expose the hydrophobic domains.27,28 Second, the targeted hydrophobic drug in ethanol was added followed by stirring for 5 min to enhance interactions between the drug and HSA. Finally, the pH value was re-adjusted to neutral with 0.1 N NaOH to induce self-assembling and encapsulate the hydrophobic drug. Then, used high-pressure homogenization with an N2-3D Nanolyzer (Gogene, Hsinchu, Taiwan) to form stabilized drug-loaded HSA NPs. An Amicon® Ultra-15 centrifugal filter (with a molecular weight (MW) cutoff of 10 kDa) was used to remove the ethanol, salt, and free drug, and then the drug-loaded HSA NPs were concentrated. The drug-loaded HSA NP concentrate was passed through 0.20-µm regenerated cellulose filtration (Phenomenex, Torrance, CA, USA) to obtain translucent dispersion with typical diameter around 150 nm. Finally, lyophilized the solution for 48 hours without cryoprotectant.
The formulation and optimal homogenizer parameters utilized in step 3 for preparing CFZ-loaded HSA NPs (CFZ/HSA NP), PTX-loaded HSA NPs (PTX/HSA NP), and CFZ/PTX-loaded HSA NPs (CFZ/PTX/HSA NP) are described below. To prepare CFZ/HSA NPs, 200 mg CFZ was dissolved in 20 mL absolute alcohol, and the dispersion was added to 200 mL 0.9% defatted HSA solution. The ratio of drug to HSA was 1:9, and a 10K psi homogenizer parameter was applied for 10 cycles. To prepare PTX/HSA NPs, 300 mg PTX was dissolved in 12 mL absolute alcohol, and the dispersion was added to 120 mL 2.25% defatted HSA solution. The ratio of drug to HSA was also 1:9, and the 20K psi homogenizer parameter was applied for 20 cycles. To prepare CFZ/PTX/HSA NPs, 60 mg CFZ and 120 mg PTX were dissolved in 18 mL absolute alcohol, and the dispersion was added to 180 mL 1% defatted HSA solution. The ratio of both drugs to HSA was 1:10, and the 10K psi homogenizer parameter was applied for 10 cycles.
Physicochemical Characterization of Human Serum Albumin Nanoparticles
The mean particle size, size distribution, zeta potential, and polydispersity index (PDI) of drug-loaded HSA NPs were measured with a Zetasizer nano ZS (Malvern, Worcestershire, UK) by scattering angle of 90° at 25°C. The drug-loaded HSA NPs were diluted with double-distilled water before the measurement, and all measurements were performed at least in triplicate. The shape and size were also observed by transmission electron microscopy (TEM), using Hitachi H-7000 (Hitachi, Tokyo, Japan). The purified NPs were diluted with water to allow clearer pictures to be taken. Samples were prepared by placing a drop on carbon-coated copper grids and sponging off the excess with filter paper. Then, the samples were stained with uranyl acetate (2% aqueous solution) for 3 minutes and dried at room temperature.28
Drug Loading and Entrapment Efficiency
To assess the entrapment efficiency of CFZ or PTX in drug-loaded HSA NPs, 10 mg lyophilized NPs was dissolved in 1 mL deionized water; then 9 mL acetonitrile was added and vortexed it for 1 minute. The solution was centrifuged at 14,000 rpm for 10 minutes. After appropriate dilution, CFZ or PTX in the supernatant was directly quantified by Waters alliance HPLC (Waters, Milford MA, USA) equipped with an Inert Sustain® C18 column (150 × 4.6 mm, particle size 5 µm, GL Sciences, Tokyo, Japan). The mobile phase was composed of acetonitrile and 0.05% formic acid aqueous solution (50:50, v/v, at a flow rate of 1 mL/min). The total analytical time for a single injection was 12 min. The injection volume was 10 μL, and chose 210-nm wavelength for detection. The column oven was kept at 35°C, and the sample cooler was maintained at 10°C. The drug loading (DL) and entrapment efficiency (EE) of nanoparticles were calculated by the following equations:
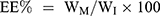
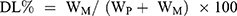
WM is the weight of the drugs in the NPs, WI is the weight of the initial feeding drug, and WP is the weight of the initial feeding HSA.
In vitro Cytotoxicity Study and Synergistic Analysis
Cell viabilities of the CFZfree (CFZ dissolved in DMSO), Sb-CFZ, CFZ/HSA NPs, PTXfree (PTX dissolved in DMSO), Sb-PTX, PTX/HSA NPs, and CFZ/PTX/HSA NPs were evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay for MIA PaCa-2 cell line obtained from ATCC. Cells were seeded at a density of 3 × 103 cells/well in 96-well plates and incubated for 24 h at 37°C with 5% CO2. Then, tumor cells were treated with different concentrations (0.01, 0.1, 1, 5, 10, 50, 100, and 1000 ng/mL) of CFZfree, Sb-CFZ, PTXfree, Sb-PTX, or drug-loaded HSA NPs. After incubation for 72 hours, 200 µL MTT (0.5 mg/mL) was added to each well for 2 hours. After removing the medium, 50 µL DMSO was added to each well and gently shaken to dissolve any purple formazan crystal. The absorbance was measured at 550 nm (Bio-Tek). The survival rate was calculated using the following formula: percentage (%) cell survival = [(mean absorbency in test wells)/(mean absorbency in control wells)] × 100. Values of the combination index (CI) were calculated by the Chou-Talalay method:29,30
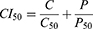
C and P denote IC50 values of CFZ and PTX in combination therapy that inhibits 50% of the cell. C50 and P50 denote doses of CFZ and PTX that inhibit 50% cells alone. Values of CI = 1, CI < 1, and CI > 1, respectively, indicate additivity, synergy and antagonism.
In vitro Release Analysis of Carfilzomib and Paclitaxel
Drugs released from the formulations were investigated in PBS (containing 0.5% Tween 80) by the dialysis method. The CFZ- or PTX-loaded HSA NPs were diluted to 0.1 or 0.2 mg/mL in 1-mL solution and then placed in a dialysis bag (OrDial D80-MWCO 6000–8000, cat. no. 60082530, Orange Scientific, Braine-l’Alleud, Belgium) against 40 mL release medium, with 100 rpm shaking speed at 37°C. Sampled 1 mL at 1, 2, 3, 4, 6, 8, 12, 24, and 48 hours, and performed the analysis using the HPLC method above.31
Maximum Tolerance Dose Study
Male BALB/c mice (BioLasco Taiwan, Yilan, Taiwan) at 7 weeks of age were randomized into 4 groups and each group contained 4 mice. For single-dose study, we used tail vein injection at 0 days. For multi-dose study, we repeated half dose of single-dose study at 0 and 1 day. If there was no obvious toxic reaction, the dosage was elevated correspondingly. The weight changes and physiological signs were observed and recorded for 5 consecutive days in the first week. During the second week, the related assessment would be performed every 2 days. The whole study continued for 15 days. It would be specified as the maximum tolerance dose if there is any event for neurotoxicity, weight loss >20% or death.
Pharmacokinetic Studies of Drug-Loaded Human Serum Albumin Nanoparticles with Intravenous Administration in Rats
Male Sprague-Dawley rats (BioLasco Taiwan, Yilan, Taiwan) at 8~10 weeks of age were used to study pharmacokinetic profiles after administration of Sb-CFZ, Sb-PTX, CFZ/HSA NPs, PTX/HSA NPs, Sb-CFZ+Sb-PTX, CFZ/HSA NPs+PTX/HSA NPs, and CFZ/PTX/HSA NPs. Rats were given a single tail vein injection of 5 mg/kg CFZ and 10 mg/kg PTX for each formulation (three or four rats per group). Blood samples were collected from the jugular vein in heparinized tubes at 0.017, 0.033, 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, and 48 h after administration. All blood samples were immediately centrifuged at 4500 rpm for 10 minutes to obtain plasma, and then stored at −80°C until analyzed by UPLC interfaced with MS (Triple Quadrupole Mass Spectrometry, TQ-XS, Waters). CFZ and PTX were extracted from the plasma as follows: 100 µL plasma was extracted with tert-butyl methyl ether (400 µL) containing an internal standard (500 ng/mL chlorpropamide (Sigma-Aldrich, St. Louis, MO, USA) and 500 ng/mL docetaxel (ScinoPharm Taiwan, Tainan, Taiwan)) by vortex-mixing for 1 min. After centrifugation at 14,000 rpm for 10 min, 300 µL organic phase was transferred to a new tube and dried at 40°C. Samples were reconstituted in 100 µL mobile phase and transferred to a new vial for the UPLC-MS/MS analysis. The measurement by UPLC-MS/MS. Chromatographic separation was performed with a Purospher® Star RP-18 end-capped column (2.1 × 50 mm, particle size 2 µm, Merck) and gradient elution (at a flow rate of 0.3 mL/min). The mobile phase comprised 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The total analytical time for a single injection was 5 minutes, and the injection volume was 2 μL. The column oven was kept at 40°C, and the sample cooler was kept at 10°C. Detection of ions was performed in the positive ionization mode with the following transitions in multiple reaction monitoring mode (MRM): 720.33 → 100.03 for CFZ, 277.06 → 174.87 for chlopropamide (the internal standard for CFZ),32 854.29 → 104.99 for PTX, and 830.40 → 549.24 for docetaxel (the internal standard for PTX). The capillary voltage was 3.0 kV, cone voltage was 30 V, desolvation temperature was 350°C, desolvation gas flow was 650 L/h, and collision gas flow was 25 L/h.
In vivo pharmacokinetic parameters including the area under the plasma concentration–time curve (AUC), the apparent volume of distribution (V), plasma clearance (CL), and elimination half-life (T1/2) of each formulation were calculated and expressed by the mean and standard deviation (SD). The AUC0-1h, AUC0-2h, AUC0-24h, and AUC0-infinity were estimated by linear trapezoidal method. Plasma clearance (CL) was calculated from the dose/AUC0-infinity. The initial half-life (T1/2,initial) and terminal half-life (T1/2,terminal) values were calculated as ln(2)/k, where k represents either the initial distribution rate constant or the terminal elimination rate constant obtained from the slope of a semilogarithmic plot of the concentration–time profile. The volume distribution (V) was estimated using a noncompartmental method provided by WinNonlin software (vers. 6.3.0.395, Pharsight®, Princeton, NJ, USA). The maximum plasma concentration (Cmax) was recorded as observed for the first sampling time point, and C0 was the concentration at t = 0 (extrapolated).
The Anti-Tumor Efficacies of Drug-Loaded Human Serum Albumin Nanoparticles in MIA PaCa-2 Tumor-Bearing Mice
C.B-17 female SCID mice, at 6~7 weeks of age (BioLasco Taiwan), were used as the tumor xenograft models. The models were established by subcutaneously inoculating MIA PaCa-2 (2×107 cells/mouse, 100-μL injection) into the right dorsal flank of each mouse. MIA PaCa-2 tumor-bearing mice with 150 mm3 tumor volumes were randomly divided into eight treatment groups (n = 5). One group of mice received an intravenous injection of saline as a control. The other groups received an injection of Sb-CFZ, Sb-PTX, CFZ/HSA NPs, PTX/HSA NPs, Sb-CFZ+Sb-PTX, CFZ/HSA NPs+PTX/HSA NPs, or CFZ/PTX/HSA NPs (equivalent to 5 mg/kg CFZ and 10 mg/kg PTX for each mouse). Administration was performed on days 0, 1, 7, 8, 14, and 15. Body weights (BWs) and tumor sizes were measured three times every week using digital calipers, and tumor volumes (mm3) were calculated. After being sacrificed by CO2 on day 46, tumors were harvested and weighed. The tumor growth inhibition (TGI) (%) was calculated as follows:33

Biodistribution Studies of Drug-Loaded Human Serum Albumin Nanoparticles in MIA PaCa-2 Tumor-Bearing Mice
Female C.B-17 SCID mice, at 6~7 weeks of age (BioLasco Taiwan), were used as the tumor xenograft models. The models were established by subcutaneously inoculating MIA PaCa-2 cells (2×107 cells/mouse, 100-μL injection) in the right dorsal flank of each mouse. On day 14 after tumor cell inoculation when tumor volumes had reached about 150 mm3, each mouse was given Sb-CFZ, Sb-PTX, CFZ/HSA NPs, PTX/HSA NPs, Sb-CFZ+Sb-PTX, CFZ/HSA NPs+PTX/HSA NPs, or CFZ/PTX/HSA NPs (equivalent to 5 mg/kg CFZ and 10 mg/kg PTX in each mouse) by an intravenous injection. After 2 and 8 h, mice were sacrificed by anesthesia and perfused with a PBS solution to remove the blood. The heart, lungs, liver, spleen, kidneys, and tumors were excised, weighed, and stored at −80°C. Tissues were homogenized by an ultrasonicator probe (VCX 750; Sonics & Materials, Newtown, CT, USA) with 5 W and three pulses for 10 s. After that, 400 µL of a PBS/0.1% heparin solution was added. Tissue homogenates (200 µL) were obtained, and drug concentrations were analyzed by UPLC/MS/MS.
Statistical Analysis
Data are presented as the mean ± SD of three different replicates. For in vivo studies, a one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons was used to test for significant differences in the longitudinal tumor volume growth over the entire experimental period among the eight treatment groups and to determine whether there was a significant interaction effect between CFZ and PTX. Significant differences between groups were indicated by *p<0.05 and **p<0.005.
Results and Discussion
Physicochemical Characteristics of Drug-Loaded Human Serum Albumin Nanoparticles
As depicted in Figure 1, the mean particle sizes of CFZ/HSA NPs, PTX/HSA NPs, and CFZ/PTX/HSA NPs were 114.5±0.6, 117.4±0.4, and 105.3±0.6 nm; PDI values were 0.144±0.007, 0.166±0.007, and 0.167±0.008; zeta potentials were −23.00±0.70, −21.1±0.58, and −21.3±0.70 mV; entrapment efficiencies (EEs, %) were 95.6±2.1%, 97.1±3.5%, and 92.7±2.6%/90.7±3.1%; and drug loadings (DLs) were 9.4±0.1%, 10.1±0.2%, and 9.1±0.4%, respectively. In addition, all zeta potentials measured were between −21.1 and −23.00 mV and indicated that the drug was encapsulated with HSA, which possessed a negative charge at a neutral pH because its PI was equivalent to 4.7.34 Figure 1 demonstrates that the three different HSA NPs were spherical, and the mean particle sizes were <200 nm under 80,000× TEM observation. Another noteworthy result was that the entrapment efficiencies for CFZ and PTX were, respectively, observed to be 92.7±2.6% and 90.7±3.1% in CFZ/PTX/HSA NPs. The calculated ratio for PTX and CFZ was 1.96; in other words, the method was suitable for preparing HSA NPs by achieving designed ratio nearly 2. Also, the high entrapment efficiency and drug loading (9.1%) indicated that HSA could act as novel and excellent nanocarriers for co-loading two drugs. In addition to the good compatibility with the two hydrophobic drugs, the preparation is simple and efficient with no cryoprotectant required.
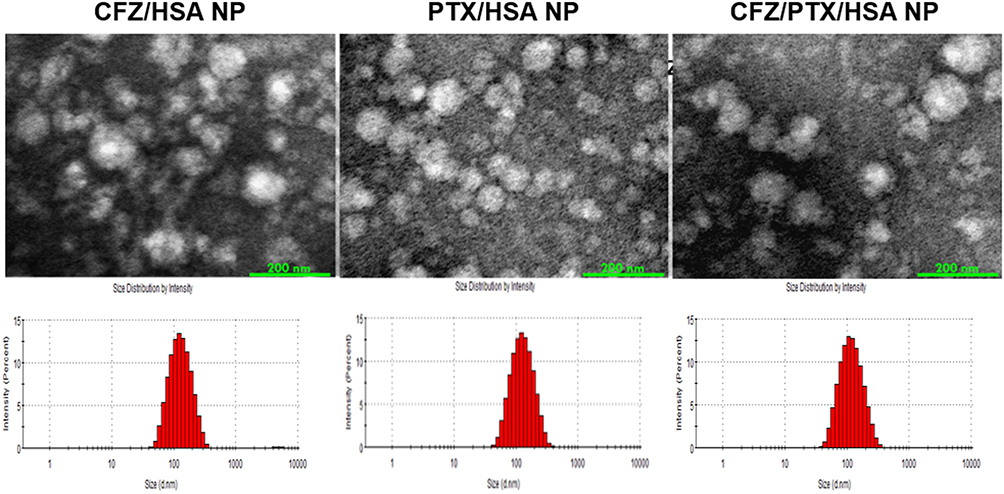 |
Figure 1 TEM image and particle size analysis of drug/human serum albumin (HSA) nanoparticles (NPs). |
Cell Cytotoxicity
The MIA PaCa-2 cell line was used as a model in the cytotoxicity study. Table 1 shows the values of the 50% inhibitory concentration (IC50) for different combinations of free drug in DMSO (CFZfree and PTXfree), two solvent-based drugs (Sb-CFZ and Sb-PTX), and two drug-loaded HSA NPs (CFZ/HSA NPs and PTX/HSA NPs). Respective IC50 values for CFZfree, Sb-CFZ, CFZ/HSA NPs, PTXfree, Sb-PTX, and PTX/HSA NPs were 8.9, 8.12, 7.89, 0.44, 0.47, and 0.86 ng/mL. The results indicated that the two model drugs maintained similar cytotoxicities in the different formulations. Compared to CFZ, PTX demonstrated a higher cytotoxicity toward the MIA PaCa-2 cell line. Table 1 also reveals the synergism of CFZ and PTX at ratios of 1:2, 1:1, and 2:1. IC50 values for free CFZ and PTX ratios of 1:2, 1:1, and 2:1 were 0.25, 1.39, and 1.00 ng/mL, respectively. According to the equation of the Chou-Talalay method, synergisms existed with free CFZ and PTX ratios of 1:2 and 2:1, and CI50 values were 0.38 and 0.83. For the Sb-CFZ and Sb-PTX combination ratios of 1:2, 1:1, and 2:1, IC50 values were 0.03, 0.37, and 0.51 ng/mL, and CI50 values were 0.05, 0.42, and 0.40, respectively. These results also revealed the synergism of Sb-CFZ and Sb-PTX. As for CFZ and PTX HSA NP ratios of 1:2, 1:1, and 2:1, IC50 values were 0.01, 0.07, and 0.66 ng/mL, and CI50 values were 0.01, 0.04, and 0.31, respectively. Figure 2 illustrates that the original product and HSA combinations exhibited synergism in all ratios examined. Since the combination ratio of 1:2 (CFZ: PTX) demonstrated the more-obvious synergic effect with IC50 value of 0.1 ng/mL and CI50 value of 0.08, the co-encapsulated ratio of 1:2 for CFZ and PTX in HSA (CFZ/PTX/HSA NPs) was chosen as the target formulation for the following assessment studies including the drug release study.
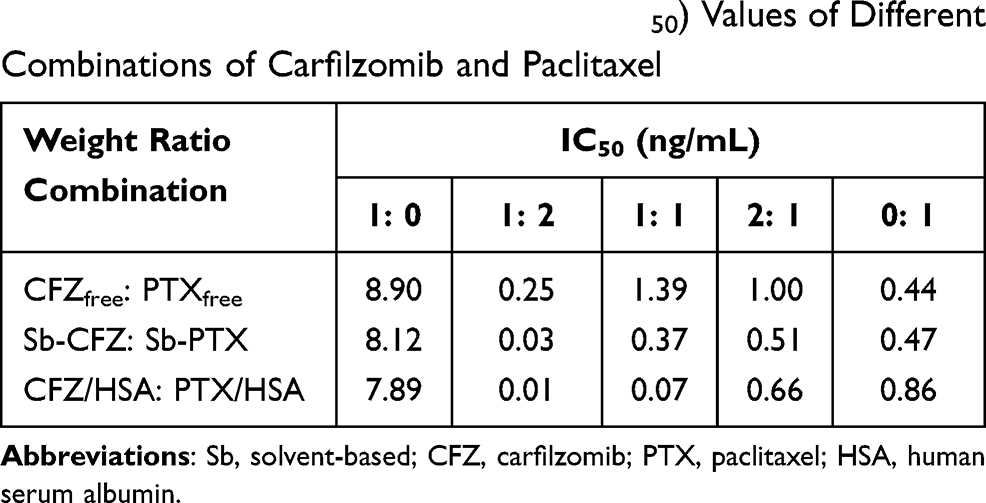 |
Table 1 50% Inhibitory Concentration (IC50) Values of Different Combinations of Carfilzomib and Paclitaxel |
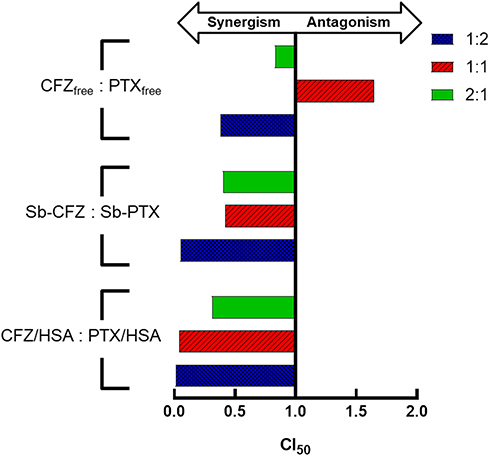 |
Figure 2 Characterization of the synergistic activity of combined carfilzomib (CFZ) and paclitaxel (PTX) treatment at different weight ratios. |
In vitro Release of Carfilzomib and Paclitaxel
Release percentages of CFZ and PTX from various formulations were assessed, and results are shown in Figure 3A and B, respectively. As shown in Figure 3A, the release of CFZ from Sb-CFZ, CFZ/HSA NPs, CFZ/HSA NPs+PTX/HSA NPs, and CFZ/PTX/HSA NPs were observed to have reached a plateau at 12~24 h with similar profiles, and release percentages at 12 h were determined to be 68.6%±7.1%, 79.4%±4.9%, 71.2%±7.9%, and 71.7%±3.8%, respectively. Furthermore, over 90% of CFZ had been released from CFZ/HSA NPs and CFZ/HSA NPs+PTX/HSA NPs at 24 h. We observed that the more-rapid release of CFZ from Sb-CFZ might be attributed to the use of the hydrophilic SBE-β-CD solubilizer to increase the solubility of CFZ in water. Similarly, the more-complete release of CFZ from the two HSA formulations was probably due to both being encapsulated in HSA NPs, which are expected to have greater surface areas for release. However, the release of CFZ from Sb-CFZ+Sb-PTX was measured at only 31.5%±7.2% at 12 and 56.3%±8.8% at 48 h, which were slower than that of CFZ released from Sb-CFZ at 12 h. This indicates that the addition of Sb-PTX to Sb-CFZ might have retarded the release of CFZ from Sb-CFZ resulting in a smaller release percentage. We suspect that by mixing Sb-CFZ with Sb-PTX, CFZ was encapsulated within the hydrophobic interior of Cremophor micelles, which was used as a solubilizing agent in the Sb-PTX formulation, causing retardation of permeation across the membrane of the dialysis bag to release CFZ.35
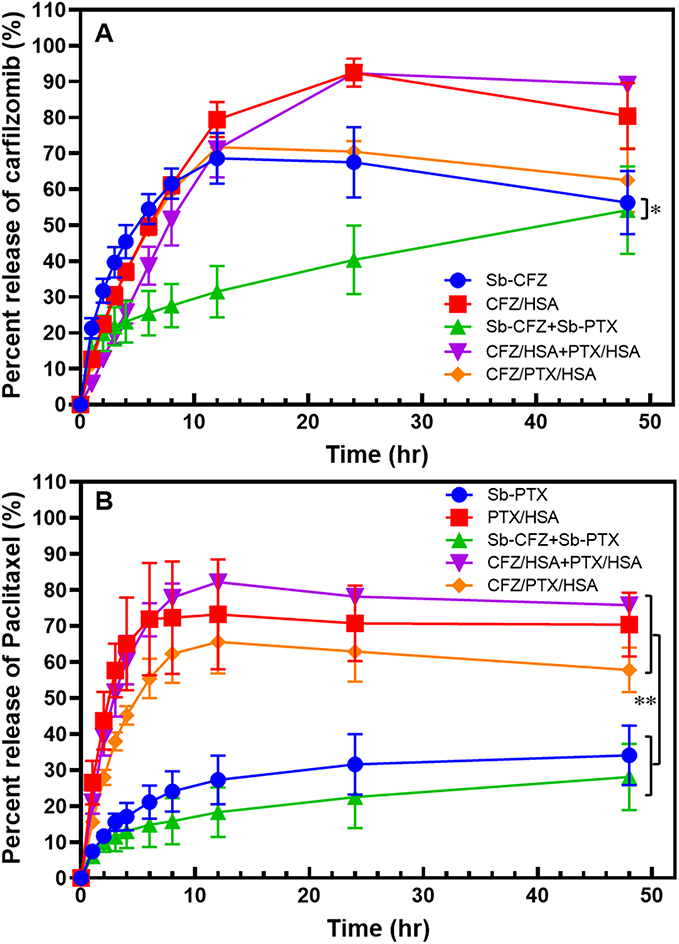 |
Figure 3 Drug release profiles of carfilzomib (CFZ, A) and paclitaxel (PTX, B). *p<0.05 and **p<0.005. |
As for the release of PTX revealed by Figure 3B, release percentages of PTX from PTX/HSA NPs, CFZ/HSA NPs+PTX/HSA NPs, and CFZ/PTX/HSA NPs were observed to have reached a plateau at 12 h with similar profiles, and release percentages at the plateau were, respectively, determined to be 73.2%±15.3%, 82.2%±0.6%, and 65.6%±8.8%. However, the release percentages of PTX from Sb-PTX and Sb-CFZ+Sb-PTX followed a gradually increasing trend, but these forms were only able to release 27.3%±6.8% and 18.3%±6.9%, respectively, at 48 h. Since both Sb-PTX and Sb-CFZ+Sb-PTX contained Cremophor as the solubilizing agent for PTX, it was expected as described above that the release of PTX trapped in Cremophor micelles would be retarded resulting in a slower release rate being observed. Therefore, CFZ and PTX were released more completely from the drug-loaded HSA NPs since both were encapsulated in HSA NPs, which were expected to have greater surface areas for release. Similarly, the greater extents of release percentages of PTX from the three HSA formulations were probably due to all of them being absorbed onto HSA in HSA NPs, which were expected to present as amorphous form to have higher solubility for increasing the extent of release.
In addition, it is worth mentioning that CFZ/PTX ratios released from co-encapsulated HSA NPs (CFZ/PTX/HSA NPs) were about 1.77~2.08 after 8 h of dissolution, which were consistent with the ratio of CFZ/PTX loaded in HSA NPs. However, the ratios of release amounts between CFZ and PTX from Sb-CFZ+Sb-PTX and CFZ/HSA NPs+PTX/HSA NPs were around 1.06~1.18 and 1.63~2.92, respectively, which did not reach the designed optimal ratio of 1:2 for synergism. It was concluded that release from the CFZ/PTX/HSA NP formulation conformed to the design combination ratio of 1:2 for CFZ and PTX to establish a potential synergistic effect.
Maximum Tolerance Dose Study
Maximum tolerance dose for drug-loaded HSA NP and two free drugs as sb-CFZ and sb-PTX was then evaluated in BALB/c mice. Major dose limiting toxicities of CFZ or PTX were determined by neurotoxicity, weight loss >20% or death. According to the result (Table 2), the established maximum tolerance doses for a single dose were: Sb-CFZ 5 mg/kg, CFZ/HSA 17.5 mg/kg, Sb-PTX 20 mg/kg, PTX/HSA 300 mg/kg, Sb-CFZ+Sb-PTX 3.75/7.5 mg/kg, CFZ/HSA+PTX/HSA 10/20 mg/kg and CFZ/PTX/HSA 10/20 mg/kg. For multi-dose were as follows: Sb-CFZ 2.5 mg/kg, CFZ/HSA 5 mg/kg, Sb-PTX 12.5 mg/kg, PTX/HSA 150 mg/kg, Sb-CFZ+Sb-PTX <2.5/5 mg/kg, CFZ/HSA+PTX/HSA 5/10 mg/kg and CFZ/PTX/HSA 5/10 mg/kg. It is demonstrated that the maximum tolerance dose for drug-loaded HSA NP is normally higher than that for solvent-based form of free drug. Compared with solution base form, the combination or co-load HSA NP has at least 2 times higher maximum tolerance dose. Dr. Ernsting reveals that the maximum tolerance dose of single-dose for Abraxane performed in BALB/c is 170 mg/kg. However, in this research, PTX/HSA shows the quite remarkable tolerability from maximum tolerance dose study (300 mg/kg).36 Moreover, Dr. He also found that HSA encapsulation could lower the systemic nervous toxicity from VM-26.37 Taking the advantages with loading multi-drug in HSA NPs, the goal of achieving higher efficacy with lower toxicity was accomplished with such a multi-drug HSA NPs technique platform.
 |
Table 2 Maximum Tolerance Dose Study for Various Combination Ratios of Carfilzomib and Paclitaxel on BALB/c Mice (n = 4) |
Pharmacokinetic Studies
Drug concentrations in plasma after a single tail vein injection of two solvent-based drugs (Sb-CFZ and Sb-PTX) and three drug-loaded HSA NPs (CFZ/HSA NPs, PTX/HSA NPs, and CFZ/PTX/HSA NPs) with respective dosing amounts of CFZ and PTX equivalent to 5 and 10 mg/kg are demonstrated in Figure 4A for CFZ and Figure 4B for PTX. Calculated pharmacokinetic parameters are listed in Table 3 for CFZ and Table 4 for PTX. As shown in Table 3, there were no significant differences among AUC0-1h, AUC0-24h, and AUC0-infinity obtained for all the various formulations after a single IV bolus administration of CFZ equivalent to 5 mg/kg. This indicates that CFZ was rapidly distributed to tissues and was quickly cleared from the systemic circulation after IV administration, resulting in the most reliable measure of the drug’s bioavailability AUC in a period of 0 to 1 h (AUC0-1h) representing nearly the entire extent of the dosing amount of CFZ entering the systemic circulation. It was reported that the in vivo potency of CFZ is determined by the total dose administered (AUC), not Cmax, since CFZ can be rapidly distributed to tissues after IV administration as demonstrated by the potent proteasome inhibition in a variety of tissues.26 Because of this, pharmacokinetic parameters of T1/2,initial (min) and AUC0-1h (hr*µg/mL) which are potentially related to the in vivo potency of CFZ were selected for comparison. Results in Table 3 demonstrate that T1/2,initial (min) and AUC0-1h (h·µg/mL) for CFZ after administration of Sb-CFZ were shorter and lower, respectively, than those for administration of CFZ/HSA NPs (12.72±2.14 vs 15.19±1.38 min and 0.222±0.034 vs 3.337±4.306 h·µg/mL), while neither of them was much different from administration of Sb-CFZ+Sb-PTX (12.72±2.14 vs 11.02±8.98 min and 0.222±0.034 vs 0.241±0.056 h·µg/mL). On the other hand, T1/2,initial (min) and AUC0-1h (h·µg/mL) for CFZ after administration of CFZ/HSA NPs were longer and much higher, respectively, than those with administration of CFZ/HSA NPs+PTX/HSA NPs (15.19±1.38 vs 9.14±2.93 min and 3.337±4.306 vs 0.055±0.009 h·µg/mL), while they were longer and much higher, respectively, than those with administration of CFZ/PTX/HSA NPs (15.19±1.38 vs 10.00±4.08 min and 3.337±4.306 vs 0.537±0.451 h·µg/mL).
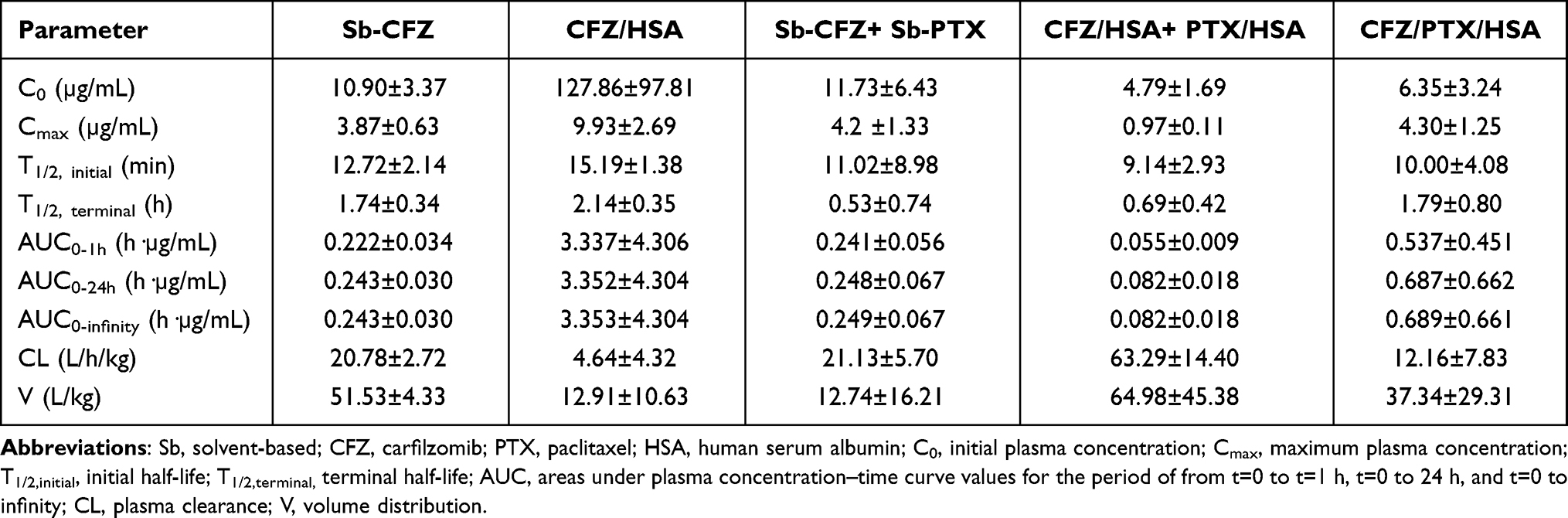 |
Table 3 Pharmacokinetic Parameters of Carfilzomib Obtained from a Single Intravenous Bolus Administration of Various Formulations (Equivalent to 5 mg/kg Carfilzomib) |
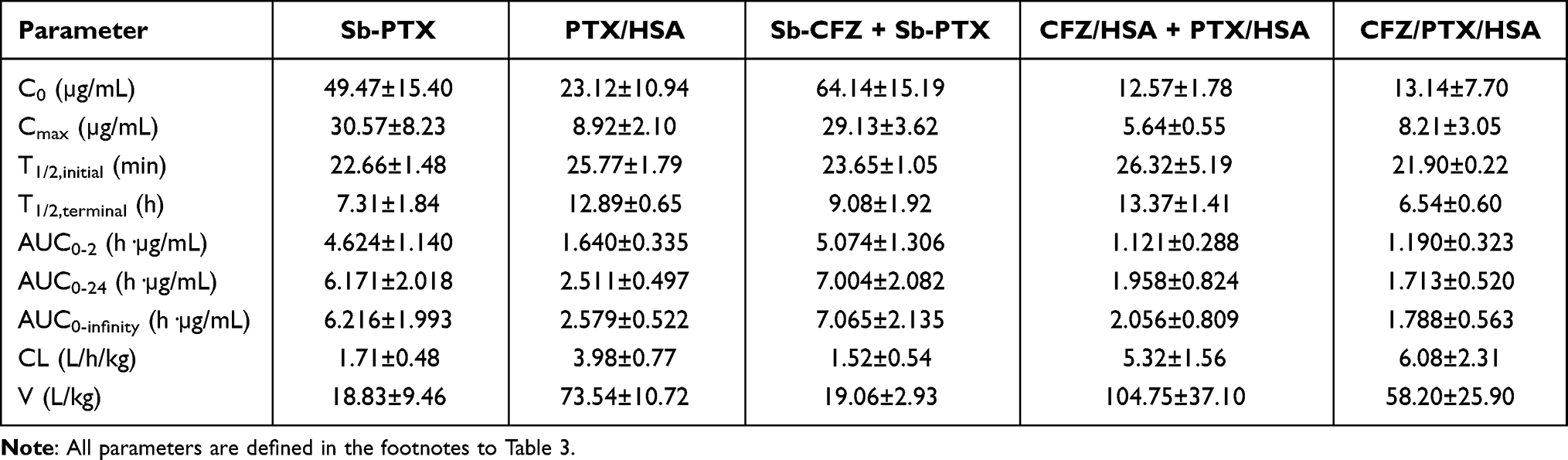 |
Table 4 Pharmacokinetic Parameters of Paclitaxel Obtained from a Single Intravenous Bolus Administration of Various Formulations (Equivalent to 10 mg/kg Paclitaxel) |
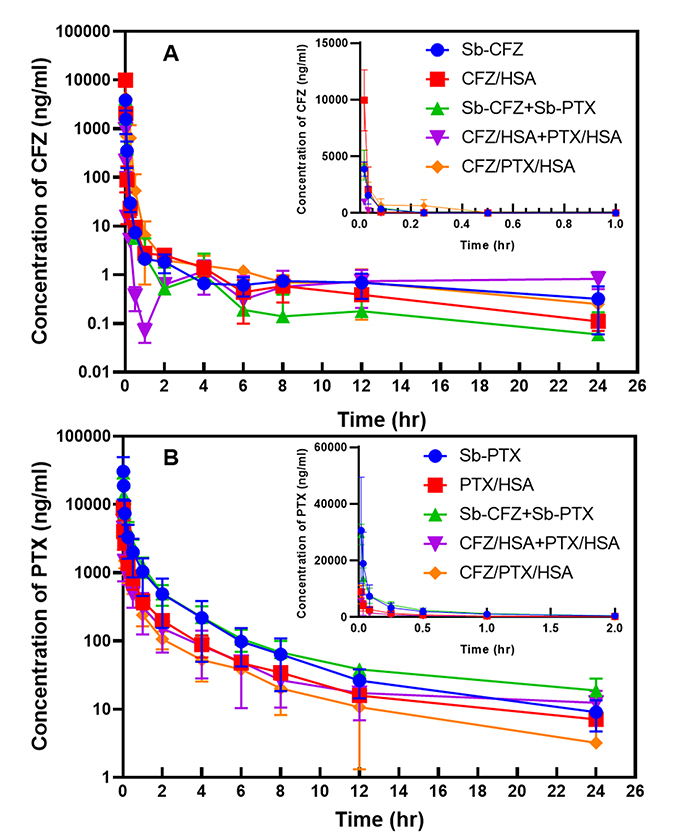 |
Figure 4 Plasma concentration–time curves of carfilzomib (CFZ, A) and paclitaxel (PTX, B) after intravenous administration at respective doses of 5 and 10 mg/kg to rats. |
As previous research reported, almost no CFZ was detected in plasma 30 min after administration with an initial half-life (T1/2,initial) of <20 min.38 By utilizing the same solvent system composed of SBE-β-CD to solubilize CFZ (Sb-CFZ), a similar T1/2,initial was observed in this study, thereby confirming the suitability of the pharmacokinetic study conducted in this research. As such, a slower terminal elimination rate (T1/2,initial) observed for administration of CFZ/HSA NPs compared to that for administration of Sb-CFZ indicates that encapsulation of CFZ with HSA somewhat protected CFZ from elimination in plasma leading to a longer T1/2,initial. With a longer T1/2,initial, it was expected to have a higher AUC0-1h as Table 3 demonstrates.
Compared to Sb-CFZ, the combined administration of the two solvent-based formulations (Sb-CFZ+Sb-PTX) resulted in a similar AUC0-1h for CFZ but with a slightly lower T1/2,initial for the CFZ distribution into tissue compartments. This might indicate that drug–drug interactions exist between CFZ and PTX that are dissolved in solvent as free solubilized forms leading to an influence on the elimination rate of CFZ but not on the AUC. On the other hand, combined administration of the two HSA NP formulations (CFZ/HSA NP+PTX/HSA NPs) could have resulted in significant influences on both T1/2,initial and AUC0-1h for CFZ compared to those for CFZ/HSA NPs. Fortunately, although administration of CFZ/PTX co-loaded HSA NPs (CFZ/PTX/HSA NPs) led to a lower AUC0-1h than that for CFZ/HSA NPs, a higher AUC0-1h than those for Sb-CFZ and Sb-CFZ+Sb-PTX was observed. This also implies that co-encapsulation of CFZ and PTX in HSA with the simultaneous protection of CFZ and PTX by HSA might minimize drug–drug interactions that existed in the plasma compartment between CFZ and PTX when presented in free forms.
Since a greater difference existed between AUC0-2h and AUC0-24h for all the various formulations of PTX administered as shown in Table 4, pharmacokinetic parameters of T1/2,terminal (h) and AUC0-24h (h·µg/mL) were selected for comparison. Results in Table 4 indicate that T1/2,terminal (h) and AUC0-24h (h·µg/mL) for PTX after administration of Sb-PTX were shorter and much higher, respectively, than those for administration of PTX/HSA NPs (7.31±1.84 vs 12.89±0.65 min and 6.171±2.018 vs 2.511±0.497 h·µg/mL), while they both insignificantly differed from those for administration of Sb-CFZ+Sb-PTX (7.31±1.84 vs 9.08±1.92 min and 6.171±2.018 vs 7.004±02.082 h·µg/mL). On the other hand, T1/2,terminal (h) and AUC0-24h (h·µg/mL) for PTX after administration of PTX/HSA NPs greatly differed from those for administration of CFZ/HSA NP+PTX/HSA NPs (12.89±0.65 vs 13.37±1.41 min and 2.511±0.497 vs 1.958±0.824 h·µg/mL), while they were longer and slightly higher, respectively, than those for administration of CFZ/PTX/HSA NPs (12.89±0.56 vs 6.54±0.60 min and 2.511±0.497 vs 1.713±0.520 h·µg/mL).
It was reported that the administration of ABI-007 (Nab-paclitaxel or Abraxane) to Sprague-Dawley rats was associated with significantly higher CL and V of PTX compared to Taxol (Sb-PTX) resulting in a shorter T1/2,terminal (h) with a reduction in the AUC0-24h.35 This was attributed to the fact that the initial dilution volume and the central V were higher for PTX formulated as ABI-007 than for PTX formulated as Taxol resulting from Cremophor (as the solubilizing agent used in solvent-based formulations) preventing the distribution of PTX to the circulation and into tissues. What we observed in the comparative pharmacokinetic analysis performed in this study conformed to data in the literature, which showed that T1/2,terminal and AUC0-24h for PTX after administration of Sb-PTX and Sb-CFZ+Sb-PTX were both shorter and much higher, respectively, than those for administration of the albumin-bound counterpart of PTX/HSA NPs and CFZ/HSA NPs+PTX/HSA NPs, while T1/2,terminal and AUC0-24h for PTX after administration of Sb-PTX and PTX/HSA NPs both insignificantly differed from those with combination administration of either solvent-based or albumin-bound counterparts of Sb-CFZ+Sb-PTX and CFZ/HSA NPs+PTX/HSA NPs. Nevertheless, the administration of the co-loaded HSA NP formulation of CFZ/PTX/HSA NPs seemed to result in an even shorter T1/2,terminal (6.54±0.60 vs 12.89±0.65, 13.37±1.41 min) but not increasing AUC0-24h (1.713±0.520 vs 2.511±0.497, 1.958±0.824 h·µg/mL) for PTX compared to that for administration of PTX/HSA NPs and CFZ/HSA NP+PTX/HSA NPs. The underlying reason for this discrepancy is currently unclear.
The Anti-Tumor Efficacy
The anti-tumor efficacies of drug-loaded HSA NPs were evaluated on MIA Paca-2 cell-xenograft mice. At 14 days after inoculation when tumor volumes had reached 150 mm3, mice were intravenously administered saline, Sb-CFZ, CFZ/HSA NPs, Sb-PTX, PTX/HSA NPs, Sb-CFZ/Sb-PTX (1:2), CFZ/HSA NPs+PTX/HSA NPs (1:2), or CFZ/PTX/HSA NPs. The administration of each formulation was performed on days 0, 1, 7, 8, 14, and 15. Tumor volumes and BWs were assessed three times a week. Tumor growth profiles after administration of the various formulations plotted against time are shown in Figure 5A. TGI (%) compared to the control saline groups was calculated on day 21 after drug administration and on day 46 at termination of the study, and the results are illustrated in Figure 5B. All formulations expressed a greater suppression of tumor growth on both days 21 and 46 than that of saline (2301 mm3). Values of TGI (%) on days 21 and 46 for the Sb-CFZ group showed no improvement compared to the CFZ/HSA NP group, whereas those for the Sb-PTX group showed greater suppression than those for the PTX/HSA NP group on both days 21 and 46. Further, values of TGI (%) on days 21 and 46 for the two combined groups (Sb-CFZ+Sb-PTX and CFZ/HSA NPs+PTX/HSA NPs) all showed increases in TGI (%) compared those of each respective individual group (Sb-CFZ+Sb-PTX vs Sb-CFZ and Sb-PTX and CFZ/HSA NP+PTX/HSA NPs vs CFZ/HSA NPs and PTX/HSA NPs). The results confirmed that a synergic effect on the treatment of MIA PaCa-2 tumors was observed for the combination of CFZ and PTX at a 1:2 ratio regardless of whether Sb-CFZ+Sb-PTZ or CFZ/HSA NP+PTX/HSA NPs were examined. Although only a slight increase in TGI (%) was observed on day 21 but not on day 46 for CFZ/PTX/HSA NPs compared to those for Sb-CFZ+Sb-PTX and CFZ/HSA NPs+PTX/HSA NPs (CFZ/PTX/HSA NPs: 110.20%±8.39% Sb-CFZ+Sb-PTX: 89.80%±21.19%, CFZ/HSA NP+PTX/HSA NPs: 78.38%±16.09%), over 100% of TGI means that tumors had obviously shrunk after treatment with CFZ/PTX/HSA NPs. This seems to indicate that the combination of CFZ and PTX at a 1:2 ratio encapsulated in HSA NPs synergistically improves tumor growth inhibition of MIA PaCa-2 cells.
 |
Figure 5 (A) MIA PaCa-2 tumor growth curve. (B) Tumor inhibition rates. (C) Tumor weights. (D) Body weight changes in tumor-bearing mice. |
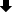
Figure 5C further shows the weights (g) of tumors excised after tumor-bearing mice were sacrificed on day 46. It clearly shows that, compared to saline (2.33±0.52 g), there was a significant anti-tumor efficacy with any combined formulations of CFZ and PTX at a 1:2 ratio of Sb-CFZ+Sb-PTX (0.69±0.12 g), CFZ/HSA NP+PTX/HSA NPs (0.91±0.33 g), and CFZ/PTX/HSA NPs (0.75±0.11 g, all p<0.05), but there were no statistically significant differences in tumor weights among the three combined formulations. This further verifies that synergistic improvement in tumor inhibition is achievable with a combination of CFZ and PTX at a 1:2 ratio loaded into solvent-based or HSA NPs. Figure 5D also reveals that the decreases in BWs of mice after administration of various formulations were all smaller than 20% for the 46-day observation period. However, a greater decrease in BW of mice was observed at several time points with the administration of Sb-CFZ+Sb-PTX. This implies that the greater decrease in BW of mice might be attributed to a higher toxicity of solvents used in the solvent-based formulations compared to HSA used in the HSA NP formulations. It could be concluded that the combination therapy of CFZ and PTX at a 1:2 ratio co-loaded in HSA NPs (CFZ/PTX/HSA NP) demonstrated optimal synergistic improvement in the growth inhibition of MIA PaCa-2 cells with less systematic toxicity.
Biodistribution Studies
To examine the biodistribution of CFZ and PTX in tumors and major organs, C.B-17 SCID mice bearing MIA CaPa-2 tumors were injected with a single IV dose of various formulations including three solvent-based drugs (Sb-CFZ, Sb-PTX, and Sb-CFZ+Sb-PTX), and four drug-loaded HSA NPs (CFZ/HSA NPs, PTX/HSA NPs, CFZ/HSA NPs+PTX/HSA NPs, and CFZ/PTX/HSA NPs) with respective dosing amounts of CFZ and PTX equivalent to 5 and 10 mg/kg. Tumor tissues and major organs were harvested at 2 or 8 h post-injection, processed to make tissue homogenates, and subsequently analyzed with respect to CFZ and PTX levels by LC-MS/MS, and results are demonstrated in Figure 6. For the biodistribution of CFZ in tumor tissues as shown by Figure 6A (2 h) and 6B (8 h), both Sb-CFZ and CFZ/HSA NP groups presented insignificant difference in CFZ levels, but both displayed significantly higher levels of CFZ than those for the Sb-CFZ+Sb-PTX group at 2 h post-dosing with an accompanying decline in the CFZ level at 8 h post-dosing for those formulations examined. An undetectable CFZ level was seen in tumors for both the CFZ/HSA NP+PTX/HSA NP and CFZ/PTX/HSA NP groups at 2 h post-dosing and for those of the Sb-CFZ+Sb-PTX, CFZ/HSA NP+PTX/HSA NP, and CFZ/PTX/HSA NP groups at 8 h post-dosing. However, the CFZ level in tumors did not seem to be correlated with the tumor growth inhibition rate (TGI %) as revealed by Figure 5B. Similar patterns of CFZ biodistributions in these major organs examined for all formulations as distributed to tumors were observed with predominant distribution to the spleen at 2 h post-dosing with an accompanying decline in the CFZ level at 8 h post-dosing for the formulations examined. Similarly, the least or undetectable CFZ levels were shown in those major organs examined for both the CFZ/HSA NP+PTX/HSA NP and CFZ/PTX/HSA NP groups at 2 and 8 h post-dosing. Since quite lower levels of CFZ (<80 ng/g) were observed in those major organs, systemic toxicity caused by the presence of CFZ might not be highly anticipated.
 |
Figure 6 Tissue distributions of carfilzomib (CFZ) and paclitaxel (PTX) at 2 (A and C) and 8 h (B and D), respectively, after intravenous administration of solvent-based (Sb)-CFZ, Sb-PTX, CFZ/human serum albumin (HSA) nanoparticles (NPs), PTX/HSA NPs, Sb-CFZ+Sb-PTX (1:2), CFZ/HSA NPs+PTX/HSA NPs (1:2), or CFZ/PTX/HSA NPs (equivalent to 5 mg/kg CFZ and 10 mg/kg PTX in each mouse). *p<0.05. |
For the biodistribution of PTX in tumors as shown by Figure 6C (2 h) and D (8 h), the Sb-PTX group presented a statistically significantly higher level of PTX than those for the PTX/HSA NP and CFZ/PTX/HSA NP groups at 2 h post-dosing (40.00±5.91 vs 19.74±4.51 and 5.72±2.85 µg/g), while those for PTX/HSA NPs and CFZ/HSA NP+PTX/HSA NPs were similar, but both were statistically higher than that for CFZ/PTX/HSA NPs (19.74±4.51 ≈ 21.60±9.54 vs 5.72±2.85 µg/g). However, a slight increase was shown in the PTX level biodistributed in tumors for the Sb-PTX group at 8 h post-dosing compared to that at 2 h post-dosing, whereas those for the remaining formulations at 8 h post-dosing (PTX/HSA NPs, Sb-CFZ+Sb-PTX, CFZ/HSA NPs+PTX/HSA NPs, and CFZ/PTX/HSA NPs) were still statistically significantly lower than that for the Sb-PTX group but showed insignificant differences among them (50.52±8.60 vs 20.62±15.08, 15.06±6.84, 15.75±3.88, and 12.98±3.93 µg/g). Similar to CFZ as described above, the PTX level distributed to tumors did not seem to be correlated with the tumor growth inhibition rate (TGI %) as revealed by Figure 5B. Similar patterns of PTX biodistributions in the major organs examined for all formulations as those distributed to tumors were observed with predominant distribution to the liver, spleen, and kidneys at 2 h post-dosing with accompanying significant declines in PTX levels at 8 h post-dosing for those formulations examined. Since quite higher levels of PTX were detected in the liver (134.7±21.9 µg/g at 2 h and 28.92±12.62 µg/g at 8 h) after administration of Sb-PTX compared to those for the other formulations examined, the higher grade of systemic toxicity caused by administration of Sb-PTX in the presence of this amount of PTX in the liver might be highly expected. It is worth noting that although both 2 and 8 h accumulations of PTX in tumors with the administration of Sb-PTX were 2~3 times more than those for the two combined groups (Sb-CFZ+Sb-PTX and CFZ/HSA NPs+PTX/HSA NPs), TGI (%) values on days 21 and 46 for the two combined groups (Sb-CFZ+Sb-PTX and CFZ/HSA NPs+PTX/HSA NPs) as revealed above all showed increases in TGI (%) compared to that for the Sb-PTX group. This seems to further confirm that the synergistic improvement in tumor growth inhibition is achievable with a combination of CFZ and PTX at a 1:2 ratio loaded into solvent-based or HSA NPs with minimal systemic toxicity.
Conclusions
It was concluded that the effective combination therapy of pancreatic cancer was enabled with treatment of CFZ and PTX co-loaded HSA NPs, which was prepared by a simple one-pot reverse self-assembly method developed in this study. The one-pot reverse self-assembly method was novel and able to optimally prepare HSA NPs loaded with hydrophobic drugs by adjusting the drug/HSA ratio and homogenization process parameters. Without using any hazardous or toxic solvent during preparation of drug-loaded HSA NPs, the one-pot reverse self-assembly method could claim to be environmentally friendly with the ability to co-encapsulate two chemodrugs in HSA NPs with the optimal ratio for synergistic therapy to inhibit tumor growth and minimize systemic toxicity compared to monotherapy. With the related data in this study, it might be able to construct a platform for combination therapy in the future.
Institutional Review Board Statement
This animal experiment was approved by the Institutional Animal Care and Use Committee of Taipei Medical University (Approval No.: LAC-2018-0419) in compliance with the Taiwanese Animal Welfare Act.
Acknowledgments
This work was supported by the Ministry of Science and Technology, Taiwan, ROC, under grant no. 107-2314-B-038-035-MY3, 108-2314-B-264-001- and 110-2221-E-264-002-.
Disclosure
The authors report no conflicts of interest with respect to this work.
References
1. American Society of Clinical Oncology. Pancreatic cancer: types of treatment. Internet. Cancer.Net Editorial Board; June 8, 2021. Available from: https://www.cancer.net/cancer-types/pancreatic-cancer/types-treatment.
2. Hidalgo M, Cascinu S, Kleeff J, et al. Addressing the challenges of pancreatic cancer: future directions for improving outcomes. Pancreatology. 2015;15(1):8–18. doi:10.1016/j.pan.2014.10.001
3. Lei F, Xi X, Batra SK, Bronich TK. Combination therapies and drug delivery platforms in combating pancreatic cancer. J Pharmacol Exp Ther. 2019;370(3):682–694. doi:10.1124/jpet.118.255786
4. Wang JP, Wu CY, Yeh YC, et al. Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: a randomized, open-label, prospective trial. Oncotarget. 2015;6(20):18162–18173. doi:10.18632/oncotarget.4216
5. Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–1703. doi:10.1056/NEJMoa1304369
6. Fraunhoffer NA, Abuelafia AM, Bigonnet M, et al. Evidencing a pancreatic Ductal Adenocarcinoma subpopulation sensitive to the proteasome inhibitor Carfilzomib. Clin Cancer Res. 2020;26(20):5506–5519. doi:10.1158/1078-0432.Ccr-20-1232
7. Kawaguchi K, Igarashi K, Murakami T, et al. MEK inhibitors cobimetinib and trametinib, regressed a gemcitabine-resistant pancreatic-cancer Patient-Derived Orthotopic Xenograft (PDOX). Oncotarget. 2017;8(29):47490. doi:10.18632/oncotarget.17667
8. Roeten MSF, Cloos J, Jansen G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother Pharmacol. 2018;81(2):227–243. doi:10.1007/s00280-017-3489-0
9. Naumann K, Schmich K, Jaeger C, Kratz F, Merfort I. Noxa/Mcl-1 balance influences the effect of the proteasome inhibitor MG-132 in combination with anticancer agents in pancreatic cancer cell lines. Anticancer Drugs. 2012;23(6):614–626. doi:10.1097/CAD.0b013e3283504e53
10. Papadopoulos KP, Burris HA
11. Deshaies RJ. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014;12(1):94. doi:10.1186/s12915-014-0094-0
12. Park JE, Chun SE, Reichel D, et al. Polymer micelle formulation for the proteasome inhibitor drug carfilzomib: anticancer efficacy and pharmacokinetic studies in mice. PLoS One. 2017;12(3):e0173247. doi:10.1371/journal.pone.0173247
13. Park JE, Park J, Jun Y, et al. Expanding therapeutic utility of carfilzomib for breast cancer therapy by novel albumin-coated nanocrystal formulation. J Control Release. 2019;302:148–159. doi:10.1016/j.jconrel.2019.04.006
14. Peng Q, Zhang S, Yang Q, et al. Preformed albumin Corona, a protective coating for nanoparticles based drug delivery system. Biomaterials. 2013;34(33):8521–8530. doi:10.1016/j.biomaterials.2013.07.102
15. Sleep D, Cameron J, Evans LR. Albumin as a versatile platform for drug half-life extension. Biochim Biophys Acta. 2013;1830(12):5526–5534. doi:10.1016/j.bbagen.2013.04.023
16. Park J, Park JE, Hedrick VE, et al. A comparative in vivo study of albumin-coated Paclitaxel Nanocrystals and Abraxane. Small. 2018;14(16):e1703670. doi:10.1002/smll.201703670
17. Stehle G, Sinn H, Wunder A, et al. Plasma protein (albumin) catabolism by the tumor itself–implications for tumor metabolism and the genesis of cachexia. Crit Rev Oncol Hematol. 1997;26(2):77–100. doi:10.1016/s1040-8428(97)00015-2
18. Desai N, Trieu V, Yao Z, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res. 2006;12(4):1317–1324. doi:10.1158/1078-0432.Ccr-05-1634
19. Guarneri V, Dieci MV, Conte P. Enhancing intracellular taxane delivery: current role and perspectives of nanoparticle albumin-bound paclitaxel in the treatment of advanced breast cancer. Expert Opin Pharmacother. 2012;13(3):395–406. doi:10.1517/14656566.2012.651127
20. Kudarha RR, Sawant KK. Albumin based versatile multifunctional nanocarriers for cancer therapy: fabrication, surface modification, multimodal therapeutics and imaging approaches. Mater Sci Eng C Mater Biol Appl. 2017;81:607–626. doi:10.1016/j.msec.2017.08.004
21. Sage H, Johnson C, Bornstein P. Characterization of a novel serum albumin-binding glycoprotein secreted by endothelial cells in culture. J Biol Chem. 1984;259(6):3993–4007. doi:10.1016/S0021-9258(17)43194-2
22. Hoogenboezem EN, Duvall CL. Harnessing albumin as a carrier for cancer therapies. Adv Drug Deliv Rev. 2018;130:73–89. doi:10.1016/j.addr.2018.07.011
23. Han H, Wang J, Chen T, Yin L, Jin Q, Ji J. Enzyme-sensitive gemcitabine conjugated albumin nanoparticles as a versatile theranostic nanoplatform for pancreatic cancer treatment. J Colloid Interface Sci. 2017;507:217–224. doi:10.1016/j.jcis.2017.07.047
24. Yu Q, Qiu Y, Li J, et al. Targeting cancer-associated fibroblasts by dual-responsive lipid-albumin nanoparticles to enhance drug perfusion for pancreatic tumor therapy. J Control Release. 2020;321:564–575. doi:10.1016/j.jconrel.2020.02.040
25. Yin T, Dong L, Cui B, et al. A toxic organic solvent-free technology for the preparation of PEGylated paclitaxel nanosuspension based on human serum albumin for effective cancer therapy. Int J Nanomedicine. 2015;10:7397–7412. doi:10.2147/ijn.S92697
26. Yang J, Wang Z, Fang Y, et al. Pharmacokinetics, pharmacodynamics, metabolism, distribution, and excretion of carfilzomib in rats. Drug Metab Dispos. 2011;39(10):1873–1882. doi:10.1124/dmd.111.039164
27. Chen RF. Removal of fatty acids from serum albumin by charcoal treatment. J Biol Chem. 1967;242(2):173–181. doi:10.1016/S0021-9258(19)81445-X
28. Cheng WJ, Chen LC, Ho HO, Lin HL, Sheu MT. Stearyl polyethylenimine complexed with plasmids as the core of human serum albumin nanoparticles noncovalently bound to CRISPR/Cas9 plasmids or siRNA for disrupting or silencing PD-L1 expression for immunotherapy. Int J Nanomedicine. 2018;13:7079–7094. doi:10.2147/ijn.S181440
29. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. doi:10.1158/0008-5472.Can-09-1947
30. Chang CE, Hsieh CM, Chen LC, et al. Novel application of pluronic lecithin organogels (PLOs) for local delivery of synergistic combination of docetaxel and cisplatin to improve therapeutic efficacy against ovarian cancer. Drug Deliv. 2018;25(1):632–643. doi:10.1080/10717544.2018.1440444
31. Cheng WJ, Lin SY, Chen M, et al. Active tumoral/tumor environmental dual-targeting by non-covalently arming with trispecific antibodies or dual-bispecific antibodies on docetaxel-loaded mPEGylated nanocarriers to enhance chemotherapeutic efficacy and Minimize systemic toxicity. Int J Nanomedicine. 2021;16:4017–4030. doi:10.2147/ijn.S301237
32. Min JS, Kim J, Kim JH, et al. Quantitative determination of carfilzomib in mouse plasma by liquid chromatography-tandem mass spectrometry and its application to a pharmacokinetic study. J Pharm Biomed Anal. 2017;146:341–346. doi:10.1016/j.jpba.2017.08.048
33. Kuo ZK, Lin MW, Lu IH, et al. Antiangiogenic and antihepatocellular carcinoma activities of the Juniperus chinensis extract. BMC Complement Altern Med. 2016;16(1):277. doi:10.1186/s12906-016-1250-6
34. Vlasova I, Saletsky A. Study of the denaturation of human serum albumin by sodium dodecyl sulfate using the intrinsic fluorescence of albumin. J Appl Spectrosc. 2009;76(4):536–541. doi:10.1007/s10812-009-9227-6
35. Sparreboom A, Scripture CD, Trieu V, et al. Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin Cancer Res. 2005;11(11):4136–4143. doi:10.1158/1078-0432.Ccr-04-2291
36. Ernsting MJ, Murakami M, Undzys E, Aman A, Press B, Li S-D. A docetaxel-carboxymethylcellulose nanoparticle outperforms the approved taxane nanoformulation, Abraxane, in mouse tumor models with significant control of metastases. J Control Release. 2012;162(3):575–581. doi:10.1016/j.jconrel.2012.07.043
37. He X, Xiang N, Zhang J, et al. Encapsulation of teniposide into albumin nanoparticles with greatly lowered toxicity and enhanced antitumor activity. Int J Pharm. 2015;487(1–2):250–259. doi:10.1016/j.ijpharm.2015.04.047
38. Kortuem KM, Stewart AK. Carfilzomib. Blood. 2013;121(6):893–897. doi:10.1182/blood-2012-10-459883
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.