")
Back to Journals » Vascular Health and Risk Management » Volume 13
Cardiovascular toxicities of BCR-ABL tyrosine kinase inhibitors in chronic myeloid leukemia: preventive strategies and cardiovascular surveillance
Authors Aghel N, Delgado DH, Lipton JH
Received 22 May 2017
Accepted for publication 27 June 2017
Published 4 August 2017 Volume 2017:13 Pages 293—303
DOI https://doi.org/10.2147/VHRM.S108874
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Nazanin Aghel,1 Diego Hernan Delgado,1 Jeffrey Howard Lipton2
1Division of Cardiology, Peter Munk Cardiac Centre, Toronto General Hospital, University Health Network,University of Toronto, 2Department of Medical Oncology and Hematology, Princess Margaret Cancer Centre, University Health Network, University of Toronto, Toronto, ON, Canada
Abstract: Tyrosine kinase inhibitors (TKIs) have revolutionized the treatment and outcomes of chronic myeloid leukemia (CML). Despite their significant impact on the management of CML, there is growing evidence that TKIs may cause cardiovascular and/or metabolic complications. In this review, we present the current evidence regarding the cardiovascular safety profiles of BCR-ABL TKIs. Methodological challenges of studies that reported the cardiovascular safety of TKIs are discussed. We also propose management strategies for cardiovascular surveillance and risk factor modification during treatment with these agents.
Keywords: BCR-ABL, tyrosine kinase inhibitor, peripheral arterial disease, cardiovascular disease, adverse event
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disorder resulting from a chromosomal translocation leading to the formation of a fusion gene, which encodes the BCR-ABL protein with protein tyrosine kinase (TK) activity.1 The treatment of CML has been transformed since the introduction of the BCR-ABL tyrosine kinase inhibitors (TKIs) that have the ability to cause arrest of growth or apoptosis in hematopoietic cells that express BCR-ABL.2
Small-molecule TKIs have revolutionized the treatment and outcomes of CML, changing it from a life-threatening disease to one with life expectancies similar to the general population for the majority of patients who are responsive to treatment.3,4 Although these treatments have radically changed the natural course of CML and many other cancers, they may result in cardiovascular and/or metabolic complications.5
Protein TKs are enzymes that catalyze the transfer of phosphate from adenosine triphosphate (ATP) to tyrosine residues on specific protein.6 TKs play a critical role in eukaryotic cellular signaling, and their dysregulation has been associated with multiple types of cancer, including CML. TKs also play a critical role in cardiovascular system, including vascular, metabolic, and myocardial physiology.7 As such, it is not surprising that inhibiting certain TKs can interfere with cardiovascular system function and cause clinical complications.
Considerable progress has been made in identifying the excess risk of cardiovascular events (CVEs) associated with exposure to TKIs in CML patients. The data on underlying mechanisms, preventive and treatment strategies however, are currently inadequate. In this review, we present current evidence regarding the cardiovascular safety profiles of BCR-ABL TKIs and propose management strategies for cardiovascular assessment and risk factor modification during treatment with these agents.
Risk factors for cardiovascular disease (CVD) in CML population
Cardiovascular epidemiological studies conducted over the past years have made important contributions to our knowledge about importance of risk factors in predicting CVEs and have led to the development of methods for estimating the individual’s global risk of CVD.8 The risk of coronary artery disease (CAD), peripheral arterial disease (PAD), stroke, and cardiovascular death can be predicted on the basis of a constellation of risk factors: dyslipidemia, hypertension, cigarette smoking, age, gender, ethnicity, obesity, family history, and physical inactivity; all have been known as traditional risk factors in the development of CVEs. The significance of these risk factors is well documented in several population-based cohorts or large-scale case–control studies.8,9 On the other hand, not all CVEs happen in people with multiple traditional risk factors, and as a matter of fact in some patients, abnormalities of inflammation or thrombosis appear to contribute. Thus, in addition to conventional risk factors, other atherothrombotic risk markers, including high-sensitivity C-reactive protein (hsCRP), and other markers of inflammation such as interleukin-1, interleukin-6, fibrinogen, and lipoprotein-associated phospholipase A 2, as well as homocysteine and lipoprotein (a), have been studied and considered as non-traditional risk factors.10
Accumulating evidence suggests that the combination of cardiovascular risk factors along with cardiovascular side effects of TKIs might contribute to CVEs in CML population. The fact that CVEs are more prevalent in CML patients who have pre-existing cardiovascular risk factors11,12 supports this notion.
CVEs in oncology trials versus cardiology trials
There are several important fundamental issues that need to be reviewed before discussing CVEs related to treatment with TKIs in CML population. These facts challenge several issues related to reporting CVEs in patients with CML.
Adverse events (AEs) in oncology trials are reported using the Common Terminology Criteria for Adverse Events (CTCAE), and these are different from outcomes as measured in cardiology trials.13 In most oncology trials, CVEs are often inconsistently defined and combined compared with cardiovascular outcome studies. An example of such definition can be found in defining CVEs in ENESTnd (Evaluating Nilotinib Efficacy and Safety in Clinical Trials – Newly Diagnosed Patients) study. In this study, PAD events were identified also by non-specific definitions such as peripheral coldness, peripheral vascular disorder, and poor peripheral circulation in addition to the standard definition of PAD.14 As a result, CVEs reported in these trials cannot be compared with cardiology trials and even among different oncology studies.
Studies reporting cardiovascular outcomes in CML patients for the most part focused on AEs that were reported in clinical trials or small cohorts of patients receiving specific type of TKI. Many studies did not consider traditional risk factors or even existence of non-traditional risk factors as a contributor to the CVEs. Most of these trials lack enough power to determine if baseline traditional risk factors of enrolled patients contributed to CVEs. The low incidence of CML makes it almost impossible to enroll large number of patients for assessing risk factors for cardiovascular outcomes. In contrast in the Cardiology era, most outcome trials can recruit large number of patients and have enough power to find association between risk factors and CVEs.8,9 Meta-analyses might address this issue. Inconsistent definition of cardiovascular outcomes and heterogeneity in reporting the CVEs in oncology trials, however, make it impossible to use the current data reliably for meta-analysis.
Almost all trials for assessing effects of a new TKI in the CML population have recruited patients some of whom have a history of CVD. When cardiac safety outcomes were reported, patients with a history of CVD were not analyzed as a separate group. Risk of future CVEs in patients with CVD is far higher than patients without CVD or patients with multiple cardiovascular risk factors.15 Measuring cardiovascular outcomes in a population consisting of patients with pre-existing CVD will be confounded by the fact that the risk of recurrent CVEs and other manifestations of atherosclerosis is higher at baseline in patients with previous history of CVD.
Methodological challenges of studies that reported the cardiovascular safety of TKIs should be acknowledged when the cardiovascular AEs of these drugs are discussed and published. Efforts are made to standardize cardiovascular outcomes in oncology trials, but unfortunately these outcomes have not been applied prospectively to the vast majority of novel anticancer medications.13 To measure the incidence and severity of CVEs and to compare the findings with cardiology studies and general population, it is vital to use the definitions for CVEs and end points that are commonly used in cardiovascular clinical trials.16
BCR-ABL inhibitors
In 2001, imatinib was the first TKI approved for the treatment of CML and has revolutionized management. Imatinib inhibits the kinase activity of the BCR-ABL fusion protein that arises from the balanced translocation which creates the Philadelphia chromosome.17 Unfortunately, ABL1 kinase can be resistant to imatinib therapy. As a result, second-generation (2G) therapies such as dasatinib, nilotinib, and bosutinib and third generation (3G) such as ponatinib were developed and approved for salvage, and now 2G TKIs (nilotinib and dasatinib) are approved as frontline therapy because of faster, deeper molecular response and less disease progression although no survival advantage.18,19
Although all approved TKIs for the treatment of CML have activity against BCR-ABL1, they are different in their potency and activity against BCR-ABL1 and against other kinases, including those involved in cardiovascular system.5,20 This can explain the reason for diversity of cardiovascular toxic effects that has been reported with different TKIs. Hence, the cardiovascular safety of each TKI needs to be addressed separately. In the next section, we review the cardiovascular safety profiles of different TKIs used in the treatment of CML.
Imatinib
Imatinib, an inhibitor of platelet-derived growth factor (PDGFR), ABL1, and KIT, was approved in 2001 for the treatment of CML.2,21 In 2006, Kerkelä et al22 reported a case series of 10 patients along with in vitro and murine studies suggesting that imatinib could cause cardiotoxicity. Clinical trials that followed large number of patients prospectively for a long period of time, however, did not show increased risk of CVEs and cardiotoxicity. For instance, long-term (11-year) follow-up of patients on imatinib in IRIS (International Randomized Study of Interferon and STI571) study showed that CVEs were rare.23 Retrospective analysis of 6 imatinib trials, including 2327 patients with median exposure of 2.4 years, revealed a congestive heart failure (CHF) incidence of 0.2%.24
Ribeiro et al25 studied 103 CML patients on imatinib after a median exposure of 28 months and found no significant difference in left ventricular ejection fraction between patients on imatinib and control group (68% [65–74] versus 69% [64–73], p=0.67)]. Similarly, Estabragh et al26 found no evidence of myocardial dysfunction by echocardiography in 59 CML patients after mean duration exposure to imatinib of 3.4 ± 1.8 years except in patients with pre-existing cardiac disease. Prospective serial MUGA scanning with an interval of 12 months in the same population of patients revealed no evidence of deterioration in myocardial function, despite continuation of imatinib. Based on available data, it appears that imatinib has little clinical impact on cardiac function.
There is a possibility that imatinib has positive metabolic and vascular effects. Imatinib has been reported to lower blood glucose levels27 and prevent the development of atherosclerotic lesions in the aorta in diabetic mice.28 Imatinib improved exercise capacity and hemodynamics in patients with advanced pulmonary arterial hypertension (PAH) in a randomized double-blind study29 and has been shown to attenuate in-stent restenosis in an animal model.30 Retrospective analysis of the IRIS study, Tyrosine Kinase Dose Optimization Study (TOPS), and ENESTnd studies has shown a lower incidence of PAD in patients treated with imatinib versus patients treated with nilotinib and lower incidence even compared with patients treated without TKIs.31 This finding raises the possibility that imatinib might be protective, but prospective studies are currently lacking.5
Nilotinib
Despite the encouraging effect of imatinib in CML, nearly 20% of patients who are on imatinib do not achieve a complete cytogenetic response, and others may have intolerable side effects or drug resistance over time.32 Nilotinib is an orally bioavailable drug with superior potency and selectivity for BCR-ABL than imatinib.18 It also inhibits the TK activity of the PDGF and c-Kit receptors.33 Nilotinib was approved and used successfully in patients with CML, inducing deeper molecular response.18,34
The first concern about vascular toxicity of nilotinib was raised in 2011, when Aichberger et al35 reported 3 cases of PAD in patients on nilotinib. In a retrospective analysis of 179 patients, 11 patients (6.2%) developed PAD involving lower limbs, 8 patients required invasive therapy (angioplasty and stent placement), and 4 patients required amputation.36 Kim et al11 prospectively assessed 159 CML patients treated with either imatinib or nilotinib for PAD, using ankle–brachial index (ABI) and duplex ultrasonography. Pathological ABI was more prevalent in patients treated with second- or first-line nilotinib compared to patients on first-line imatinib. It is important to note, however, that distribution of patients with previous history of CAD was uneven in this study between the imatinib and nilotinib groups: pre-existing CAD was 2 times more prevalent in nilotinib group compared to imatinib group. Prevalence of PAD is quite higher in patients with pre-existing CVD,37 and patients with established CVD experience 2–3 times higher event rates when even compared with patients with multiple risk factors.15 Baseline ABI and duplex ultrasonography of patients before exposure to TKIs was not available in this study and all other studies that reported CVEs in patients receiving TKIs.
Five-year update of the randomized ENESTnd trial14 also reported higher CVEs in nilotinib arm than imatinib arm. Framingham multivariate cardiovascular risk scores were measured and were predictive of patients’ risk of developing a CVE during nilotinib therapy. This can be interpreted as the fact that drivers of patients’ CVEs during treatment with nilotinib might be their baseline cardiovascular risk factors. One might argue that if CVEs in nilotinib group can be predicted by patients’ Framingham risk score, then there is a possibility that lower CVEs in patients on imatinib arm might be due to potential cardiovascular protective effect of imatinib. As discussed before, retrospective analysis of IRIS, TOPS, and ENESTnd studies has shown a lower incidence of PAD in patients treated with imatinib versus patients treated with nilotinib and lower incidence even compared with CML patients treated without TKIs.31 The result of this study also supports the idea that imatinib might be cardio protective in CML patients. Further prospective and well-designed studies would be needed to investigate this possibility.
Electrocardiograms (ECGs) performed during the Phase I clinical trial of nilotinib revealed a 5–15 ms prolongation in the corrected QT interval in a subgroup of patients.34 However, during the ENESTnd trial, patients were monitored closely and no patient developed a QT interval corrected for heart rate of >500 ms.18 No torsade or malignant arrhythmia was also reported after long-term follow-up.14
Unfortunately, in most of these studies with nilotinib, CVEs were not reported according to the American College of Cardiology/American Heart Association (ACC/AHA) guidelines16 for clinical outcomes in cardiology trials, and as mentioned before, these outcomes were also inconsistently defined and combined. Meta-analysis of cardiac AEs related to BCR-ABL TKIs has been recently published.38,39 The heterogeneity of cardiovascular outcomes of reviewed studies in the meta-analysis makes it almost impossible to anticipate a summary that represents cardiovascular AEs of BCR-ABL TKIs.
The exact relationship between CVEs and nilotinib therapy and the exact mechanism for CVEs still remain hypothetical. Increased blood glucose40 and cholesterol levels41 have been observed with exposure to nilotinib and proposed as a mechanism for increased risk of CVEs in patients on nilotinib.
Dasatinib
Dasatinib inhibits many imatinib-resistant BCR-ABL forms, and it shows ~325-fold more potency than imatinib at inhibiting BCR-ABL.42 It also inhibits several other kinases, including members of the Src family.43 Dasatinib is now approved for frontline CML therapy, based on superior 12-month complete cytogenetic response rates compared with imatinib.19
Pleural effusion is a common side effect of dasatinib and occurs in up to 28% of patients.44 The mechanism of dasatinib-related pleural effusion is poorly understood, but it does not appear to be cardiovascular in etiology. It has been hypothesized that this AE may be immune-mediated, based on reports of high lymphocyte (large granular/natural killer) counts in pleural fluid.45
In 2012, the French Pulmonary Hypertension Registry reported 9 patients with PAH related to dasatinib.46 Before initiation of dasatinib, previous treatments included interferon in 6 patients, hydroxyurea in 6 patients, cytarabine in 3 patients, and imatinib in 8 patients. Clinical, functional, and hemodynamic improvements were observed after dasatinib was discontinued. One patient died with refractory right heart failure, and another unexplained sudden death happened in this population. Based on the result of Registry, it was estimated that PAH happens in at least 0.45% of individuals who are chronically exposed to dasatinib.46 The 7-year follow-up report of the study “dasatinib in imatinib-resistant or -intolerant CML patients” indicated also PAH in <3% of 670 CML patients who were followed up for 7 years and arterial ischemic events occurred in <4% of patients.47 Drug-related pulmonary hypertension, assessed and estimated only by Doppler echocardiography, was reported in 14 dasatinib-treated patients (5%) and 1 imatinib-treated patient (0.4%) in final 5-year follow-up report of Dasatinib Versus Imatinib Study in Treatment-Naïve Chronic Myeloid Leukemia Patients (DASISION) trial.44 Right heart catheterization (RHC) to confirm PAH was performed only in 1 patient (which did not confirm PAH), and therefore, the possibility of PAH in the remaining 13 patients remains unproven.
Bosutinib
Bosutinib is a dual Src and ABL TKI with minimal activity against PDGFR or c-KIT.48 Bosutinib is a 2G TKI and has demonstrated efficacy in prospective clinical trials as second- and third-line therapy in CML patients with resistance or intolerance to other TKIs.49 Retrospective analyses of 2 large clinical trials of bosutinib demonstrated no significant differences in overall incidence of CVEs between bosutinib and imatinib in the first-line setting. Incidence of CVEs during long-term bosutinib therapy was relatively low in the first-line (Phase III study) and second- and third-line (Phase I/II study) settings.50 Of note, bosutinib-treated patients with CVEs were mostly managed with concomitant medication without a need for discontinuing or interrupting the treatment for CML with bosutinib. Nine deaths were reported with bosutinib in this retrospective study. Five deaths were due to cerebrovascular events, and 4 patients died as a result of CVEs. It is important to note that the median age of the overall patient population in both studies was lower than the average CML patient age in the western world at diagnosis.50
Ponatinib
Ponatinib, the only 3G TKI available, is a potent oral TKI that blocks mutated BCR-ABL, including the gatekeeper mutant T315I, which is resistant to other TKIs.51 Ponatinib received accelerated approval in December 2012. Data obtained in October 2013 from the ongoing PACE trial (Ponatinib Ph-Positive Acute Lymphoblastic Leukemia and CML Evaluation)12 showed that after a median follow-up of 24 months, ponatinib was associated with an increased cumulative incidence of CVEs. At 12 months, 6% of patients experienced cardiovascular AEs, 3% experienced cerebrovascular AEs, and 4% experienced peripheral AEs. At a median follow-up of 24 months, cumulative cardiovascular, cerebrovascular, and peripheral AEs increased to 10%, 7%, and 7%, respectively. These AEs resulted in a partial clinical hold on ongoing trials of ponatinib and the termination of the EPIC trial, the frontline randomized trial of ponatinib versus imatinib (Ponatinib in Newly Diagnosed Chronic Myeloid Leukemia).52 At EPIC trial termination, median follow-up for the entire trial population was 5.1 months. More CVEs were detected during the follow-up with ponatinib compared with imatinib.
In October 2013, the sale of this medication was temporarily suspended due to CVEs reported during the single-arm Phase II PACE trial. After consideration of the lack of any other durable effective treatments (except for bone marrow transplant and in some cases omacetaxine53) available for T315I mutant CML and patients who are resistant to multiple TKIs, sale of ponatinib was resumed in January 2014 with a black box warning about increased risk of arterial and venous occlusive events.
The exact pathophysiology of the CVEs (thrombosis, accelerated atherosclerosis, or another vascular event, such as a vasospasm) triggered by exposure to ponatinib remains unclear.7 The higher rate of events in patients with multiple risk factors and history of CVD may suggest an atherosclerotic cause for the events. Retrospective data from 3 clinical trials (N=671) to determine the impact of ponatinib dose intensity revealed association between dose intensity and risk of CVEs.54
More recently, lower doses either initially or in step-down are being studied to assess efficacy and safety of different doses of ponatinib in CML treatment (Ponatinib in Patients with Resistant Chronic Phase Chronic Myeloid Leukemia [CML] to characterize the Efficacy and Safety of a Range of Doses [OPTIC], https://clinicaltrials.gov/ct2/show/NCT02467270). Given the high incidence of CVEs reported in ponatinib trials, risks and benefits of treatment with ponatinib need to be carefully assessed and shared with the patient.55,56
Risk estimation and screening
Atherosclerotic CVD is common in the general population and in CML patients.57 Risk models identify patients who are likely to develop CVD within a defined period. For example, 10-year risk for CVD in the Framingham model8 in the general population. A number of multivariate risk models (Table 1) have been developed for estimating the risk of initial CVEs in healthy, asymptomatic individuals. It is important to note that some of these risk models have not measured important CVD outcomes such as stroke, heart failure, or development of symptomatic PAD. Regarding PAD, only a few of these risk models included intermittent claudication8 or CVD death due to PAD58 as their end points (Table 1).
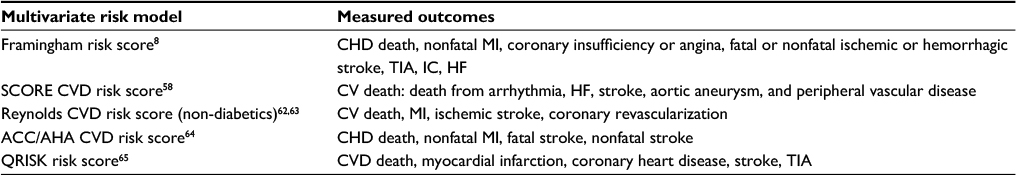 | Table 1 Multivariate risk models, estimation of cardiovascular risk in an individual patient without known CVD Abbreviations: CVD, cardiovascular disease; CHD, coronary heart disease; MI, myocardial infarction; TIA, transient ischemic attack; IC, intermittent claudication; HF, heart failure; CV, cardiovascular; ACC/AHA, American College of Cardiology/American Heart Association. |
Recently, these risk models have been used to predict CVEs in CML patients receiving nilotinib.59,60 It is important to note that the cardiovascular end points selected for these studies were quite different from what the SCORE multivariable risk model58 could predict. SCORE CVD risk estimator essentially estimates risk of cardiovascular death due to CVD, and risk of non-fatal events cannot be predicted by this model. As a result, these risk models have not been properly tested in CML patients.
It is recommended to screen for symptomatic CAD, PAD, or CHF before starting patients on TKIs.13 If there is no history of CVD, then application of the risk estimation tool to CML patients without CVD could help oncologists to risk stratify patients before starting TKI (Figure 1). Selecting the most appropriate risk estimation model depends on the population under the study and the anticipated outcome of interest (Table 1). Global risk of future CVEs then should be measured for every patient, and modifiable risk factors need to be treated aggressively based on the guidelines. Patients at increased risk of PAD based on known cardiovascular risk factors should undergo a comprehensive assessment and a review of symptoms to assess for exertional leg symptoms, including claudication or other walking impairment, ischemic rest pain, and non-healing wounds.61 For patients who will receive a TKI that is known to increase risk of CVEs and especially PAD, routine ABI can be considered before starting TKI. The new guideline on the management of PAD61 in general population recommends (class IIa) routine ABI for patients at increased risk of PAD including the following: 1-Age ≥65 years, 2-Age 50–64 years with risk factors for atherosclerosis, and 3-Age <50 years with diabetes mellitus and 1 additional risk factor, or 4-Patients with atherosclerotic disease in another vascular bed. These recommendations can also be applied to CML patients for a comprehensive cardiovascular risk assessment before starting TKI.
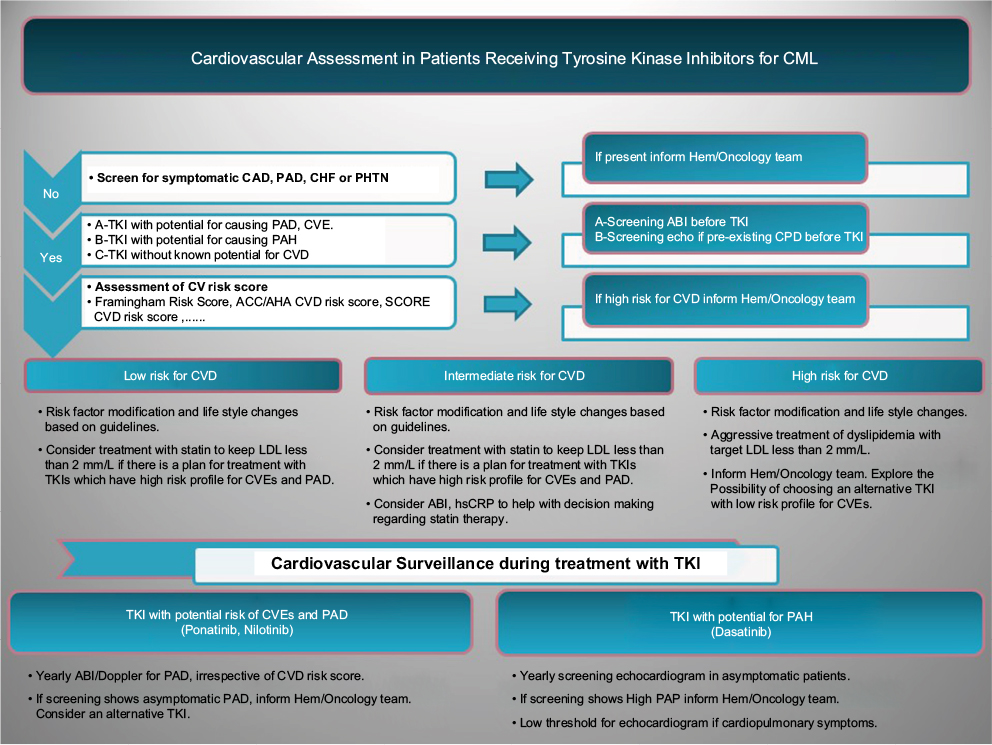 | Figure 1 Proposed cardiovascular assessment and surveillance in patients receiving TKIs for CML. Abbreviations: TKIs, tyrosine kinase inhibitors; CML, chronic myeloid leukemia; CAD, coronary artery disease; PAD, peripheral arterial disease; CHF, congestive heart failure; PHTN, pulmonary hypertension; CVE, cardiovascular event; PAH, pulmonary artery hypertension; CV, cardiovascular; CVD, cardiovascular disease; Hem, hematology; ABI, ankle–brachial index; CPD, cardiopulmonary disease; ACC/AHA, American College of Cardiology/American Heart Association; LDL, low-density lipoprotein; hsCRP, high-sensitivity C-reactive protein. |
Currently, there is no evidence-based recommendation for screening patients who will receive dasatinib. Obtaining a detailed past medical history and a careful clinical assessment will help identify patients who might have high pulmonary artery pressure (PAP) at baseline, for example, patients with a history of lung disease or thromboembolic pulmonary disease, connective tissue disorders, and congenital heart disease (Figure 1). Echocardiogram with Doppler studies would provide non-invasive assessment and estimation of PAP before starting dasatinib in CML patients. Based on the expert opinion, routine echocardiography is not recommended but low threshold for performing echocardiogram in patients with cardiopulmonary symptoms who will be started on dasatinib is warranted.5
It is reasonable to perform an ECG before initiating TKIs as all may be associated with QT prolongation. Electrolyte abnormalities should be corrected and concomitant administration of drugs that prolong QT should be avoided.
Preventive strategies
The key element in the prevention of CVD in the general population is management of modifiable cardiovascular risk factors. CML patients have high prevalence of both pre-existing cardiovascular risk factors and diseases.57 Among people with CML, however, total cardiovascular risk cannot be solely attributed to cardiovascular risk factors as treatment with TKIs might also increase the chance of CVEs.
BCR-ABL kinase inhibitors also inhibit kinases other than the target that can result in off-target toxicities. Off-target toxicities are generally due to inhibition of other unintended targets. Cardiovascular toxicity can be a combination of on-target and an off-target side effect of TKIs,66 for which every patient treated with these TKIs should be at risk. However, there is variability in cardiovascular toxicity in different patients while receiving the same TKI. This suggests that cardiovascular toxicity of TKIs might be as a result of interactions among cardiovascular risk factors, genetic predisposition of individuals, and certain targets that each TKI affects.67
Currently, the prevention of CVD is generally similar in patients with or without CML. The increased risk in CML patients, which results from multiple interacting factors, including cardiac safety profile of TKIs emphasizes the need for developing an evidence-based primary prevention guideline in this population. Application of the multivariate risk model in CML patients at baseline can recognize future risk of CVEs and help aggressively manage modifiable cardiovascular risk factors. Both ACC/AHA expert committee report64 and Canadian guidelines for the management of dyslipidemia68 recommend that quantitative risk assessment should occur first in the general population, and if a risk-based decision is uncertain for the treatment of dyslipidemia, then assessment of hsCRP, coronary artery calcium score, or ABI may be considered to help with decision making. These recommendations have not been validated in CML population but can help with aggressive management of modifiable risk factors. When using these risk models, care should be taken to consider that “low risk “does not mean “no risk” and risk factor modification and life style changes need to be provided to all patients (Figure 1). There is growing evidence that using measures such as ABI,69 calcium score,70 or hsCRP10 will identify patients who would benefit from lipid lowering but still based on current multivariable risk estimators are not candidate for such treatment.68
Certain TKIs such as ponatinib or nilotinib may increase risk of CVEs in CML patients. In spite of recognizing exposure to these medications as an emerging possible risk for development of CVD, there is not enough evidence to recommend lipid lowering as a primary prevention in these patients with lipid targets similar to the patients with high risk for future CVD. Modifiable risk factors such as obesity, hypertension, dyslipidemia, diabetes, and cigarette smoking should be aggressively managed in these patients. Some experts have proposed that receiving these TKIs may constitute a high risk and that patients on these medications should routinely be placed on lipid-lowering therapy. This issue remains controversial, as statins are not free of side effects, and their effect in slowing progression of atherosclerosis in patients on TKIs has not been adequately studied. Despite the lack of recognition of these TKs as a formal risk factor for CVD, lipid lowering with a statin can be an effective primary prevention strategy for patients who can tolerate the medication and are at high risk for CVEs. It is important to note that especially for patients on ponatinib, who usually have failed several TKIs and do not have many options if ponatinib has to be discontinued, the importance of primary prevention becomes more essential.
Daily low-dose aspirin has proved to be beneficial in preventing recurrent CVEs in patients with a history of CVD. The role of aspirin for primary prevention in patients with no CVD, however, is subject of ongoing debate. Meta-analyses of aspirin primary prevention trials reported a 10%–13% relative reduction in combined serious CVEs, but there was increased risk of intracranial and gastrointestinal bleeding. Assessing the net benefit of aspirin use in primary prevention is challenging because of the difficulty in balancing the consequences of ischemic and bleeding events.71 In the CML population, use of aspirin for primary prevention is more challenging due to the increased risk of bleeding that may be associated with certain TKIs such as dasatinib which inhibits platelet aggregation.72
Surveillance and therapeutic strategies
The exact incidence of CVEs in long-term TKI-treated patients with CML and risk factors predisposing to its development is unknown. At present, it is not possible to differentiate between the CVEs that are caused by the drug from the one that would happen as a result of patients’ risk factors. The exact pathophysiology of the vascular events also remains unclear.7
The potential for cardiac biomarkers such as B-type natriuretic peptide and troponin to detect cardiotoxicity has been explored, but it is currently unclear whether these biomarkers will be able to identify patients who are at risk of CVEs while receiving BCR-ABL TKI or provide prognostic information.73 Dyspnea, fatigue, peripheral edema, and pleural effusion can be side effects of treatment with TKI rather than heart failure. For example, pleural effusion is a common side effect of dasatinib, and fluid retention can be due to imatinib. Given the shared symptoms and signs in both CML patients who are receiving TKI and patients who suffer from CHF, it is likely that B-type natriuretic peptide and NT-proBNP might be of clinical value in screening and excluding heart failure as a cause for these symptoms and signs. Further studies in the CML population are needed.
Surveillance ABI for patients who are receiving ponatinib or nilotinib have been recommended by experts but evidence is lacking for this recommendation. European LeukemiaNet recommends surveillance ABI (or duplex ultrasonography) every 6–12 months in patients on ponatinib or nilotinib.74 Surveillance echocardiography has been recommended for patients who are on dasatinib to monitor PAP. The exact interval for performing a surveillance echo is unknown, and currently there are no evidence-based recommendations. European Society of Cardiology (ESC) recently recommended echocardiography every 3 months while patients are receiving dasatinib; however, there are no data to support an optimal time interval between the echocardiograms.75
Current practice in those patients who experience CVEs while receiving TKI consists of reassessment of treatment plan that includes interruption, dose reduction, and alternative TKI. In the event of CVEs, patients might not be able to receive the same TKI that controlled their leukemia and the treatment might become compromised. Early recognition of AEs and appropriate intervention can help more patients benefit from long-term treatment with TKIs. This magnifies the importance of primary prevention in current practice, which will help with the prevention of CVD and the chance to continue with the current cancer treatment.
Treatment strategies are not different from general population, except for consideration of drug interaction between TKIs and certain cardiology drugs. BCR-ABL TKIs are inhibitors of CYP3A4 and can increase plasma levels of medications metabolized by this enzyme, for example, diltiazem, verapamil, simvastatin, and atorvastatin. The same drugs mentioned can themselves increase plasma levels of imatinib, dasatinib, and nilotinib by inhibition of P-glycoprotein (Pgp).76,77 Pravastatin, rosuvastatin, atenolol, ramipril, candesartan, furosemide, and hydrochlorothiazide have no interactions with imatinib, dasatinib, and nilotinib and can be used safely with these TKIs.77
Dasatinib has been described as a drug with a potential to cause PAH.46 PAH is an uncommon disease due to progressive remodeling of the distal pulmonary arteries. Pulmonary hypertension, however, is common and may result from several different disorders such as left heart disease, pulmonary disease, and chronic thromboembolic diseases. PAH is characterized by increase in pulmonary vascular resistance and increased PAP, which can eventually cause right ventricular failure.78 Dasatinib can cause precapillary PAH (Group 1) that is due to a primary elevation of pressure in the pulmonary arterial system.79 Patients suspected of having PAH due to dasatinib should be referred to a cardiologist for comprehensive evaluation. This evaluation is aimed at confirming the presence of PAH and identifying and excluding other potential causes for increased PAP. Investigation can be started by performing an echocardiogram to assess PAP. Although PAP can be estimated on echocardiography, the definitive diagnosis of PAH requires RHC. PAH is defined as an increase in mean PAP ≥25 mmHg at rest and pulmonary capillary wedge pressure (PCWP) <15 mmHg, assessed by RHC.80 Early diagnosis of PAH is challenging but is essential in patients on dasatinib since improvement in PAP is usually observed after withdrawal of dasatinib.46 If dasatinib-induced PAH is confirmed by RHC, dasatinib should be discontinued.79 Given the fact that novel treatment options currently exist for PAH, collaborative care and involving pulmonary hypertension team are essential to optimize the care in patients with dasatinib-induced PAH.
Management of dasatinib-induced pleural effusion includes dose interruptions, dose reductions, or permanent discontinuation. Diuretics and steroids are also used in some cases.81 The majority of patients who developed PAH in dasatinib studies had pleural effusion at the time of diagnosis.79 Therefore, we would suggest low threshold for performing an echocardiogram to assess PAP in patients who develop pleural effusion.
Based on available data, there have been several expert recommendations on cardiovascular surveillance in patients receiving treatment with TKIs in CML population.13,56,82 The basis of all these recommendations are aggressive management of modifiable risk factors and cardiovascular surveillance during the treatment based on specific risk profile of each TKI. Clinicians are advised to collect information about the presence of cardiovascular risk factors, past cardiovascular history and check about medications that might have interactions with TKIs. Drugs that do not potentially worsen underlying conditions should be chosen, everything else being equal. However, if it is necessary to choose one with added risk, management of co-morbidities may reduce the chances of AEs.
Conclusion
The cumulative cardiovascular effects of TKIs and cardiovascular risk factors seem to drive the CVEs in CML population. Assessment of the cardiovascular safety profile of different TKIs is an essential task before starting treatment. Understanding the mechanisms of cardiovascular toxicities will be critical for developing future evidence-based preventive strategies. More efforts also should be taken to standardize cardiovascular outcome measurements in oncology trials. Given the excess risk of CVD in patients with CML and challenges that these patients face in cancer treatment after cardiovascular event, careful cardiovascular risk stratification and surveillance should be an essential part of management in CML patients.
Acknowledgment
Work for this project is supported by our patients through the Friends for Life Fund, Princess Margaret Hospital Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
Epstein FH, Faderl S, Talpaz M, et al. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341(3):164–172. | ||
Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037. | ||
Gambacorti-Passerini C, Antolini L, Mahon F-X, et al. Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib. J Natl Cancer Inst. 2011;103(7):553–561. | ||
Bower H, Bjorkholm M, Dickman PW, Hoglund M, Lambert PC, Andersson TM-L. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J Clin Oncol. 2016;34(24):2851–2857. | ||
Moslehi JJ, Deininger M. Tyrosine kinase inhibitor-associated cardiovascular toxicity in chronic myeloid leukemia. J Clin Oncol. 2015;33(35):4210–4218. | ||
Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;3532353:172–187. | ||
Bellinger AM, Arteaga CL, Force T, et al. Cardio-oncology. Circulation. 2015;132(23):2248–2258. | ||
D’Agostino RB, Vasan RS, Pencina MJ, et al. General cardiovascular risk profile for use in primary care: the Framingham heart study. Circulation. 2008;117(6):743–753. | ||
Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364(9438):937–952. | ||
Kaptoge S, Di Angelantonio E, Pennells L, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367(14):1310–1320. | ||
Kim TD, Rea D, Schwarz M, et al. Peripheral artery occlusive disease in chronic phase chronic myeloid leukemia patients treated with nilotinib or imatinib. Leukemia. 2013;27(6):1316–1321. | ||
Cortes JE, Kim D-W, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome–positive leukemias. N Engl J Med. 2013;369(19):1783–1796. | ||
Li W, Croce K, Steensma DP, McDermott DF, Ben-Yehuda O, Moslehi J. Vascular and metabolic implications of novel targeted cancer therapies. J Am Coll Cardiol. 2015;66(10):1160–1178. | ||
Hochhaus A, Saglio G, Hughes TP, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30(5):1044–1054. | ||
Steg PG, Bhatt DL, Wilson PWF, et al. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA. 2007;297(11):1197. | ||
Hicks KA, Tcheng JE, Bozkurt B, et al. 2014 ACC/AHA key data elements and definitions for cardiovascular endpoint events in clinical trials. J Am Coll Cardiol. 2015;66(4):403–469. | ||
Wood AJJ, Savage DG, Antman KH. Imatinib mesylate — a new oral targeted therapy. N Engl J Med. 2002;346(9):683–693. | ||
Saglio G, Kim D-W, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362(24):2251–2259. | ||
Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362(24):2260–2270. | ||
Rix U, Hantschel O, Dürnberger G, et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood. 2007;110(12):4055–4063. | ||
Buchdunger E, Zimmermann J, Mett H, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56(1):100–104. | ||
Kerkelä R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006;12(8):908–916. | ||
Hochhaus A, Larson RA, Guilhot F, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376(10):917–927. | ||
Hatfield A, Owen S, Pilot PR. In reply to “Cardiotoxicity of the cancer therapeutic agent imatinib mesylate”. Nat Med. 2007;13(1):13–13. | ||
Ribeiro AL, Marcolino MS, Bittencourt HNS, et al. An evaluation of the cardiotoxicity of imatinib mesylate. Leuk Res. 2008;32(12):1809–1814. | ||
Estabragh ZR, Knight K, Watmough SJ, et al. A prospective evaluation of cardiac function in patients with chronic myeloid leukaemia treated with imatinib. Leuk Res. 2011;35(1):49–51. | ||
Agostino NM, Chinchilli VM, Lynch CJ, et al. Effect of the tyrosine kinase inhibitors (sunitinib, sorafenib, dasatinib, and imatinib) on blood glucose levels in diabetic and nondiabetic patients in general clinical practice. J Oncol Pharm Pract. 2011;17(3):197–202. | ||
Lassila M, Allen TJ, Cao Z, et al. Imatinib attenuates diabetes-associated atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24(5):935–942. | ||
Shah AM, Campbell P, Rocha GQ, et al. Effect of imatinib as add-on therapy on echocardiographic measures of right ventricular function in patients with significant pulmonary arterial hypertension. Eur Heart J. 2015;36(10):623–632. | ||
Masuda S, Nakano K, Funakoshi K, et al. Imatinib mesylate-incorporated nanoparticle-eluting stent attenuates in-stent neointimal formation in porcine coronary arteries. J Atheroscler Thromb. 2011;18(12):1043–1053. | ||
Giles FJ, Mauro MJ, Hong F, et al. Rates of peripheral arterial occlusive disease in patients with chronic myeloid leukemia in the chronic phase treated with imatinib, nilotinib, or non-tyrosine kinase therapy: a retrospective cohort analysis. Leukemia. 2013;27(6):1310–1315. | ||
Deininger M, O’Brien SG, Guilhot F, et al. International randomized study of interferon vs STI571 (IRIS) 8-year follow-up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood. 2009;114(22):1–1. | ||
Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7(4):399. | ||
Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–2551. | ||
Aichberger KJ, Herndlhofer S, Schernthaner G-H, et al. Progressive peripheral arterial occlusive disease and other vascular events during nilotinib therapy in CML. Am J Hematol. 2011;86(7):533–539. | ||
Le Coutre P, Rea D, Abruzzese E, et al. Severe peripheral arterial disease during nilotinib therapy. J Natl Cancer Inst. 2011;103(17):1347–1348. | ||
Poredos P, Jug B. The prevalence of peripheral arterial disease in high risk subjects and coronary or cerebrovascular patients. Angiology. 2007;58(3):309–315. | ||
Douxfils J, Haguet H, Mullier F, Chatelain C, Graux C, Dogné J-M. Association between BCR-ABL tyrosine kinase inhibitors for chronic myeloid leukemia and cardiovascular events, major molecular response, and overall survival. JAMA Oncol. 2016;2(5):625. | ||
Chai-Adisaksopha C, Lam W, Hillis C. Major arterial events in patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors: a meta-analysis. Leuk Lymphoma. 2016;57(6):1300–1310. | ||
Racil Z, Razga F, Drapalova J, et al. Mechanism of impaired glucose metabolism during nilotinib therapy in patients with chronic myelogenous leukemia. Haematologica. 2013;98(10):e124–e126. | ||
Rea D, Mirault T, Cluzeau T, et al. Early onset hypercholesterolemia induced by the 2nd-generation tyrosine kinase inhibitor nilotinib in patients with chronic phase-chronic myeloid leukemia. Haematologica. 2014;99(7):1197–1203. | ||
Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305(5682):399–401. | ||
Lombardo LJ, Lee FY, Chen P, et al. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47(27):6658–6661. | ||
Cortes JE, Saglio G, Kantarjian HM, et al. Final 5-year study results of DASISION: the dasatinib versus imatinib study in treatment-naïve chronic myeloid leukemia patients trial. J Clin Oncol. 2016;34(20):2333–2340. | ||
Quintas-Cardama A, Kantarjian H, O’Brien S, et al. Pleural effusion in patients with chronic myelogenous leukemia treated with dasatinib after imatinib failure. J Clin Oncol. 2007;25(25):3908–3914. | ||
Montani D, Bergot E, Günther S, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125(17):2128–2137. | ||
Shah NP, Rousselot P, Schiffer C, et al. Dasatinib in imatinib-resistant or -intolerant chronic-phase, chronic myeloid leukemia patients: 7-year follow-up of study CA180-034. Am J Hematol. 2016;91(9):869–874. | ||
Remsing Rix LL, Rix U, Colinge J, et al. Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid leukemia cells. Leukemia. 2009;23(3):477–485. | ||
Khoury HJ, Cortes JE, Kantarjian HM, et al. Bosutinib is active in chronic phase chronic myeloid leukemia after imatinib and dasatinib and/or nilotinib therapy failure. Blood. 2012;119(15):3403–3412. | ||
Cortes JE, Jean Khoury H, Kantarjian H, et al. Long-term evaluation of cardiac and vascular toxicity in patients with Philadelphia chromosome-positive leukemias treated with bosutinib. Am J Hematol. 2016;91(6):606–616. | ||
Cortes JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367(22):2075–2088. | ||
Lipton JH, Chuah C, Guerci-Bresler A, et al. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17(5):612–621. | ||
Cortes JE, Kantarjian HM, Rea D, et al. Final analysis of the efficacy and safety of omacetaxine mepesuccinate in patients with chronic- or accelerated-phase chronic myeloid leukemia: results with 24 months of follow-up. Cancer. 2015;121(10):1637–1644. | ||
Dorer DJ, Knickerbocker RK, Baccarani M, et al. Impact of dose intensity of ponatinib on selected adverse events: multivariate analyses from a pooled population of clinical trial patients. Leuk Res. 2016;48:84–91. | ||
Poch Martell M, Sibai H, Deotare U, Lipton JH. Ponatinib in the therapy of chronic myeloid leukemia. Expert Rev Hematol. 2016;9(10):923–932. | ||
Breccia M, Pregno P, Spallarossa P, et al. Identification, prevention and management of cardiovascular risk in chronic myeloid leukaemia patients candidate to ponatinib: an expert opinion. Ann Hematol. 2017;96(4):549–558. | ||
Hoffmann VS, Baccarani M, Hasford J, et al. The EUTOS population-based registry: incidence and clinical characteristics of 2904 CML patients in 20 European Countries. Leukemia. 2015;29(6):1336–1343. | ||
Conroy RM, Pyörälä K, Fitzgerald AP, et al. Estimation of ten-year risk of fatal cardiovascular disease in Europe: the SCORE project. Eur Heart J. 2003;24(11):987–1003. | ||
Rea D, Mirault T, Raffoux E, et al. Usefulness of the 2012 European CVD risk assessment model to identify patients at high risk of cardiovascular events during nilotinib therapy in chronic myeloid leukemia. Leukemia. 2015;29(5):1206–1209. | ||
Breccia M, Molica M, Zacheo I, Serrao A, Alimena G. Application of systematic coronary risk evaluation chart to identify chronic myeloid leukemia patients at risk of cardiovascular diseases during nilotinib treatment. Ann Hematol. 2015;94(3):393–397. | ||
Gerhard-Herman MD, Gornik HL, Barrett C, et al. 2016 AHA/ACC guideline on the management of patients with lower extremity peripheral artery disease: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. J Am Coll Cardiol. 2017;69(11):e71–e126. | ||
Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: the Reynolds Risk Score. JAMA. 2007;297(6):611–619. | ||
Ridker PM, Paynter NP, Rifai N, Gaziano JM, Cook NR. C-reactive protein and parental history improve global cardiovascular risk prediction: the Reynolds Risk Score for men. Circulation. 2008;118(22):2243–51, 4p following 2251. | ||
Goff DC, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk. Circulation. 2014;129(25 suppl 2):S49–S73. | ||
Hippisley-Cox J, Coupland C, Vinogradova Y, Robson J, May M, Brindle P. Derivation and validation of QRISK, a new cardiovascular disease risk score for the United Kingdom: prospective open cohort study. BMJ. 2007;335(7611):136. | ||
Force T, Kolaja KL. Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes. Nat Rev Drug Discov. 2011;10(2):111–126. | ||
Brown S-A, Nhola L, Herrmann J. Cardiovascular toxicities of small molecule tyrosine kinase inhibitors: an opportunity for systems-based approaches. Clin Pharmacol Ther. 2017;101(1):65–80. | ||
Anderson TJ, Gr J, Pearson GJ, et al. 2016 Canadian Cardiovascular Society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in the adult. Can J Cardiol. 2016;32(11):1263–1282. | ||
Ankle Brachial Index Collaboration, Fowkes FGR, Murray GD, et al. Ankle brachial index combined with Framingham risk score to predict cardiovascular events and mortality. JAMA. 2008;300(2):197. | ||
Polonsky TS, McClelland RL, Jorgensen NW, et al. Coronary artery calcium score and risk classification for coronary heart disease prediction. JAMA. 2010;303(16):1610–1616. | ||
Capodanno D, Angiolillo DJ. Aspirin for primary cardiovascular risk prevention and beyond in diabetes mellitus. Circulation. 2016;134(20):1579–1594. | ||
Quintás-Cardama A, Kantarjian H, Ravandi F, et al. Bleeding diathesis in patients with chronic myelogenous leukemia receiving dasatinib therapy. Cancer. 2009;115(11):2482–2490. | ||
Witteles RM, California S. Biomarkers as predictors of cardiac toxicity from targeted cancer therapies. J Card Fail. 2016;22(6):459–464. | ||
Steegmann JL, Baccarani M, Breccia M, et al. European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronic myeloid leukaemia. Leukemia. 2016;30(8):1648–1671. | ||
Zamorano JL, Lancellotti P, Rodriguez Muñoz D, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines. Eur Heart J. 2016;37(36):2768–2801. | ||
van Leeuwen RWF, van Gelder T, Mathijssen RHJ, Jansman FGA. Drug–drug interactions with tyrosine-kinase inhibitors: a clinical perspective. Lancet Oncol. 2014;15(8):e315–e326. | ||
Haouala A, Widmer N, Duchosal MA, Montemurro M, Buclin T, Decosterd LA. Drug interactions with the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib. Blood. 2011;117(8):e75–e87. | ||
McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol. 2015;65(18):1976–1997. | ||
Shah NP, Wallis N, Farber HW, et al. Clinical features of pulmonary arterial hypertension in patients receiving dasatinib. Am J Hematol. 2015;90(11):1060–1064. | ||
Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25):D42–D50. | ||
Cortes JE, Jimenez CA, Mauro MJ, Geyer A, Pinilla-Ibarz J, Smith BD. Pleural effusion in dasatinib-treated patients with chronic myeloid leukemia in chronic phase: identification and management. Clin Lymphoma Myeloma Leuk. 2017;17(2):78–82. | ||
Breccia M, Arboscello E, Bellodi A, et al. Proposal for a tailored stratification at baseline and monitoring of cardiovascular effects during follow-up in chronic phase chronic myeloid leukemia patients treated with nilotinib frontline. Crit Rev Oncol Hematol. 2016;107:190–198. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.