Back to Journals » Risk Management and Healthcare Policy » Volume 19
Cardiovascular Mortality in Amyloidosis: Long-Term Trends, Disparities, and Projections in the United States
Authors Chen Z, Tian Y, Zhang Y ![]()
Received 7 March 2026
Accepted for publication 28 June 2026
Published 9 July 2026 Volume 2026:19 607682
DOI https://doi.org/10.2147/RMHP.S607682
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Gulsum Kaya
Zhan Chen,1 Ying Tian,2 Yue Zhang3
1Department of Vascular Surgery, Beijing Haidian Hospital, Beijing, People’s Republic of China; 2Department of Hematology, Beijing Chao-Yang Hospital, Capital Medical University, Beijing, People’s Republic of China; 3Department of Scientific Research, Beijing Haidian Hospital, Beijing, People’s Republic of China
Correspondence: Yue Zhang, Email [email protected] Ying Tian, Email [email protected]
Background: Cardiac involvement is a major determinant of prognosis in systemic amyloidosis, but population-level trends in cardiovascular mortality remain insufficiently characterized.
Methods: We performed a retrospective, population-based analysis of U.S. adults aged ≥ 45 years with amyloidosis using death certificate data from the Centers for Disease Control and Prevention Wide-ranging Online Data for Epidemiologic Research (CDC WONDER) database from 1999 through 2023. Cardiovascular disease (CVD)–related deaths were identified using ICD-10 codes and classified into heart failure (HF), atrial fibrillation/flutter (AF), ischemic heart disease (IHD), or other CVD categories. Age-adjusted mortality rates (AAMRs) were calculated using the 2000 U.S. standard population. Joinpoint regression assessed temporal trends, and autoregressive integrated moving average (ARIMA) models projected mortality through 2033.
Results: A total of 35,222 CVD-related deaths occurred among adults with amyloidosis during the study period, yielding an overall AAMR of 1.12 per 100,000 population. Cardiovascular mortality increased nearly threefold, from 0.80 per 100,000 in 1999 to 2.27 per 100,000 in 2023, with a marked acceleration beginning in the late 2010s. HF consistently accounted for the largest proportion of CVD-related deaths. Mortality rates were higher among males, older adults, and non-Hispanic Black individuals, with demographic and geographic disparities widening over time. Similar upward trajectories were observed across U.S. Census regions and levels of urbanization. Projections indicated continued increases in cardiovascular mortality through 2033.
Conclusion: Cardiovascular mortality among U.S. adults with amyloidosis has increased substantially over the past 25 years, with recent acceleration and persistent population-level disparities. These findings highlight a growing public health burden and support earlier detection, equitable access to specialized care, and continued national surveillance.
Keywords: amyloidosis, cardiac amyloidosis, cardiovascular mortality, population-based study, health disparities, CDC WONDER
Background
Amyloidosis comprises a heterogeneous group of protein misfolding disorders characterized by extracellular deposition of insoluble fibrils in multiple organs, resulting in progressive organ dysfunction and excess mortality.1,2 Cardiac involvement represents the most consequential clinical manifestation of systemic amyloidosis and is the dominant determinant of prognosis.3,4 Once myocardial infiltration occurs, patients face a substantially elevated risk of heart failure (HF), atrial arrhythmias, ischemic complications, and premature cardiovascular death.5
Importantly, amyloidosis subtypes differ substantially in epidemiology, clinical trajectory, and prognosis. Light-chain amyloidosis (AL amyloidosis) is driven by an underlying plasma cell dyscrasia and may progress rapidly, particularly when cardiac involvement is present, whereas transthyretin amyloidosis (ATTR amyloidosis) is more strongly age-dependent and includes both wild-type and hereditary forms with distinct demographic and genetic distributions. These subtype-specific differences may influence population-level mortality trends, because death certificate data do not reliably distinguish AL amyloidosis from ATTR amyloidosis. Recent meta-analytic evidence further supports the prognostic importance of cardiac involvement. In patients with multiple myeloma, concomitant cardiac AL amyloidosis has been associated with markedly higher all-cause mortality and substantially shorter survival.6 In addition, systematic evidence in ATTR amyloidosis has demonstrated important subtype-specific differences in prevalence and clinical outcomes.7 These findings reinforce the biological plausibility and clinical relevance of focusing on cardiovascular mortality in amyloidosis.
During the past two decades, the clinical landscape of amyloidosis has evolved considerably. Advances in noninvasive diagnostic techniques and increasing clinical awareness have led to more frequent recognition of cardiac involvement, particularly transthyretin amyloid cardiomyopathy among older adults.8–10 In parallel, the emergence of disease-modifying therapies has reshaped expectations regarding the natural history of amyloidosis, raising the possibility that earlier diagnosis and treatment could ultimately translate into improved cardiovascular outcomes at the population level.11–14 Whether these advances have meaningfully altered long-term cardiovascular mortality patterns, however, remains unclear.
Current epidemiologic evidence addressing this question is limited. Prior investigations have primarily focused on selected clinical cohorts, referral-based registries, or hospitalization data, often emphasizing overall survival or HF-related outcomes.15,16 Although these studies have established the high cardiovascular risk associated with amyloidosis, they provide limited insight into national long-term mortality trends, temporal inflection points, and the evolving contribution of specific cardiovascular causes of death, such as HF, atrial fibrillation (AF), and ischemic heart disease (IHD). Moreover, population-level evaluations of demographic and geographic disparities in cardiovascular mortality among individuals with amyloidosis remain sparse. Social determinants of health and therapeutic inequities may further shape the contemporary burden of cardiovascular mortality in amyloidosis. Delayed diagnosis, limited access to advanced cardiac imaging, uneven availability of amyloidosis specialty centers, differences in insurance coverage, and barriers to disease-modifying therapies may contribute to disparities in detection, treatment initiation, and outcomes. These factors are particularly relevant because recent diagnostic and therapeutic advances may not translate into uniform population-level benefit if access remains unequal across racial, socioeconomic, geographic, and urban–rural groups. Evaluating mortality patterns across demographic and geographic strata may therefore help identify populations in which earlier detection, equitable care delivery, and ongoing surveillance are most urgently needed.
National mortality surveillance data provide a unique opportunity to address these gaps. The Centers for Disease Control and Prevention Wide-ranging Online Data for Epidemiologic Research (CDC WONDER) database captures all U.S. death certificates over multiple decades, enabling systematic assessment of cause-specific mortality across time, demographic groups, and geographic regions.17 This approach allows for characterization of long-term trends and disparities in cardiovascular mortality that cannot be adequately assessed using clinical or administrative datasets alone.
Accordingly, we conducted a population-based analysis of U.S. adults aged 45 years and older with amyloidosis using CDC WONDER data from 1999 through 2023. The objectives of this study were to characterize long-term trends in cardiovascular mortality within a rare but increasingly prevalent cardiomyopathic condition, identify temporal changes in the pace of mortality increase, examine disparities by age, sex, race and ethnicity, geographic region, and urbanization level, and delineate the evolving distribution of cardiovascular causes of death, including HF, AF, and IHD. In addition, we applied time-series modeling to project future cardiovascular mortality patterns. By providing a comprehensive national perspective spanning 25 years, this study aims to clarify the contemporary burden of cardiovascular mortality in amyloidosis and to inform strategies for earlier detection, equitable care delivery, and ongoing population-level surveillance.
Methods
Study Design and Data Source
We conducted a retrospective, population-based analysis using data from the CDC WONDER Multiple Cause-of-Death database, covering the period from January 1, 1999, through December 31, 2023. This publicly accessible database includes all death certificates from the 50 U.S. states and the District of Columbia, with causes of death coded according to the International Classification of Diseases, Tenth Revision (ICD-10). Each record contains the underlying cause of death, up to 20 contributing causes, and detailed demographic and geographic information. Annual population denominators were obtained from U.S. Census intercensal and postcensal population estimates.18
Study Population
We included all decedents aged ≥45 years with any diagnosis of amyloidosis (ICD-10 codes E85.0–E85.9) listed as either an underlying or contributing cause of death. The age threshold was selected to focus on adults at higher risk for cardiac involvement in systemic amyloidosis while ensuring adequate sample sizes for stratified analyses. Within this amyloidosis cohort, cardiovascular disease (CVD)–related deaths were identified when any CVD-related ICD-10 code appeared anywhere on the death certificate, either as the underlying cause of death or as a contributing cause of death. This definition was intended to capture deaths with documented cardiovascular involvement among decedents with amyloidosis, rather than deaths for which CVD was necessarily the sole or principal cause. CVD subtypes included HF (I50), AF (I48), IHD (I20–I25), and other CVD (remaining codes within I00–I99, excluding HF, AF, and IHD). Subtype-specific analyses for light-chain amyloidosis and transthyretin amyloidosis were not performed because amyloidosis subtypes are not consistently or reliably captured in death certificate data, and the available ICD-10 amyloidosis codes do not allow definitive separation of AL amyloidosis from ATTR amyloidosis at the population level. Strata with suppressed case counts (<10 deaths), as designated by CDC WONDER, were excluded from stratified analyses.
Demographic and Geographic Variables
Race and ethnicity were categorized according to U.S. Office of Management and Budget standards as non-Hispanic (NH) White, NH Black or African American, Hispanic/Latino, and NH Other (including American Indian/Alaska Native and Asian/Pacific Islander).19 Geographic variables included U.S. Census region (Northeast, Midwest, South, West) and urbanization level based on the 2013 National Center for Health Statistics (NCHS) Urban–Rural Classification Scheme (large metropolitan, medium/small metropolitan, nonmetropolitan/rural).20 State-year strata with suppressed death counts were excluded from corresponding rate calculations.
Classification of Cardiovascular Mortality
Because multiple cardiovascular diagnoses may be listed on a single death certificate, we applied a mutually exclusive hierarchical classification algorithm to assign each amyloidosis-related CVD death to a single primary subtype. Based on clinical relevance in amyloid cardiomyopathy, the hierarchy prioritized HF, followed by AF, IHD, and other CVD. Deaths listing HF were classified as HF regardless of additional cardiovascular codes; among remaining cases, AF was assigned next, followed by IHD and then other CVD.
This hierarchy was specified a priori to reflect the clinical phenotype of cardiac amyloidosis, in which HF commonly represents the dominant manifestation and final common pathway of advanced myocardial infiltration. AF was prioritized next because atrial arrhythmias are frequent and clinically consequential in restrictive amyloid cardiomyopathy, followed by IHD and other CVD categories. This approach ensured that subtype categories were nonoverlapping and collectively exhaustive. It was intended to create mutually exclusive categories for descriptive trend analyses, rather than to assign a definitive causal mechanism of death.
Statistical Analysis
Crude mortality rates (CMRs) and age-adjusted mortality rates (AAMRs) per 100,000 population were calculated with 95% confidence intervals (CIs) using direct standardization to the U.S. 2000 standard population. Subgroup-specific AAMRs were estimated by sex, race/ethnicity, Census region, and urbanization level, while age-specific rates were reported as crude rates.
Temporal trends in AAMRs from 1999 to 2023 were evaluated using log-linear joinpoint regression (Joinpoint Regression Program, version 5.1.0.0; National Cancer Institute). Models allowed up to four joinpoints, with statistically significant changepoints identified using Monte Carlo permutation testing (4,499 permutations; α = 0.05). Annual percent changes (APCs) were estimated for each segment, and average annual percent changes (AAPCs) were calculated as weighted averages across segments. Parallelism tests were used to compare trends across subgroups.
Predictive Time-Series Modeling
To characterize potential future patterns, autoregressive integrated moving average (ARIMA) models were fitted to annual AAMRs. Stationarity was assessed using the augmented Dickey–Fuller test, with log transformation or differencing applied as appropriate. Optimal ARIMA (p,d,q) models were selected using the corrected Akaike Information Criterion (AICc).21 Model adequacy was confirmed through residual diagnostics, including autocorrelation function plots, partial autocorrelation function plots, and the Ljung–Box test.22,23 The final model generated 10-year forecasts (2024–2033) with 95% prediction intervals. Mortality patterns during 2020–2021 were highlighted to contextualize potential disruptions related to the COVID-19 pandemic. These projections were intended to characterize potential future population-level mortality patterns rather than to generate individual-level risk predictions.
The COVID-19 pandemic period was treated as a contextual disruption rather than as a formally modeled causal intervention. Because only a limited number of post-pandemic observations were available through 2023, interrupted time-series or segmented forecasting models were not used as the primary approach to estimate pandemic-specific effects.
Statistical Software
All analyses were conducted using R version 4.4.2 (R Foundation for Statistical Computing, Vienna, Austria). Time-series analyses used the forecast and tseries packages. Temporal trend analyses were performed using the Joinpoint Regression Program (version 5.1.0.0; National Cancer Institute).24 Statistical significance was defined as two-sided P < 0.05. Subgroup analyses were exploratory and hypothesis-generating; no adjustments for multiple comparisons were applied. Estimates for smaller racial and ethnic subgroups, particularly Hispanic/Latino and NH Other populations, were interpreted cautiously because of smaller death counts and greater statistical uncertainty.
Ethical Considerations
This study was submitted to and reviewed by the Beijing Haidian Hospital Medical Ethics Committee, Beijing, China, in accordance with the requirement for local ethics review of research involving human data, and was approved by the committee (approval No. 2026–070; project No. M202667). The study used publicly available, de-identified, aggregate mortality data from the CDC WONDER Multiple Cause-of-Death database, and no identifiable individual-level information was accessed. Therefore, individual informed consent was waived/not required. All analyses complied with CDC WONDER data-use policies and followed the STROBE reporting guidelines.
Results
Overall Mortality Trends
From 1999 to 2023, a total of 35,222 CVD–related deaths occurred among U.S. adults aged ≥45 years with amyloidosis, corresponding to an overall AAMR of 1.12 per 100,000 population (95% CI: 1.06–1.18; Table 1). Over the 25-year period, CVD-related mortality increased substantially, with an AAPC of 4.26% (95% CI: 3.98–4.68%).
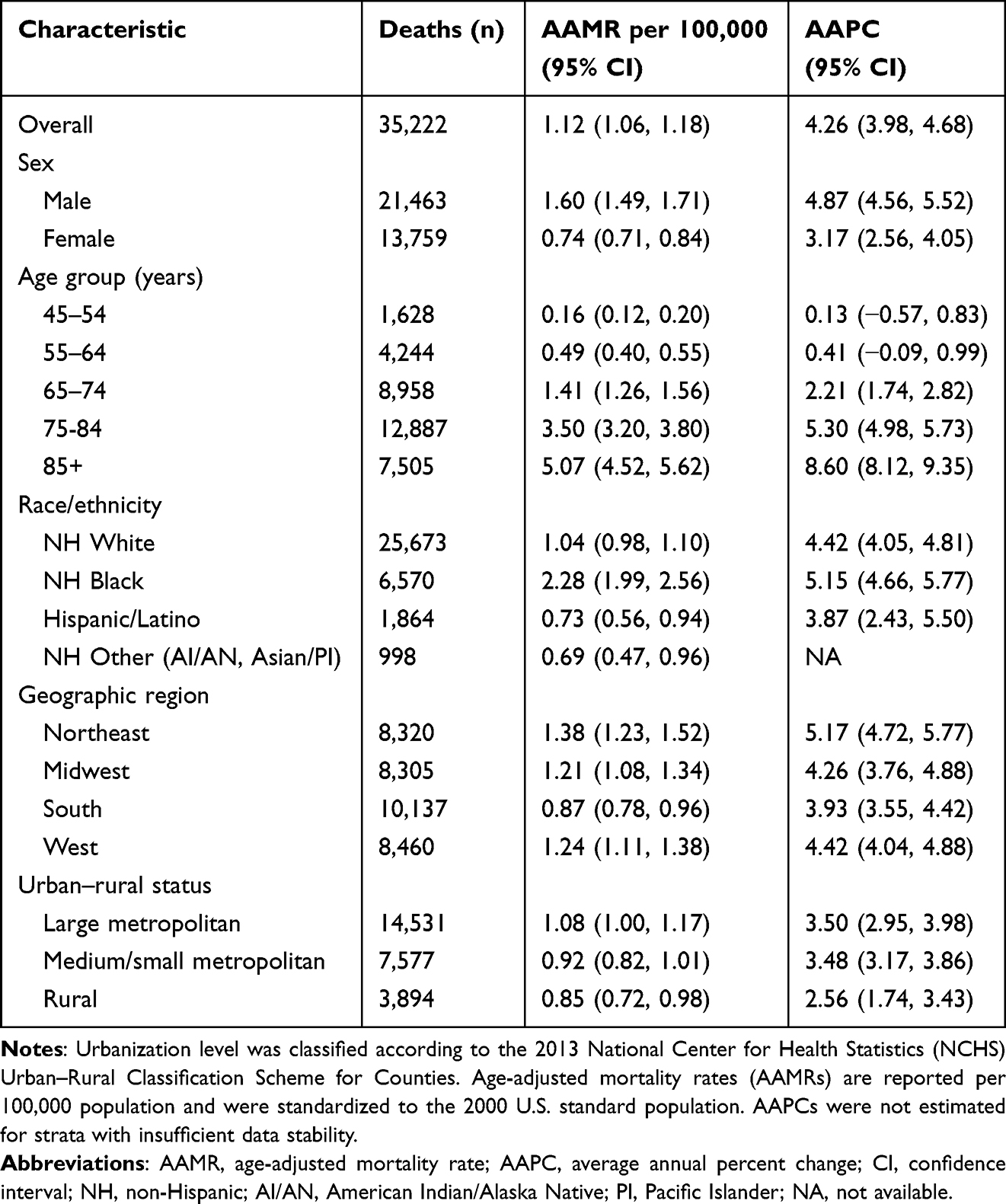 |
Table 1 Distribution and Temporal Trends in Cardiovascular Disease–Related Mortality Among Adults with Amyloidosis in the United States, 1999–2023 |
Annual AAMRs increased from 0.80 per 100,000 in 1999 to 2.27 per 100,000 in 2023, representing an approximately 2.8-fold increase (Supplemental Table 1). Joinpoint regression identified four distinct temporal phases (Figure 1 and Supplemental Table 2). Mortality rates remained stable from 1999 to 2011 (APC: 0.14%; P = 0.75), followed by a gradual increase from 2011 to 2018 (APC: 6.41%; P = 0.09), a marked acceleration from 2018 to 2021 (APC: 15.04%; P < 0.001), and a continued increase from 2021 to 2023 (APC: 6.65%; P < 0.001). A pronounced increase was observed during 2020–2021, temporally coinciding with the COVID-19 pandemic period.
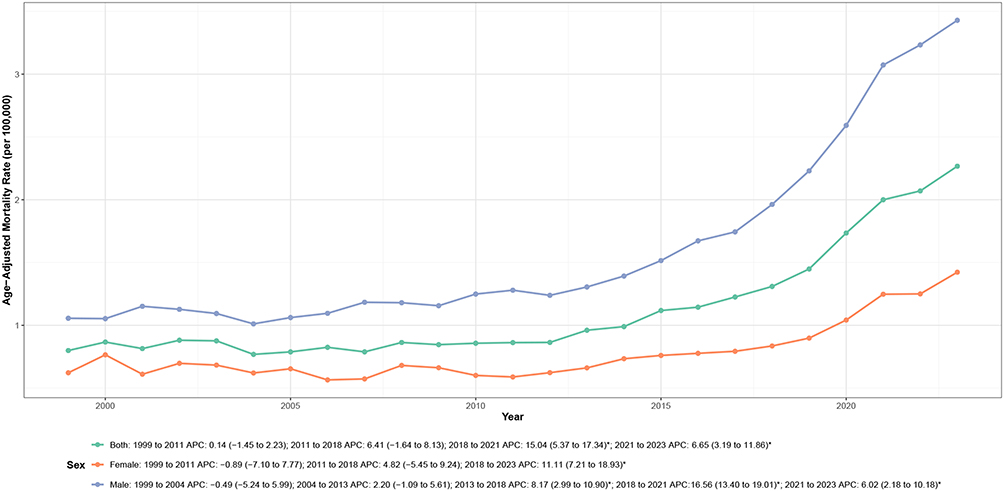 |
Figure 1 Overall and sex-specific trends in age-adjusted cardiovascular disease–related mortality among adults with amyloidosis in the United States, 1999–2023. Notes: APC indicates annual percent change. Mortality rates are expressed per 100,000 population and age-adjusted to the 2000 U.S. standard population. Asterisks (*) denote statistically significant trends (P < 0.05) identified by joinpoint regression analysis. |
Sex-Specific Mortality Trends
Throughout the study period, males consistently exhibited higher CVD-related mortality rates than females, accounting for 61.0% of all CVD-related deaths among adults aged ≥45 years with amyloidosis (Table 1). Mortality rates increased significantly over time in both sexes, with males demonstrating a higher AAPC over the 1999–2023 period.
Joinpoint regression analyses revealed distinct sex-specific temporal patterns (Figure 1 and Supplemental Table 2). Among males, mortality rates were relatively stable during the early study period, followed by marked acceleration beginning in the mid-2010s, with a pronounced increase from 2018 to 2021 and sustained elevation through 2023. In contrast, females exhibited a more gradual upward trend through 2018, followed by significant acceleration from 2018 to 2023, with less pronounced peak acceleration than that observed among males.
Age-Specific Mortality Trends
Mortality rates increased progressively with age, with the oldest age group bearing the greatest burden (Table 1, Figure 2 and Supplemental Table 3). The AAMR among adults aged ≥85 years was approximately 32-fold higher than that among adults aged 45–54 years.
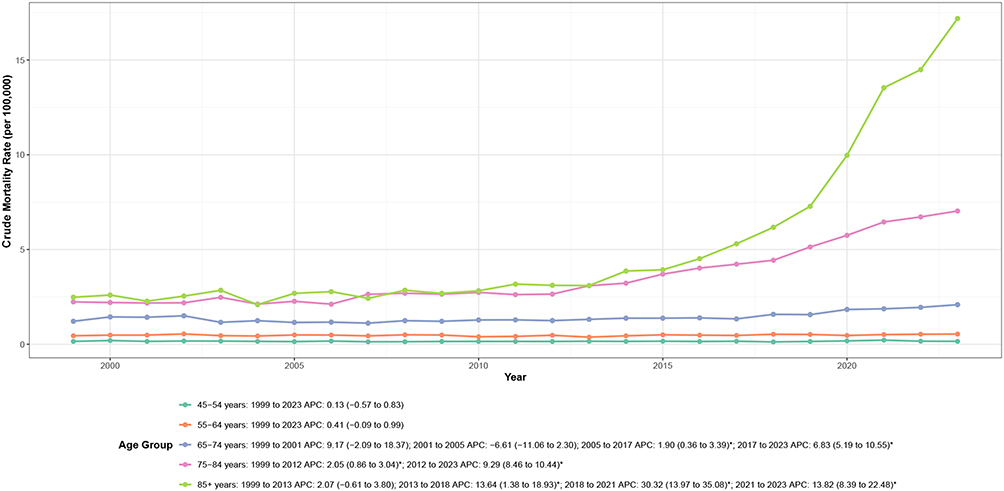 |
Figure 2 Age-specific trends in cardiovascular disease–related mortality among adults with amyloidosis in the United States, 1999–2023. Notes: APC indicates annual percent change. Mortality rates are crude rates expressed per 100,000 population within each age group. Asterisks (*) denote statistically significant trends (P < 0.05) identified by joinpoint regression analysis. |
Temporal trends differed substantially across age groups (Figure 2 and Supplemental Table 2). Adults aged 45–64 years exhibited largely stable mortality rates over the study period, whereas older age groups demonstrated progressively steeper increases. Adults aged 65–74 years experienced gradual increases from 2005 to 2017, followed by significant acceleration thereafter (APC: 6.83%; P < 0.001). Similar but more pronounced patterns were observed among adults aged 75–84 years, with sustained increases from 1999 onward and marked acceleration after 2012 (APC: 9.29%; P < 0.001). The most pronounced increases occurred among adults aged ≥85 years, particularly during 2018–2021 (APC: 30.32%; P < 0.001).
Racial and Ethnic Disparities
Substantial racial and ethnic disparities were evident in CVD-related mortality among adults with amyloidosis (Table 1, Figure 3 and Supplemental Table 4). NH Black adults exhibited the highest AAMR (2.28 per 100,000), more than twice the rate observed among NH White adults (1.04 per 100,000). These disparities widened substantially over the study period, with NH Black adults experiencing an approximately 3.5-fold increase in mortality rates, compared with 2.9-fold and 1.9-fold increases among NH White and Hispanic/Latino adults, respectively.
 |
Figure 3 Race and ethnicity–specific trends in age-adjusted cardiovascular disease–related mortality among adults with amyloidosis in the United States, 1999–2023. Notes: APC indicates annual percent change; NH denotes non-Hispanic. Mortality rates are expressed per 100,000 population and age-adjusted to the 2000 U.S. standard population. Asterisks (*) denote statistically significant trends (P < 0.05) identified by joinpoint regression analysis. |
Temporal patterns varied markedly by race and ethnicity (Figure 3 and Supplemental Table 2). NH Black adults experienced stable rates through 2012, followed by marked acceleration thereafter (APC: 9.92%; P < 0.001). NH White adults demonstrated a triphasic pattern: stable rates from 1999 to 2011, moderate increases from 2011 to 2017 (APC: 5.89%; P = 0.01), and marked acceleration from 2017 to 2023 (APC: 12.02%; P < 0.001). Hispanic/Latino adults showed stable rates through 2016, followed by significant increases thereafter (APC: 10.64%; P < 0.001). Notably, all racial and ethnic groups experienced pronounced mortality surges during 2020–2021, temporally coinciding with the COVID-19 pandemic.
Geographic Region-Specific Mortality Trends
CVD-related mortality rates varied substantially across U.S. Census regions (Table 1, Figure 4 and Supplemental Table 5). The Northeast exhibited the highest AAMR (1.38 per 100,000), followed by the West (1.24 per 100,000) and Midwest (1.21 per 100,000), while the South had the lowest rate (0.87 per 100,000).
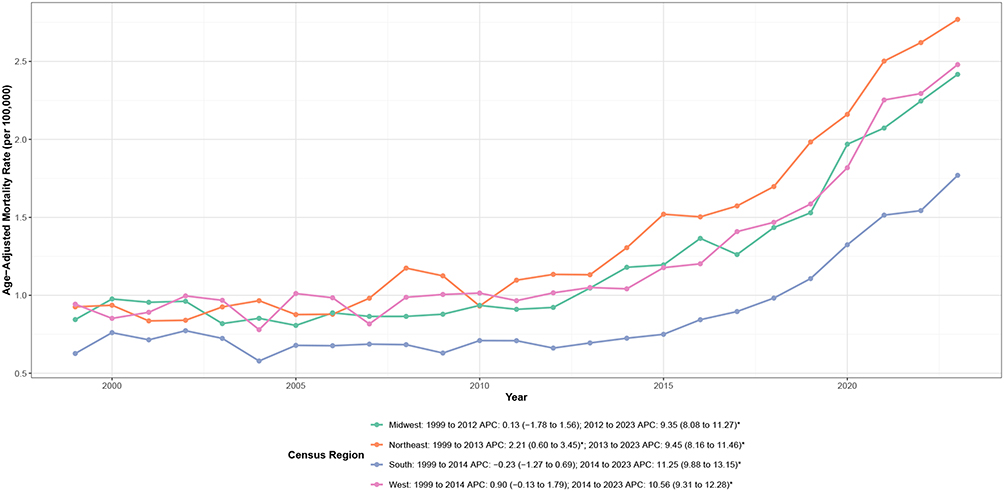 |
Figure 4 U.S. Census region–specific trends in age-adjusted cardiovascular disease–related mortality among adults with amyloidosis, United States, 1999–2023. Notes: APC indicates annual percent change. Mortality rates are expressed per 100,000 population and age-adjusted to the 2000 U.S. standard population. Asterisks (*) denote statistically significant trends (P < 0.05) identified by joinpoint regression analysis. |
Temporal patterns demonstrated remarkable consistency across all regions despite baseline differences (Figure 4 and Supplemental Table 2). All four regions exhibited stable or slowly increasing rates through the early-to-mid 2010s, followed by marked acceleration thereafter. Joinpoint analyses identified region-specific inflection points occurring between 2012 and 2014: Midwest (2012), Northeast (2013), and South and West (both 2014). Following these inflection points, all regions experienced substantial acceleration with APCs ranging from 9.35% in the Midwest to 11.25% in the South (all P < 0.001). Notably, the South demonstrated the steepest acceleration despite having the lowest baseline rates, suggesting a convergence pattern in regional mortality burdens.
Urban-Rural Mortality Trends
Mortality patterns differed modestly across urbanization levels (Table 1, Figure 5 and Supplemental Table 6). Large metropolitan areas exhibited the highest AAMR (1.08 per 100,000), followed by medium/small metropolitan areas (0.92 per 100,000) and rural counties (0.85 per 100,000). All urbanization categories demonstrated increasing trends over time, with AAPCs ranging from 2.56% in rural counties to 3.50% in large metropolitan areas.
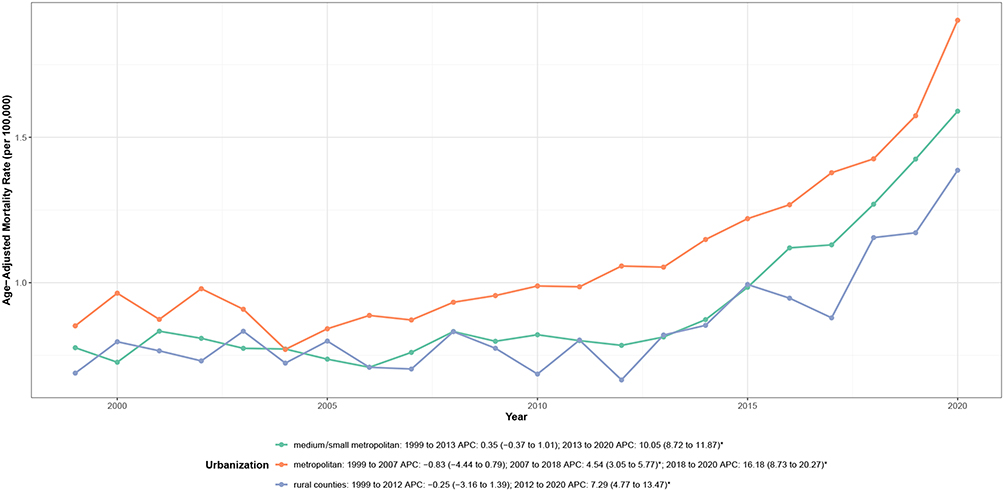 |
Figure 5 Urbanization level–specific trends in age-adjusted cardiovascular disease–related mortality among adults with amyloidosis, United States, 1999–2023. Notes: APC indicates annual percent change. Mortality rates are expressed per 100,000 population and age-adjusted to the 2000 U.S. standard population. Asterisks (*) denote statistically significant trends (P < 0.05) identified by joinpoint regression analysis. |
Temporal patterns revealed important heterogeneity by urbanization level (Figure 5 and Supplemental Table 2). Large metropolitan areas demonstrated a triphasic pattern: stable rates from 1999 to 2007, gradual increases from 2007 to 2018 (APC: 4.54%; P < 0.001), and marked acceleration from 2018 to 2020 (APC: 16.18%; P < 0.001). Medium/small metropolitan areas showed stable rates through 2013, followed by significant acceleration thereafter (APC: 10.05%; P < 0.001). Rural counties exhibited a biphasic pattern with stable rates through 2012, followed by acceleration from 2012 to 2020 (APC: 7.29%; P < 0.001).
Temporal Distribution of Cardiovascular Disease Subtypes
The composition of CVD-related deaths among adults with amyloidosis evolved substantially between 1999 and 2023 (Figure 6 and Supplemental Table 7). HF consistently accounted for the largest proportion of CVD-related deaths and increased in both absolute numbers and relative contribution over time, whereas the proportional contribution of other cardiovascular causes declined. AF and IHD also increased, particularly in later years. Figure 6 Temporal trends in cardiovascular disease subtypes among adults with amyloidosis in the United States, 1999–2023. (A–J) show annual death counts and proportional distributions of cardiovascular disease (CVD) subtypes, stratified by population subgroup. (A and B) show the total population; (C and D), males; (E and F), females; (G and H), adults aged <65 years; and Panels (I and J), adults aged ≥65 years. For each subgroup, the left panel shows annual death counts and the right panel shows proportional composition. CVD subtypes include heart failure, atrial fibrillation/flutter, ischemic heart disease, and other cardiovascular causes. Notes: Proportions are calculated within each calendar year and sum to 100%. Death counts represent the absolute number of cardiovascular disease–related deaths among decedents with amyloidosis. Data are derived from the CDC WONDER Multiple Cause-of-Death database. Figure 6 Continued.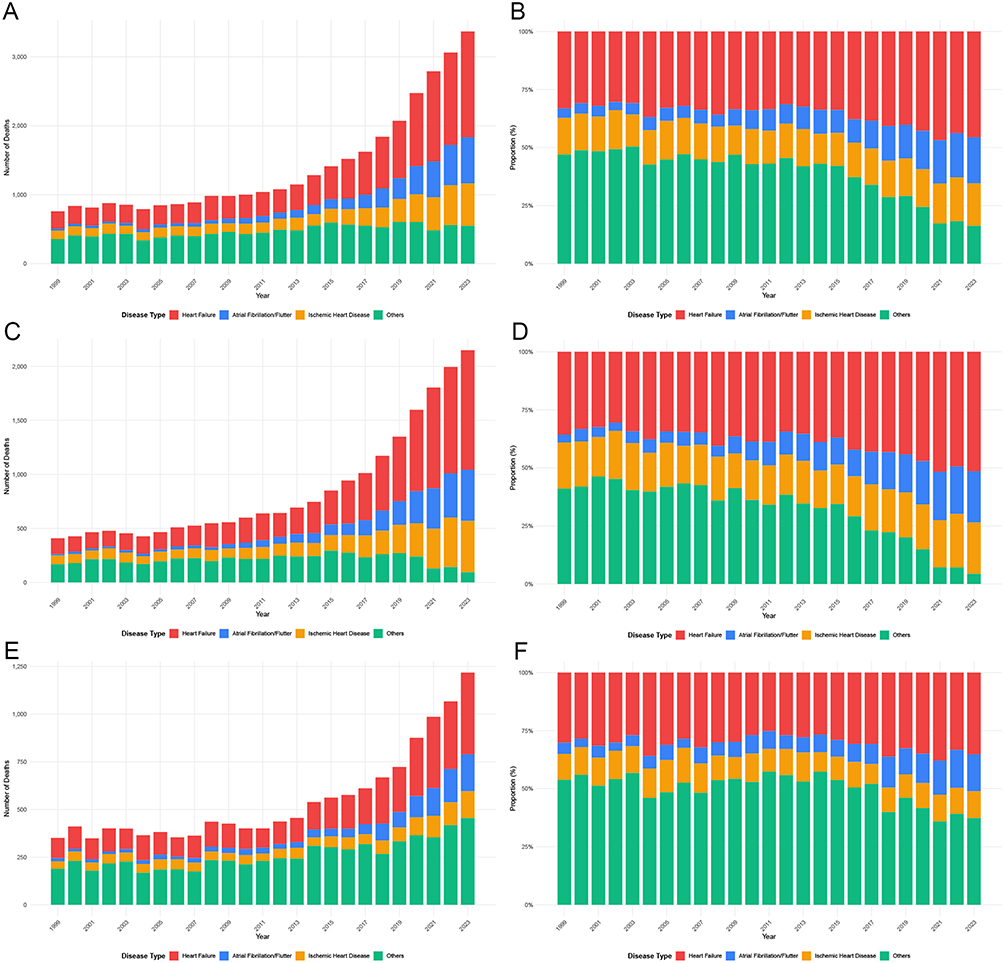
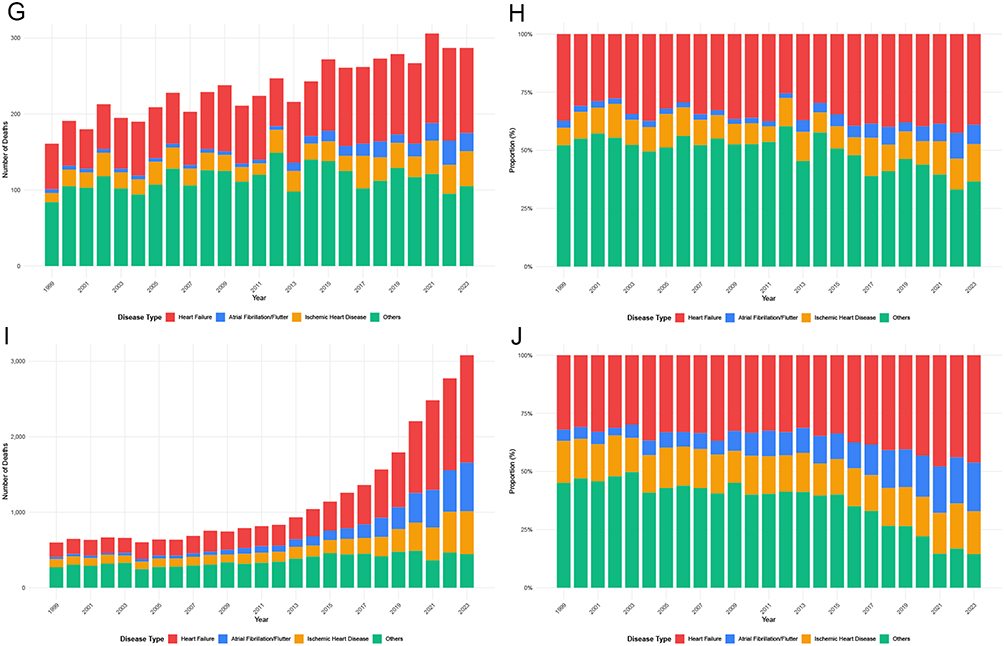
Sex- and age-stratified analyses demonstrated broadly similar temporal patterns, with HF predominating across all subgroups. However, females exhibited a more pronounced proportional increase in AF over time, and adults aged <65 years showed relatively higher contributions from IHD compared with older age groups, among whom HF was markedly dominant throughout the study period.
State-Level Geographic Distribution and Temporal Trends
In 2023, marked geographic heterogeneity was observed in state-level CVD–related mortality among adults aged ≥45 years with amyloidosis in the United States (Figure 7 and Supplemental Table 8). Absolute death counts were highest in several populous states, particularly Texas, Florida, California, and New York, whereas lower counts were observed in less populous states and those with suppressed values due to small numbers.
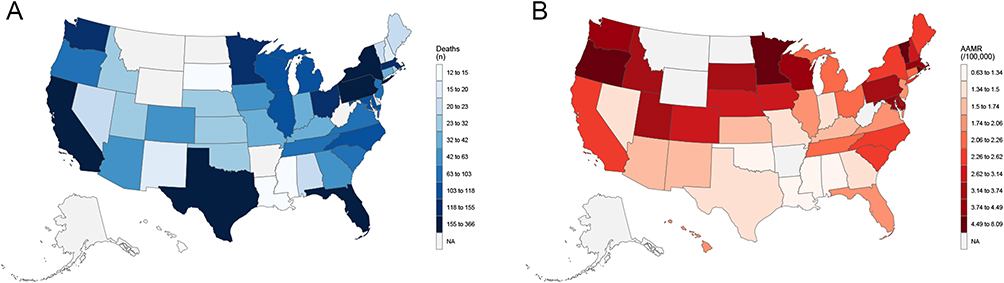 |
Figure 7 State-level cardiovascular disease–related mortality burden among adults with amyloidosis in the United States, 2023. (A) shows state-level death counts, and (B) shows age-adjusted mortality rates. Notes: AAMR indicates age-adjusted mortality rate. Mortality rates are expressed per 100,000 population and age-adjusted to the 2000 U.S. standard population. States with fewer than 10 deaths are suppressed and labeled as NA to ensure confidentiality. |
In contrast, AAMRs demonstrated a distinct spatial pattern not strictly aligned with population size. The highest AAMRs (>3.74 per 100,000) were concentrated in multiple states across the Northeast and Midwest, with moderately elevated rates along parts of the Pacific coast, whereas relatively lower rates were observed across much of the South and Mountain West regions.
Joinpoint regression analyses revealed substantial between-state variability in long-term temporal trends in CVD-related mortality from 1999 to 2023 (Supplemental Table 2). Several large states exhibited prolonged periods of stable mortality followed by significant acceleration in more recent years. In California, mortality rates remained stable from 1999 to 2014, followed by a significant increase from 2014 to 2023 (APC: 9.56%; P < 0.001). Florida demonstrated a similar pattern, with nonsignificant changes through 2015 and marked acceleration thereafter (APC: 12.71%; P < 0.001). Pennsylvania showed one of the steepest recent increases, with a pronounced rise beginning in 2014 (APC: 14.66%; P < 0.001). Illinois and Ohio experienced significant increases beginning in 2009 and 2007, respectively, while New York exhibited a sustained upward trend throughout the entire study period (APC: 4.75%; P < 0.001).
Southern and Southwestern states also demonstrated notable late-period accelerations. In Texas, mortality rates remained stable through 2015, followed by a significant increase from 2015 to 2023 (APC: 11.17%; P < 0.001). Collectively, these findings indicate pronounced state-level disparities in both the magnitude and timing of increases in CVD-related mortality among adults with amyloidosis, with many states exhibiting sharp accelerations beginning in the mid-to-late 2010s.
ARIMA-Based Mortality Forecasting
To project future CVD–related mortality among adults with amyloidosis, ARIMA modeling was applied to annual AAMRs from 1999 to 2023. The observed time series demonstrated a sustained upward trajectory over the study period, with a noticeable acceleration during 2020–2021 corresponding to the COVID-19 pandemic (Figure 8).
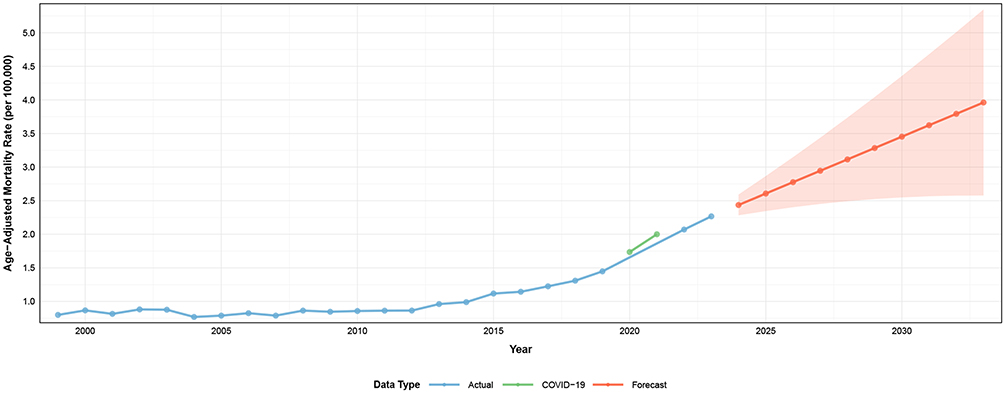 |
Figure 8 ARIMA-based projections of cardiovascular disease–related mortality among adults with amyloidosis in the United States, 1999–2033. Observed age-adjusted mortality rates from 1999 to 2023 are shown along with ARIMA model–based projections for 2024–2033 and corresponding 95% prediction intervals. Notes: ARIMA indicates autoregressive integrated moving average. Mortality rates are expressed per 100,000 population and age-adjusted to the 2000 U.S. standard population. The COVID-19 pandemic period (2020–2021) is highlighted to distinguish pandemic-related perturbations from underlying long-term trends. |
The original series was non-stationary and achieved stationarity after second-order differencing. The optimal ARIMA (0,2,1) model was selected based on minimization of information criteria and satisfactory diagnostic performance. Residual analyses indicated adequate model fit, with no evidence of significant autocorrelation.
Ten-year projections estimated a continued increase in CVD-related mortality through 2033 (Figure 8 and Supplemental Table 9). The forecasted AAMR was 2.44 per 100,000 population (95% CI: 2.28–2.59) in 2024 and increased progressively to 3.96 per 100,000 (95% CI: 2.58–5.35) by 2033. Prediction intervals widened substantially over time, particularly after 2030, reflecting increasing uncertainty in long-term forecasts. The COVID-19 period (2020–2021) was highlighted in the temporal visualization to delineate pandemic-related deviations from the underlying long-term upward trend.
Discussion
This 25-year population-based analysis demonstrates a marked and sustained increase in cardiovascular mortality among U.S. adults with amyloidosis, highlighting a growing and underrecognized public health burden, with a nearly threefold rise between 1999 and 2023 and a pronounced acceleration beginning in the late 2010s. Notably, this pattern was remarkably consistent across sex, age, race and ethnicity, geographic region, and urbanization level, underscoring the growing population-level impact of cardiac involvement in contemporary amyloidosis. HF accounted for the majority of cardiovascular deaths throughout the study period, while AF and IHD contributed increasingly over time, reflecting an evolving and multifaceted cardiac phenotype.
These findings extend prior epidemiologic studies that largely focused on selected clinical cohorts, hospitalization-based samples, or specific amyloidosis subtypes. Previous work has documented rising recognition of cardiac amyloidosis, particularly transthyretin-related disease, and increasing HF hospitalizations among older adults with amyloidosis.15,25,26 However, comprehensive population-level assessments of long-term cardiovascular mortality trends across demographic and geographic strata have remained limited. By leveraging nationally representative mortality data spanning a quarter century, our analysis demonstrates convergent temporal acceleration across diverse subgroups, suggesting that shared national-level forces, rather than subgroup-specific phenomena, are driving contemporary mortality patterns.
The acceleration observed in the mid-to-late 2010s likely reflects the convergence of diagnostic, demographic, and biological factors. The widespread adoption of technetium-99m pyrophosphate scintigraphy beginning around 2015, along with heightened clinical awareness following landmark consensus statements and evolving diagnostic algorithms, substantially improved the detection of cardiac involvement, particularly transthyretin amyloid cardiomyopathy.8,13,27 Concurrently, advances in systemic amyloidosis therapy have extended survival,14,28 potentially allowing progressive myocardial infiltration to emerge as the dominant determinant of late mortality.
Ascertainment bias should also be considered when interpreting the observed temporal increase. Greater clinical awareness of cardiac amyloidosis, broader use of noninvasive diagnostic pathways, improved access to nuclear scintigraphy in selected settings, and increased recognition of amyloidosis on death certificates may have contributed to higher detection and reporting over time. Therefore, part of the observed rise in CVD-related mortality involving amyloidosis may reflect improved diagnostic ascertainment rather than a true increase in disease incidence or case fatality alone. Nevertheless, the magnitude, persistence, and consistency of the increase across multiple demographic and geographic strata suggest that improved ascertainment is unlikely to fully explain the observed national trends.
Several additional factors support this interpretation. The acceleration extended beyond the initial period of diagnostic diffusion, and similar upward trajectories were observed despite marked regional variation in access to advanced imaging and specialized centers, arguing against recognition bias as the sole explanation. Population aging represents a critical underlying driver, as the U.S. population aged 65 years and older expanded substantially during the study period,18 and both major amyloidosis subtypes exhibit strong age-dependent incidence.15 In addition, the rising prevalence of cardiometabolic multimorbidity likely amplifies vulnerability to cardiovascular decompensation once myocardial infiltration is established, while delayed access to specialized care limits opportunities for timely diagnosis and early intervention.
The continued rise in population-level cardiovascular disease–related mortality involving amyloidosis in the era of expanding diagnostic and therapeutic options warrants further investigation.29–33 However, because this study did not include individual-level data on diagnostic testing, treatment exposure, treatment timing, or therapeutic access, these findings should not be interpreted as evidence that recent therapeutic advances failed to reduce mortality. Rather, the observed trends highlight the need to examine whether diagnostic and therapeutic advances are reaching affected populations equitably and early enough to influence population-level outcomes.31,32
The predominance of HF as the leading cardiovascular subtype should be interpreted in the context of our mutually exclusive hierarchical classification strategy. Because HF was prioritized over AF and IHD when multiple cardiovascular codes appeared on the same death certificate, the relative contribution of HF may have been reinforced by this classification approach. This hierarchy was chosen a priori because HF is a central clinical manifestation of amyloid cardiomyopathy and often represents the final common pathway of advanced cardiac involvement. Therefore, subtype-specific proportions should be interpreted as mutually exclusive categories assigned according to this predefined hierarchy, rather than as the full burden of each cardiovascular condition listed on death certificates. The predominance of HF as the leading cause of cardiovascular death aligns with the restrictive cardiomyopathy characteristic of amyloid infiltration.34,35 Progressive extracellular deposition results in ventricular stiffening, impaired diastolic filling, reduced ventricular compliance, and ultimately fixed stroke-volume physiology. In contrast to other cardiomyopathies in which systolic dysfunction predominates early, cardiac amyloidosis frequently presents with preserved or only mildly reduced ejection fraction, thereby limiting the effectiveness of conventional guideline-directed HF therapies.36 Once clinically overt HF develops, prognosis deteriorates substantially. Together, these pathophysiologic features help explain why HF remains the dominant cardiovascular cause of death in amyloidosis and underscore the importance of population-level strategies emphasizing earlier recognition rather than late-stage management.
The increasing contribution of atrial arrhythmias likely reflects extensive atrial infiltration, fibrosis, and chamber dilation, which together create a substrate for electrical instability.37,38 In patients with restrictive physiology, loss of coordinated atrial contraction can precipitate abrupt hemodynamic decompensation, while the exceptionally high thromboembolic risk associated with cardiac amyloidosis further contributes to mortality.39,40 The more pronounced proportional increase in atrial arrhythmia–related mortality among females represents a novel observation and suggests potential sex-specific differences in disease manifestation, recognition, or management that warrant further investigation.
Rising IHD mortality among individuals with amyloidosis likely reflects mechanisms extending beyond epicardial atherosclerosis. Amyloid deposition within intramural coronary vessels leads to microvascular obstruction and impaired coronary flow reserve, resulting in myocardial ischemia even in the absence of obstructive coronary artery disease.41,42 This microvascular dysfunction is further exacerbated by increased myocardial mass and elevated myocardial oxygen demand, creating a persistent supply–demand mismatch. Together, these mechanisms highlight the expanding cardiovascular complexity of amyloidosis, in which multiple interrelated processes contribute to adverse outcomes. From a population health perspective, recognition of these nontraditional ischemic pathways has important implications for cardiovascular risk stratification and surveillance in individuals with amyloidosis.
The pronounced age gradient observed in this analysis, with dramatically higher mortality among the oldest adults, reflects cumulative amyloid deposition, progressive cardiac dysfunction, and diminishing physiologic reserve.43 The particularly steep acceleration among adults aged 85 years and older during the late 2010s suggests heightened vulnerability to both disease progression and healthcare disruptions. In contrast, relatively stable mortality among middle-aged adults may reflect later disease manifestation or persistent underdiagnosis in younger populations. Given that disease-modifying therapies confer the greatest benefit when initiated early, these findings reinforce the importance of diagnostic vigilance across the age spectrum.
Sex-based differences in mortality further highlight important epidemiologic patterns. The consistently higher mortality among males likely reflects the higher prevalence of transthyretin amyloidosis in men,15,34,44 whereas underrecognition of infiltrative cardiomyopathy in women presenting with atypical phenotypes may contribute to delayed diagnosis. The increasing relative contribution of atrial arrhythmias among females over time underscores the need for heightened attention to rhythm surveillance and anticoagulation strategies in this population.
The most concerning finding of this analysis is the marked and widening racial disparity in cardiovascular mortality, with NH Black adults experiencing more than twice the mortality rate of NH White adults. While the higher prevalence of the p.Val142Ile transthyretin variant among individuals of African ancestry contributes to elevated baseline risk,45–48 genetic factors alone cannot explain the progressive widening of disparities over time. Structural barriers, including delayed diagnosis, reduced access to advanced imaging and specialized centers, higher comorbidity burden, and inequitable access to disease-modifying therapies, are likely central contributors.49,50 Without deliberate efforts to address these systemic inequities, ongoing diagnostic and therapeutic advances risk exacerbating existing disparities rather than mitigating them.51
Geographic analyses revealed substantial heterogeneity in baseline mortality rates, yet temporal patterns were strikingly similar across regions. Inflection points clustered tightly in the mid-2010s, followed by parallel acceleration across all Census regions, strongly suggesting that national-level forces rather than region-specific factors are driving contemporary trends.27 Urban–rural gradients were modest. Lower observed mortality rates in rural areas may reflect differences in diagnostic recognition, death certificate coding, access to specialized diagnostic resources, or true differences in disease burden.52 Because CDC WONDER does not include individual-level diagnostic or healthcare access data, these explanations remain speculative and require further evaluation in clinically adjudicated datasets.
The pronounced mortality surge observed during 2020–2021 highlights heightened vulnerability among individuals with cardiac amyloidosis during the COVID-19 pandemic. Limited cardiac reserve, susceptibility to inflammatory and thrombotic complications, and disruptions in access to specialized care likely contributed synergistically.53–55 The disproportionate pandemic impact on medically underserved communities may have further amplified existing racial and socioeconomic disparities.56,57 Importantly, mortality rates did not return to pre-pandemic baseline but continued rising through 2023, raising concern that the pandemic may have accelerated underlying trajectories or produced lasting downstream effects on disease progression and healthcare delivery.
These findings may help inform clinical awareness and public health prioritization. In particular, they suggest the potential value of improved recognition of cardiac involvement in amyloidosis, especially among populations with higher observed mortality rates or limited access to specialized care. However, because the present analysis is based on mortality surveillance data, it cannot directly evaluate the effectiveness of screening, diagnostic pathways, or therapeutic interventions. Future research using clinically adjudicated cohorts, registry-linked data, or datasets with validated amyloidosis subtype information is needed to distinguish cardiovascular mortality patterns in AL amyloidosis from those in ATTR amyloidosis. Such subtype-specific analyses would help identify whether prevention, diagnostic, and therapeutic efforts should be prioritized differently across these clinically distinct diseases.
Several limitations warrant consideration. Death certificate data are subject to potential misclassification of both amyloidosis and cardiovascular causes, particularly in earlier years when clinical awareness was more limited. The validity of amyloidosis diagnoses based on ICD-10 codes E85.0–E85.9 could not be independently verified in this database. Although these codes are appropriate for national mortality surveillance, death certificate–based ascertainment may be affected by underdiagnosis, coding variation, and incomplete recognition of amyloidosis, particularly in earlier years. Future validation using individual-level data linked to electronic health records, amyloidosis registries, pathology reports, genetic testing, cardiac imaging, treatment records, and the National Death Index would help determine whether the observed temporal and subgroup patterns reflect true differences in disease burden, differences in diagnostic recognition, or both. In addition, our definition of CVD-related death was based on the presence of any CVD-related ICD-10 code anywhere on the death certificate, including both underlying and contributing causes of death. Therefore, some included deaths may represent cases in which cardiovascular disease coexisted with amyloidosis but was not necessarily the principal driver of death. Accordingly, our findings should be interpreted as mortality involving both amyloidosis and CVD on death certificates, rather than strictly cardiovascular-attributable mortality. Although an underlying-cause-only sensitivity analysis may help bound the influence of broader multiple-cause-of-death coding, such a restrictive definition may underestimate the burden of amyloidosis-associated cardiovascular mortality because amyloidosis may contribute substantially to death without being selected as the underlying cause. Future studies comparing multiple-cause and underlying-cause definitions, ideally with individual-level clinical validation, are warranted. Furthermore, the distribution of cardiovascular subtypes was influenced by the mutually exclusive hierarchical classification algorithm. Because HF was prioritized over AF and IHD when multiple cardiovascular diagnoses were present, HF may be overrepresented relative to AF or IHD in subtype-specific analyses. Thus, subtype proportions should be interpreted as hierarchy-assigned categories rather than direct estimates of the total frequency of each cardiovascular condition among decedents with amyloidosis. The absence of individual-level clinical data precludes assessment of amyloidosis subtype, cardiac biomarker stage, disease severity, treatment exposure, and care pathways, fundamentally limiting causal inference. In particular, we could not conduct separate analyses for AL amyloidosis and ATTR amyloidosis because death certificate data do not consistently record amyloidosis subtype, and ICD-10 coding in CDC WONDER does not reliably distinguish these clinically distinct entities. Consequently, the observed mortality patterns should be interpreted as aggregate population-level trends among decedents with amyloidosis rather than subtype-specific estimates for AL or ATTR amyloidosis. We cannot distinguish whether rising mortality reflects increasing disease incidence, worsening case-fatality rates, or both. Temporal increases may partially reflect improved diagnostic recognition rather than true increases in disease burden, though the magnitude, consistency, and persistence of trends across diverse subgroups suggest that substantial genuine increases have occurred. Suppression of small cell counts necessitated exclusion of some strata from analyses, potentially introducing selection bias. Although the 2020–2021 period was highlighted to contextualize potential pandemic-related disruptions, this study did not formally isolate the causal effect of COVID-19 on amyloidosis-associated cardiovascular mortality. Interrupted time-series or segmented forecasting approaches may be useful in future analyses as additional post-pandemic years become available, but the limited number of post-pandemic observations in the current dataset constrained reliable estimation of pandemic-specific level or slope changes. Finally, mortality projections assume continuation of historical patterns and cannot account for transformative changes in clinical practice that may fundamentally alter future trajectories. These limitations underscore the critical need for prospective studies that link diagnostic information, genetic testing results, treatment exposure, and longitudinal outcomes.
Conclusions
In conclusion, cardiovascular disease–related mortality involving amyloidosis among U.S. adults has risen substantially over the past 25 years, representing a growing national public health challenge characterized by accelerating trends and persistent disparities. These findings highlight the need for continued population-level surveillance and further studies to clarify the relative contributions of true disease burden, diagnostic ascertainment, healthcare access, and therapeutic uptake to observed mortality patterns. Future studies incorporating validated amyloidosis subtype information are also needed to distinguish cardiovascular mortality patterns in AL amyloidosis and ATTR amyloidosis and to better define subtype-specific priorities for prevention, diagnosis, and treatment.
Abbreviations
AAMR, Age-adjusted mortality rate; AAPC, Average annual percent change; AF, Atrial fibrillation/flutter; AIC, Akaike Information Criterion; AL, Light-chain amyloidosis; APC, Annual percent change; ARIMA, Autoregressive integrated moving average; ATTR, Transthyretin amyloidosis; CDC, Centers for Disease Control and Prevention; CI, Confidence interval; CMR, Crude mortality rate; CVD, Cardiovascular disease; HF, Heart failure; ICD-10, International Classification of Diseases, Tenth Revision; IHD, Ischemic heart disease; NCHS, National Center for Health Statistics; NH, Non-Hispanic; U.S., United States; WONDER, Wide-ranging Online Data for Epidemiologic Research.
Data Sharing Statement
The datasets generated and/or analyzed during the current study are available on the CDC WONDER Database, https://wonder.cdc.gov/.
Ethics Approval and Consent to Participate
This study was reviewed and approved by the Beijing Haidian Hospital Medical Ethics Committee (approval No. 2026-070). Because this study used only publicly available, de-identified, aggregate mortality data from the CDC WONDER Multiple Cause-of-Death database and no identifiable individual-level information was accessed, individual informed consent was waived/not required.
Author Contributions
All authors made a significant contribution to the work reported, whether in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors declare no competing interests in this work.
References
1. Benson MD, Buxbaum JN, Eisenberg DS, et al. Amyloid nomenclature 2020: update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2020;27(4):217–17. doi:10.1080/13506129.2020.1835263
2. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6):583–596. doi:10.1056/NEJMra023144
3. Pinney JH, Smith CJ, Taube JB, et al. Systemic amyloidosis in England: an epidemiological study. Br J Haematol. 2013;161(4):525–532. doi:10.1111/bjh.12286
4. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45–59.
5. Ruberg FL, Maurer MS. Cardiac amyloidosis due to transthyretin protein: a review. JAMA. 2024;331(9):778–791. doi:10.1001/jama.2024.0442
6. Novo G, Stabile F, Di Lisi D, et al. The impact of cardiac amyloidosis on patients with multiple myeloma: a systematic review and meta-analysis. Cardiooncology. 2026;12(1):32. doi:10.1186/s40959-025-00435-1
7. Antonopoulos AS, Panagiotopoulos I, Kouroutzoglou A, et al. Prevalence and clinical outcomes of transthyretin amyloidosis: a systematic review and meta-analysis. Eur J Heart Fail. 2022;24(9):1677–1696. doi:10.1002/ejhf.2589
8. Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–2412. doi:10.1161/CIRCULATIONAHA.116.021612
9. Bokhari S, Castaño A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. (99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging. 2013;6(2):195–201. doi:10.1161/CIRCIMAGING.112.000132
10. Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203–1212. doi:10.1161/CIRCULATIONAHA.108.843334
11. Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15(7):387–404. doi:10.1038/s41582-019-0210-4
12. Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30(36):4541–4549. doi:10.1200/JCO.2011.37.7614
13. Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis. A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur J Heart Fail. 2021;23(4):512–526. doi:10.1002/ejhf.2140
14. Gonzalez-Lopez E, Maurer MS, Garcia-Pavia P. Transthyretin amyloid cardiomyopathy: a paradigm for advancing precision medicine. Eur Heart J. 2025;46(11):999–1013. doi:10.1093/eurheartj/ehae811
15. Gilstrap LG, Dominici F, Wang Y, et al. Epidemiology of cardiac amyloidosis-associated heart failure hospitalizations among fee-for-service medicare beneficiaries in the United States. Circ Heart Fail. 2019;12(6):e005407. doi:10.1161/CIRCHEARTFAILURE.118.005407
16. Witteles RM, Bokhari S, Damy T, et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail. 2019;7(8):709–716. doi:10.1016/j.jchf.2019.04.010
17. Centers for Disease Control and Prevention, National Center for Health Statistics. Multiple cause of death 1999-2023 on CDC WONDER online database. Available from: https://wonder.cdc.gov/mcd.html.
18. United States Census Bureau. Population estimates program. Available from: https://www.census.gov/programs-surveys/popest.html.
19. Office of Management and Budget. Revisions to the standards for the classification of federal data on race and ethnicity. Fed Regist. 1997;62(210):58781–58790.
20. United States Census Bureau. Census regions and divisions of the United States. Available from: https://www2.census.gov/geo/pdfs/maps-data/maps/reference/us_regdiv.pdf.
21. Hyndman RJ, Khandakar Y. Automatic time series forecasting: the forecast package for R. Journal of Statistical Software. 2008;27(3):1–22. doi:10.18637/jss.v027.i03
22. Hyndman RJ, Athanasopoulos G. Forecasting: Principles and Practice.
23. NIST/SEMATECH e-Handbook of statistical methods. Box-Ljung Test. Available from: https://www.itl.nist.gov/div898/handbook/pmc/section4/pmc4481.htm.
24. National Cancer Institute. Joinpoint regression program, version 5.1.0.0. statistical methodology and applications branch, surveillance research program, National Cancer Institute; 2023. Available from: https://surveillance.cancer.gov/joinpoint/.
25. Porcari A, Bussani R, Merlo M, et al. Incidence and characterization of concealed cardiac amyloidosis among unselected elderly patients undergoing post-mortem examination. Front Cardiovasc Med. 2021;8:749523. doi:10.3389/fcvm.2021.749523
26. González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585–2594. doi:10.1093/eurheartj/ehv338
27. Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2-evidence base and standardized methods of imaging. J Nucl Cardiol. 2019;26(6):2065–2123. doi:10.1007/s12350-019-01760-6
28. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020;136(23):2620–2627. doi:10.1182/blood.2020006913
29. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi:10.1056/NEJMoa1805689
30. Damy T, Garcia-Pavia P, Hanna M, et al. Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and long-term extension study. Eur J Heart Fail. 2021;23(2):277–285. doi:10.1002/ejhf.2027
31. Elliott P, Drachman BM, Gottlieb SS, et al. Long-Term survival with tafamidis in patients with transthyretin amyloid cardiomyopathy. Circ Heart Fail. 2022;15(1):e008193. doi:10.1161/CIRCHEARTFAILURE.120.008193
32. Kazi DS, Bellows BK, Baron SJ, et al. Cost-Effectiveness of tafamidis therapy for transthyretin amyloid cardiomyopathy. Circulation. 2020;141(15):1214–1224. doi:10.1161/CIRCULATIONAHA.119.045093
33. Falk RH, Alexander KM, Liao R, Dorbala S. AL (Light-Chain) cardiac amyloidosis: a review of diagnosis and therapy. J Am Coll Cardiol. 2016;68(12):1323–1341. doi:10.1016/j.jacc.2016.06.053
34. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC State-of-the-Art review. J Am Coll Cardiol. 2019;73(22):2872–2891. doi:10.1016/j.jacc.2019.04.003
35. Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142(1):e7–e22. doi:10.1161/CIR.0000000000000792
36. Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286–1300. doi:10.1161/CIRCULATIONAHA.111.078915
37. Barbhaiya CR, Kumar S, Baldinger SH, et al. Electrophysiologic assessment of conduction abnormalities and atrial arrhythmias associated with amyloid cardiomyopathy. Heart Rhythm. 2016;13(2):383–390. doi:10.1016/j.hrthm.2015.09.016
38. Donnellan E, Wazni OM, Hanna M, et al. Atrial fibrillation in transthyretin cardiac amyloidosis: predictors, prevalence, and efficacy of rhythm control strategies. JACC Clin Electrophysiol. 2020;6(9):1118–1127. doi:10.1016/j.jacep.2020.04.019
39. Feng D, Edwards WD, Oh JK, et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation. 2007;116(21):2420–2426. doi:10.1161/CIRCULATIONAHA.107.697763
40. Cappelli F, Tini G, Russo D, et al. Arterial thrombo-embolic events in cardiac amyloidosis: a look beyond atrial fibrillation. Amyloid. 2021;28(1):12–18. doi:10.1080/13506129.2020.1798922
41. Mueller PS, Edwards WD, Gertz MA. Symptomatic ischemic heart disease resulting from obstructive intramural coronary amyloidosis. Am J Med. 2000;109(3):181–188. doi:10.1016/S0002-9343(00)00471-X
42. Dorbala S, Vangala D, Bruyere J, et al. Coronary microvascular dysfunction is related to abnormalities in myocardial structure and function in cardiac amyloidosis. JACC Heart Fail. 2014;2(4):358–367. doi:10.1016/j.jchf.2014.03.009
43. Tanskanen M, Peuralinna T, Polvikoski T, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40(3):232–239. doi:10.1080/07853890701842988
44. Lane T, Fontana M, Martinez-Naharro A, et al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation. 2019;140(1):16–26. doi:10.1161/CIRCULATIONAHA.118.038169
45. Quarta CC, Buxbaum JN, Shah AM, et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372(1):21–29. doi:10.1056/NEJMoa1404852
46. Jacobson DR, Alexander AA, Tagoe C, et al. The prevalence and distribution of the amyloidogenic transthyretin (TTR) V122I allele in Africa. Mol Genet Genomic Med. 2016;4(5):548–556. doi:10.1002/mgg3.231
47. Damrauer SM, Chaudhary K, Cho JH, et al. Association of the V122I hereditary transthyretin amyloidosis genetic variant with heart failure among individuals of African or Hispanic/Latino ancestry. JAMA. 2019;322(22):2191–2202. doi:10.1001/jama.2019.17935
48. Parcha V, Malla G, Irvin MR, et al. Association of transthyretin Val122Ile variant with incident heart failure among black individuals. JAMA. 2022;327(14):1368–1378. doi:10.1001/jama.2022.2896
49. Alexander KM, Orav J, Singh A, et al. Geographic disparities in reported US amyloidosis mortality from 1979 to 2015: potential underdetection of cardiac amyloidosis. JAMA Cardiol. 2018;3(9):865–870. doi:10.1001/jamacardio.2018.2093
50. Miller P, Elias P, Einstein AJ, Maurer MS, Ahmed GY, Poterucha TJ. Socioeconomic disparities are associated with delayed access to tafamidis in transthyretin cardiac amyloidosis. Circ Heart Fail. 2024;17(12):e012075. doi:10.1161/CIRCHEARTFAILURE.124.012075
51. Madu EC, Mezue K. Uneven burden of cardiac amyloidosis in people of African descent - global imbalance in resources and access. BMC Glob Public Health. 2023;1(1):15. doi:10.1186/s44263-023-00016-3
52. Shankar B, Yanek L, Jefferson A, et al. Race and socioeconomic status impact diagnosis and clinical outcomes in transthyretin cardiac amyloidosis. JACC CardioOncol. 2024;6(3):454–463. doi:10.1016/j.jaccao.2024.05.001
53. Madjid M, Safavi-Naeini P, Solomon SD, Vardeny O. Potential effects of coronaviruses on the cardiovascular system: a review. JAMA Cardiol. 2020;5(7):831–840. doi:10.1001/jamacardio.2020.1286
54. Wadhera RK, Shen C, Gondi S, Chen S, Kazi DS, Yeh RW. Cardiovascular deaths during the COVID-19 pandemic in the United States. J Am Coll Cardiol. 2021;77(2):159–169. doi:10.1016/j.jacc.2020.10.055
55. Brannagan TH, Auer-Grumbach M, Berk JL. ATTR amyloidosis during the COVID-19 pandemic: insights from a global medical roundtable. Orphanet J Rare Dis. 2021;16(1):204. doi:10.1186/s13023-021-01834-0
56. Wilder JM. The disproportionate impact of COVID-19 on Racial and Ethnic Minorities in the United States. Clin Infect Dis. 2021;72(4):707–709. doi:10.1093/cid/ciaa959
57. Wadhera RK, Wadhera P, Gaba P, et al. Variation in COVID-19 hospitalizations and deaths across New York City boroughs. JAMA. 2020;323(21):2192–2195. doi:10.1001/jama.2020.7197
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.