")
Back to Journals » International Journal of Nanomedicine » Volume 14
Carbon Nanotube Reinforced Hydroxyapatite Nanocomposites As Bone Implants: Nanostructure, Mechanical Strength And Biocompatibility
Authors Lawton K, Le H , Tredwin C, Handy RD
Received 4 June 2019
Accepted for publication 4 September 2019
Published 1 October 2019 Volume 2019:14 Pages 7947—7962
DOI https://doi.org/10.2147/IJN.S218248
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Kiruthika Lawton,1,2 Huirong Le,3,4 Christopher Tredwin,1 Richard D Handy5,6
1Peninsula School of Medicine and Dentistry, Plymouth University, Plymouth PL4 8AA, UK; 2School of Pharmacy, Centre for Biomolecular Sciences, University of Nottingham, Nottingham, UK; 3School of Mechanical Engineering & Built Environment, University of Derby, Derby DE22 3AW, UK; 4The Future Lab, Tsinghua University, Beijing, China; 5School of Biological and Marine Sciences, Plymouth University, Plymouth PL4 8AA, UK; 6Department of Nutrition, Cihan University, Erbil, Kurdistan Region, Iraq
Correspondence: Huirong Le
School of Mechanical Engineering & Built Environment, University of Derby, Markeaton Street, Derby DE22 3AW, UK
Tel +44 1332 13 3259 3599
Email [email protected]
Richard D Handy
School of Biological and Marine Sciences, Plymouth University, Plymouth PL4 8AA, UK
Email [email protected]
Purpose: Hydroxyapatite (HA) is a biologically active ceramic which promotes bone growth, but it suffers from relatively weak mechanical properties. Multi-walled carbon nanotubes (MWCNTs) have high tensile strength and a degree of stiffness that can be used to strengthen HA; potentially improving the clinical utility of the bone implant.
Methods: HA was precipitated by the wet precipitation method in the presence of pristine (p) or functionalised (f) MWCNTs, and polyvinyl alcohol (PVA) or hexadecyl trimethyl ammonium bromide (HTAB) as the surfactant. The resulting composites were characterised and the diametral tensile strength and compressive strength of the composites were measured. To determine the biocompatibility of the composites, human osteoblast cells (HOB) were proliferated in the presence of the composites for 7 days.
Results: The study revealed that both the MWCNTs and surfactants play a crucial role in the nucleation and growth of the HA. Composites made with f-MWCNTs were found to have better dispersion and better interaction with the HA particles compared to composites with p-MWCNTs. The mechanical strength was improved in all the composites compared to pure HA composites. The biocompatibility study showed minimal LDH activity in the media confirming that the composites were biocompatible. Similarly, the ALP activity confirmed that the cells grown on the composites containing HTAB were comparable to the control whereas the composites containing PVA surfactant showed significantly reduced ALP activity.
Conclusions: The study shows that the composites made of f-MWCNTs HTAB are stronger than pure HA composites and biocompatible making it a suitable material to study further.
Keywords: calcium phosphate, carbon nanotubes, osteoblast cells, LDH assay, alkaline phosphatase
Introduction
A unique feature of bone is that it has apparently contradictory properties of stiffness yet flexibility, and lightness yet strength. Bone provides mechanical support for the body and also protects the vital organs; but also enables physiological processes such as erythropoiesis in the marrow, serves as a reservoir for minerals, and takes part in acid–base balance.1 However, many circumstances call for bone grafting owing to bone defects from traumatic or from non-traumatic destruction. Synthetic nanostructured hydroxyapatite (HA) has long been an attractive choice for the partial replacement of bone because of its similarity to the mineral component of natural bone. Synthetic HA has shown excellent biocompatibility in vitro with cultured osteoblasts, as well as other cells, that readily grow on HA composites or HA-coated surfaces.2 However, synthetic HA does not mimic all the mechanical, chemical or biological properties of natural bone. A major disadvantage of synthetic HA is its poor mechanical properties, such as brittleness and low tensile strength; thus the clinical uses have been limited particularly in load-bearing applications or where the section of bone replacement is large.3 Natural HA in bone and synthetic HA also differ in their chemical composition. Natural HA contains other ions: mainly CO32− and traces of Na+, Mg2+, Fe2+, Cl−, F−, whereas synthetic HA is usually stoichiometric, with a chemical composition of Ca10(PO4)6(OH)2.4 Regardless of the precise chemistry, HA plays an important role in bone repair including osteoblast cell adhesion, proliferation and in bone remodelling.
To overcome the mechanical problem of synthetic HA not imparting all the desired properties of natural bone, it has been usual to present the synthetic HA as part of a composite material for bone replacement. Composites can be defined as a material consisting of two or more different components, which are combined with the aim of improving the mechanical and/or biological properties of the overall material.5,6 Over the years, synthetic HA microcomposites have been developed with various metals (titanium, stainless steel, titanium alloys),7,8 other ceramics (bioglass, tricalcium phosphate, alumina, zirconia)9,10 and polymers (collagen, gelatine, chitosan, poly lactic-co-glycolic acid (PLGA), polylactic acid (PLA) and polymethyl methacrylate (PMMA)). However, these composites suffer from either very strong or very poor mechanical properties [e.g., Stainless steel, PLGA, PLA,11,12]; induce an adverse immune response from the host bone [e.g., Calcium sulphate13]; or lack strong interfacial bonding between the constituents such that the composite is not durable or satisfactorily homogenous for clinical use.14 Recently, with advances in nanotechnology, nanocomposites have gained much interest and might overcome some of these shortcomings of the micron scale composites by allowing the manipulation of the composite constituents at the nano level.
Nanocomposites can be defined as a heterogeneous combination of two or more materials in which at least one of those materials should be on a nanometer scale.5 Since natural bone has a nanoscale structure made of several chemicals, designing bone grafts in the form of nanocomposite is perceived to be beneficial over single-phase and microcomposite materials. Nanocomposites are usually made of two components; the basic matrix and the reinforcement. In bone implants, the reinforcement serves to improve the strength of the nanocomposite by blocking the growth of cracks in them.15 This can be achieved by employing materials that have a high aspect ratio. One such reinforcement is carbon nanotubes (CNTs).
CNTs with their intrinsic properties including: a high tensile strength (approximately ×100 times greater than steel); high flexibility and elasticity; high electrical and thermal conductivity and a low thermal expansion coefficient; and a high aspect ratio; is an ideal reinforcement material.16 Therefore, in the case of HA-CNTs nanocomposites, a synthetic HA matrix reinforced with CNTs can be anticipated to be biocompatible and strong.
There are many reports on the development of HA matrix composites reinforced with multi-walled carbon nanotubes (MWCNTs) as reviewed by Lahiri et al,17 and Shin et al.18 Previous studies focussed on the HA matrix composites reinforced with MWCNTs through various processes such as sol–gel precipitation, spark plasma deposition or ball milling.19,20 It was found that the addition of CNTs does improve the mechanical strength of the HA composites but requires further improvement for load-bearing devices. The dispersion of CNTs has a significant effect on the mechanical properties. Most researchers used functionalised CNTs with strong acids or surfactants21,22 to obtain a homogenous dispersion of the CNTs in the matrix. The surface modifications of CNTs also have a significant effect on the subsequent nucleation and growth of HA crystals during the sol–gel precipitation process. However, there are few reports on the interplay of surface modifications and surfactants on the mechanical properties. In addition, controversy exists on the biocompatibility of the CNTs. Some studies have reported that CNTs increase the adhesion and proliferation of osteoblasts and fibroblasts,19,23,24 although the biocompatibility with these cells may depend on the diameter and surface energy of the CNTs. In contrast, CNTs have also been reported to reduce the proliferation of osteoblast cells and are toxic to them.25–28 Hence, although CNTs have remarkable properties, their application in nanocomposites for bone implants is still uncertain owing to the difficulty in obtaining a homogenous composite that would be durable and clinically robust, as well as disagreement on biocompatibility of the composites, especially for bone cells.
The overall aims of the current study were therefore to provide a HA-MWCNT composite that would be acceptable, in principal, for clinical use. Initial experiments explored the best approaches to preparing a homogeneous composite by using functionalised and pristine MWCNTs and various dispersing agents to explore nucleation of the HA and growth of the composite. Then, after testing the resulting composites for their mechanical properties, the best materials were selected for biocompatibility studies. These latter experiments explored cell health in vitro using cultured human osteoblasts (HOB) cells.
Materials And Methods
In order to study the effect of MWCNTs in synthetic HA nanocomposites, 4 different types of nanocomposite powders were produced using pristine (p, as produced) and functionalised (f, oxidised with Nitric acid) MWCNTs with two types of surfactants – polyvinyl alcohol (PVA) and hexadecyl trimethyl ammonium bromide (HTAB), in combinations as follows: 1. p-MWCNTs-PVA, (HA + pristine MWCNTs + PVA); 2. f-MWCNTs-PVA, (HA + functionalized MWCNTs + PVA); 3. p-MWCNTs-HTAB, (HA + pristine MWCNTs + HTAB); 4. f-MWCNTs-HTAB, (HA + functionalized MWCNTs + HTAB). Pure HA powder (without the presence of MWCNTs or surfactants) was produced in house and used as a control. The nanocomposite powders were then mixed with PVA, or HTAB as appropriate, to produce the final composites. Analytical grade reagents were used throughout the production process.
Functionalization Of MWCNTs
MWCNTs were functionalised to improve their dispersivity in aqueous media and to compare against the pristine (not functionalised) nanotubes. Commercially supplied MWCNTs (Shenzhen Nanotech Port, China) with a diameter of 10–30 nm, length 5–15 μm and ≥95% purity (manufacturer’s information) were functionalised in house using concentrated nitric acid (Sigma Aldrich, UK) following an established protocol.29 Briefly, 0.3 g of the as-received MWCNTs was mixed with 25 mL of 14.5 mol/L concentrated nitric acid and refluxed at room temperature for 48 hrs. The resulting dispersion was then filtered through a whatman® glass microfiber filter paper (WHA1827070, Sigma Aldrich, Irvine, UK) with pore size 1.5 µm and washed in deionised water until neutral pH was achieved. The sample was then dried in a vacuum oven (Edwards High Vacuum Ltd, Crawley, Sussex) at 40°C for 24 hrs. Where measurements could be made using TEM, the pristine MWCNTs had a length ranging between 2 and 10 μm and diameters between 19 and 25 nm. The functionalised MWCNTs in comparison were less tangled, and the tubes had a curled appearance (Figure S1). The functionalised MWCNTs were fragmented compared to the pristine material, with lengths between 0.8 −2 μm and diameters of 13–24 nm.
Preparation Of HA-MWCNTs Powder
The various HA-MWCNT nanocomposites were prepared using the sol–gel technique. To produce the nanocomposite powders, HA must be nucleated on the walls of the MWCNTs in the presence of the surfactant. In a typical experiment, to produce 10 g of the nanocomposite powder, 0.005 g of PVA (MW- 70,000–100,000, Sigma Aldrich, UK) or 0.005 g of HTAB (Sigma Aldrich, UK) was added to 100 mL ultrapure water. Then, 0.05 g of pristine or functionalized MWCNTs was added to the mixture which was sonicated at 50–60 Hz (Metason 120T, Struers, UK) for 30 mins at room temperature until all the MWCNTs were dispersed. HA was nucleated into the dispersion by the addition of 200 mL of 0.1 mol/L calcium acetate and 0.06 mol/L of ammonium phosphate to obtain a final calcium: phosphate ratio of 10:6, normally expressed as 1.67. The reaction occurs as follows:
5Ca (CH3COO)2·H2O + 3(NH4)2H2PO4→Ca5 (PO4)3(OH) + 3NH4 (CH3CO2) + 7CH3CO2H
The solution was stirred at the rate of 900–1000 rpm using a magnetic stirrer (dial setting 9, Hotplate/stirrer, RCT basic, IKA Oxford, UK). While stirring, the pH of the solution was checked periodically and maintained at 9.5–10 by the addition of 6 mol/L sodium hydroxide solution. The solution was stirred for 1 hr and left to mature for 24 hrs at 40°C. The resulting dispersion containing the HA nucleated on the MWCNTs was then filtered through a Whatman® filter paper (WHA1001070, Sigma Aldrich, Irvine, UK) with a pore size of 11 µm. The precipitate collected was washed with ultrapure water and subsequently dried under a vacuum at 60°C for 24 hrs. The dry precipitate was manually crushed with a glass pestle and mortar to make a fine powder, and was then sintered at 100°C for 8 hrs. In order to obtain HA alone, the process was conducted exactly as above, but without the addition of the MWCNTs.
HA-MWCNTs Composites
The nanocomposite powders obtained from the above method were used to prepare the final composites which were produced following an established protocol in our laboratory.30 Briefly, 1 g of the calcined nanocomposite powder was mixed with 1 mL of 20% PVA at room temperature. The mixture was packed into cylindrical moulds. Different sizes of moulds were used for mechanical and biocompatibility test; to comply with the international testing standards on mechanical testing (see below) and to ensure that the composites fitted in 24 well plates for cell culture work, respectively. The ends of the moulds were blocked with short aluminium cylinders and then pressure was applied on top of the cylinders using mechanical mini pellet press (GS03940, Specac, UK). The pressure applied on each sample was 26 MPa, which was held for 1 min; then the aluminium cylinders were removed from the moulds. The samples were placed in an oven at 40°C for 48 hrs to dry the composite. Afterwards, the samples were carefully removed from the moulds and stored at room temperature, until required for mechanical testing and biocompatibility studies (see below)
Characterisation Of The Composite Materials
Physico-chemical characterisation was performed at several steps in the synthesis of the composites including; the original materials and their functionalised forms in dispersions, as well as the resulting powers that were subsequently compressed into pellets to make the final composite. In addition to the manufacturer’s information, the materials were examined by electron microscopy for their appearance and primary size. X-ray diffraction (XRD) is the most common technique to determine crystal structures and atomic spacing; and was used to identify the phase purity of the produced nanocomposite powders. Fourier transform infrared spectroscopy (FTIR) analysis was performed to detect the functional groups and to characterise covalent bonding.
To confirm the primary dimensions (length, tube diameter) and morphology (straightness, branches and bundles), the MWCNTs as supplied by the manufacturer were examined by transmission electron microscopy (TEM). Batches of the functionalized MWCNTs prior to making the nanocomposite powder were also examined; followed by the nanocomposite powders, especially for morphology of the HA crystal. Briefly, in separate runs, approximately 0.05 g of the material to be analysed (pristine MWCNTs, functionalised MWCNTs and nanocomposite powders) were dispersed separately in distilled water and sonicated for 5 mins to create a stock dispersion. All electron microscope observations were made on 3 sub-samples of each stock. A drop of the relevant dispersion was placed on a copper grid, then air dried, and subsequently observed at an accelerating voltage of 120 kV using a high resolution transmission electron microscope (TEM, JEOL 1400, JEOL ltd, Japan). On each specimen, 30 random images were collected for quantification of dimensions of the materials. The images were processed using image J software (Windows version 1.48) to measure the length and diameter of the tubes. The same process was used to analyse the shape, size and growth of the HA crystals around MWCNTs powder.
Slight changes in the Ca:P ratio of the reaction mixture and/or sintering temperature during the production of nanocomposite powders can yield other types of calcium phosphates such as tri- and tetra-calcium phosphate. Hence, the phase purity of the sintered nanocomposite powders was determined by XRD. The patterns were obtained by an automated Philips powder diffractometer. Briefly, each specimen was irradiated using copper anode K (alpha) X-ray (CuKα, wavelength 1.5418 Å) generated by 45 kV and 40 mA of current in the cathode-ray source. All data were collected in a 2ϴ scan mode in the range of 20–40° using a step width of 0.03 Å and a 1 sec count time (see31 for discussion of XRD parameters).
The quality of the materials was also examined by FTIR. Briefly, FTIR involves shining a beam of light that contains many frequencies at the specimen, and then determining which frequencies are absorbed by the specimen. The pattern of the absorbed frequencies is characteristic of the molecular structure of the specimen (review,32). FTIR was used to especially determine the functional groups in the nanocomposite powders and the final composites which were measured for every new batch that was produced. A Bruker αP FTIR spectrometer (Bruker, UK) was used for the measurements in attenuated total reflection (ATR) mode with an ATR accessory utilizing diamond prism. Spectra were recorded using small amount (approximately 2–5 mg) of each sample (starting powders and the final composites), enough to cover the prism. The prepared nanocomposite powders did not require further processing, whereas the final composites were ground in a glass pestle and mortar to obtain a fine powder. Each spectrum was the result of 16 accumulated scans at 4 cm−1 resolution on the same sample.
Mechanical Testing
Mechanical tests were performed to determine the tensile and compressive strength (CS) of the produced composites. To determine the tensile strength, the diametral tensile strength (DTS) method was used. Diametral tensile strength is a property described by the American National Standards Institute (ANSI) for characterizing dental composites. For each material, five replicates of the final composites were prepared as explained in the “HA-MWCNTs composites” section to evaluate CS and DTS. The dimensions of the composites were 6.0 mm diameter × 12.0 mm height for the CS test and 6.0 mm diameter × 3.0 mm height for the DTS test. The specimens were tested under compressive load in a universal testing machine (3300 single column Instron, UK) at a crosshead speed of 1.0 mm/min for CS and 0.5 mm/min for the DTS test. For the CS test, the specimens were placed in a vertical position, with the force incident on the long axis of the specimen. For the DTS measurements, load was applied vertically on the lateral portion of the cylinder, producing tensile stress perpendicular to the vertical plane passing through the centre of the specimen.33 The DTS was calculated as follows:
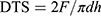
where F is the load applied; d is the diameter of the composite; h is the height of the composite; π = 3.14 (constant).
Experimental Design And Preparation Of The In Vitro Study
In vitro cultures of human osteoblasts were used to demonstrate the biocompatibility of the finished composites. Human osteoblast cells (406-05A; Sigma Aldrich, UK) were grown in Dulbecco’s modified eagle medium (DMEM; product number 11530596; Thermo Fisher scientific, Loughborough, UK) supplemented with 10% Fetal bovine serum (FBS, product number 11563397; Thermo Fisher scientific, Loughborough, UK) and 1% Antibiotic-antimycotic (contains penicillin, streptomycin and Gibco Amphotericin B; product number 15240–096; Thermo Fisher scientific, Loughborough, UK), referred to hereafter as “DMEM”. Cells were sub-cultured in 25 cm2 cell culture flasks until the required amount were available.
The experimental design involved exposing the osteoblast cells to the composites. Before exposing the cells, the composites were sterilized using Gamma radiation (dosage: 36.42–40.72). Then, using Sterile forceps, the sterile composites (d = 15mm) were carefully placed at the bottom of 24 well plate (flat bottom sterile, tissue culture treated polystyrene microplate; 662,160, Greiner bio-one, Stonehouse, UK). The cell culture plate containing all the controls and treatments, and was the unit of replication in the study design. Composites made of pure HA served as a positive control, while HOB cells grown in wells without the presence of any composites served as negative controls on each cell culture plate. In addition to these controls, the treatments were: p-MWCNTs-PVA, f-MWCNTs-PVA, p-MWCNTs-HTAB, f-MWCNTs-HTAB. Since the unit of replication was the cell culture plate with each plate contained all the materials and controls, a total of nine plates were prepared to give n = 9 replicates. To ensure the cells used were representative, the experiments were repeated on three separate days with different aliquots of cells from the stock cultures (i.e., triplicate runs of triplicate plates with all material to make nine replicates). Each well was seeded with 1.2×104 cells/well and the microplates were incubated for seven days at 37°C under 5% CO2 in atmospheric air to allow the cells to grow. The DMEM media was changed at 24 and 96 hrs to ensure the culture conditions were not limiting. Changes in pH and presence of lactate dehydrogenase activity (LDH) were measured from the collected media at the three time points (1st, 4th and 7th day). In addition, six of the replicate plates were used to make cell homogenates for biochemistry and three plates were left intact for electron microscope observation on the cells (see below).
For biochemistry, cell homogenates were prepared in hypo-osmotic buffer at the end of the experiment according to Gitrowski et al34. Briefly, cells were carefully washed twice with 2 mL of isosmotic sucrose buffer (300 mmol/L sucrose, 0.1 mmol/L ethylenediaminetetraacetic acid (EDTA), 20 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), buffered to pH 7.4 with a few drops of 2 mol/L Trizma base). The cells attached to each composite were then treated with 1 mL of a lysis buffer (hypo-osmotic version of the buffer above consisting of 30 mmol/L of sucrose). The lysis buffer containing the cell homogenate was sonicated for 30 s to ensure the lysed sample was mixed well. Fresh aliquots of the cell homogenate were used for the assessment of LDH, alkaline phosphatase activity (ALP), the protein content and the electrolyte composition (Na+, K+, Ca2+, P+ and Mg2+) of the cells.
Biochemical Indicators Of Cell Health
The presence of lactate dehydrogenase activity in the external media (DMEM) was used to assess the viability of the cells in terms of membrane integrity (i.e., membrane leak of this normally cytosolic enzyme). In addition, the LDH activity in the cell homogenates was also measured to indicate the metabolic status of the cells. Briefly, LDH activity was measured according to Campbell et al,35 with minor modification. The collected DMEM, and cell homogenates, were gently centrifuged for 1 min at approximately 13,000 ×g (Heraeus pico 17 centrifuge, Thermo electron crop, UK) to remove debris. Then, 100 μL of the supernatant from each sample was added to 2.9 mL of a reaction mixture consisting of 2800 μL of 6 mmol/L pyruvate in 50 mmol/L of phosphate buffer (pH 7.4) and 100 μL of 6 mmol/L nicotinamide adenine dinucleotide (NADH, Melford Laboratories Ltd, Suffolk, UK), directly in a 3 mL cuvette. The change in absorbance was measured immediately over 2 mins at 340 nm (Helios β Spectrophotometer, UK). LDH activity was calculated using the extinction coefficient of NADH at 340 nm of 6.3 mmol/L and a 1 cm path length. The cell homogenate LDH was normalized with the homogenate protein content.
The total protein content in cell homogenates was measured using the Pierce BCA protein assay kit (Product number: 23227, Thermo Fisher scientific, Loughborough UK). The assay was performed according to the manufacturer’s instructions. Briefly, a working reagent was prepared by mixing reagents A and B in a 50:1 ratio. Twenty μL of the cell homogenate (in triplicate) was transferred to a 96-well microplate, to which 200 μL of the working reagent was added and carefully mixed. Plates were covered and incubated at 37°C for 30 mins and absorbances were read at 562 nm on microplate reader (OptiMax Tunable microplate reader, Molecular devices, UK) against bovine serum albumin protein standards.
Alkaline phosphatase activity in the homogenates was also measured. This enzyme is involved in the calcification of bone and is therefore of functional significance to osteoblasts as developing bone cells. Alkaline phosphatase activity is, therefore, useful to assess the health and functionality of osteoblast. The activity of alkaline phosphatase enzyme was measured using a colorimetric assay based on the hydrolysis of para-nitrophenylphosphate (pNPP) to p-nitrophenol (PNP), a yellow-coloured substrate.36 Briefly, 65 μL of the cell homogenate was added to 595 μL of the reagent assay (265 μL of 0.1 mol/L glycine buffer + 330 μL of 0.5 mmol/L pNPP in glycine buffer). The appearance of p-nitrophenol was measured spectrophotometrically at 405 nm (Helios β Spectrophotometer, Thermo scientific, England). ALP activity was calculated using an extinction coefficient of 18.3 mmol/L for a path length of 1 cm. The cell homogenate ALP activity was normalized with cell protein content as above.
Electrolytes In The Culture Media And Cell Homogenates
The total concentration of Na+, K+, Ca2+, P and Mg2+ was determined in the DMEM media after 1, 4 and 7 days, and in the cell homogenates at the end of the experiment. The electrolyte measurements were made in the context of the osmotic health (ie, electrolytes involved in cell volume control and integrity of the cells), as well to inform on the presence of the minerals needed for bone formation. For electrolyte analysis, 400 µL of the external media from each well was acidified with 20 µL of 70% nitric acid. For the cell homogenate, 800 µL from each sample was taken and acid digested with 1 mL of 70% nitric acid and left overnight for complete acid digestion. Then, Na+, K+ and Ca2+ P and Mg2+ of each sample were determined using inductively coupled plasma optical emission spectroscopy (ICP-OES, iCAP 7400 RADIAL, Hemel Hempstead, UK) against matrix-matched standards. Sample runs included blanks to correct for instrument drift and procedural blanks to correct for background metal concentrations. The concentration of electrolytes was expressed as mmol/L in the media, and as µmol/mg cell protein in the cell homogenates.
Cell Morphology
Morphology (shape and appearance) of the cells was regularly observed by light microscope to determine the health of the cells. The DMEM media appeared normal with no loss of the pH indicator or excessive cell debris. Light microscopy observations showed no signs of deterioration of the cells such as; necrosis, detachment of cells from the substrate, granularity around the nucleus or obvious disruption of the cell membrane (i.e., no membrane blebs or cell swelling). At the end of the experiment, the presence and health of the cells were determined using a scanning electron microscope (SEM, JEOL JSM −5600LV, JEOL ltd, Japan). For logistical reasons, a separate run (n = 3 replicates) was done for the SEM work. After the media was removed, samples were washed twice with phosphate-buffered saline (PBS) and fixed using 2.5% glutaraldehyde in 0.05 mol/L cacodylate buffer at pH 7.4 for 2 hrs. Fixed samples were dehydrated through a series of ethanol and then critical point dried. Samples were mounted on conducting carbon stubs and coated with gold in a sputter coater (EMITECH K550, Quorum Technologies, UK). SEM images were collected using a 15 kV accelerating voltage. The observations were conducted systematically, starting at a lower magnification (×30) to examine the distribution of the cells on the composites, and then at a higher magnification (×1000) to observe the morphology of the cell membrane, organelles and nucleus as well as to determine the attachment of cells on the composites.
Statistics
All data are presented as mean  standard error and were analysed using statgraphics software for windows (version XVI.I). After descriptive statistics to determine normality, skewness or kurtosis, parametric data were analysed by ANOVA. Briefly, after a variance check (Levene’s test) and one-way ANOVA was conducted followed by Tukey’s multiple range test to identify the locations of any differences. The differences between the treatments and controls at each time point, and time effects within treatment were evaluated using one-way analysis of variance (ANOVA). Non- parametric data were analysed by the Kruskal–Wallis test. For treatment × time effects, a two-way ANOVA was also applied to the data. All statistical analysis used the default 95% confidence limit for statistical significance.
standard error and were analysed using statgraphics software for windows (version XVI.I). After descriptive statistics to determine normality, skewness or kurtosis, parametric data were analysed by ANOVA. Briefly, after a variance check (Levene’s test) and one-way ANOVA was conducted followed by Tukey’s multiple range test to identify the locations of any differences. The differences between the treatments and controls at each time point, and time effects within treatment were evaluated using one-way analysis of variance (ANOVA). Non- parametric data were analysed by the Kruskal–Wallis test. For treatment × time effects, a two-way ANOVA was also applied to the data. All statistical analysis used the default 95% confidence limit for statistical significance.
Results
Physico-Chemical Characterization Of The Dispersed Powders And Final Composites
The prepared dispersions of each of the nanocomposite powders were examined for morphology by electron microscopy and characterised by XRD and FTIR. In addition, the final composites which were made by mixing the powders with PVA and compressed into pellets were also studied by FTIR. TEM analysis (Figure 1) of the nanocomposite powders dried from the stock dispersions shows the presence of nano HA in all the samples. The HA crystals exhibited the typical needle-like morphology as expected in the pure HA control, p-MWCNTs-PVA and f-MWCNTs-PVA nanocomposite powders (Figure 1A–C, respectively). The crystal structure of nanocomposite powders made in the presence of HTAB surfactant (p-MWCNTs-HTAB and f-MWCNTs-HTAB; Figure 1D and E) show short nano-rod like structures. The difference in length of the overall powder rod-like HA structure is detailed in Table 1 with the pure HA powder exhibiting maximum size and the powders containing HTAB surfactant exhibiting the least size. Figure 1 shows that, in all the treatments, the HA had nucleated and grown around the MWCNTs.
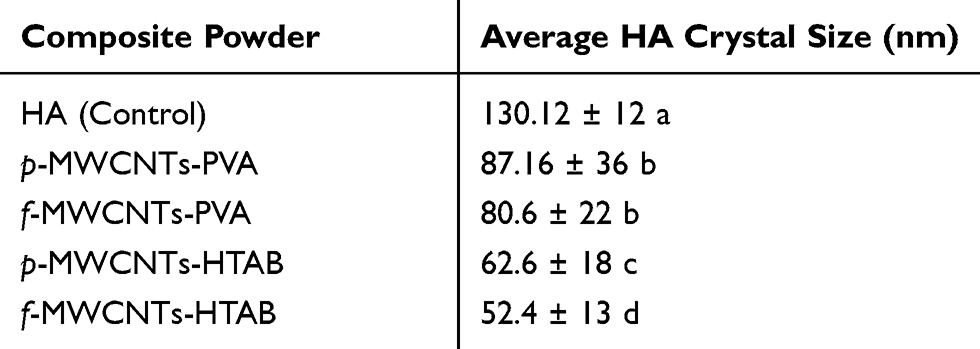 |
Table 1 Average HA Crystal Size Of The Prepared Nanocomposite Powders* |
 |
Figure 1 Representative transmission electron microscopy images of stock dispersions of the prepared composite powders depicting the difference in HA particle structure and interaction with the CNTs (n = 3 samples/treatment for each batch); (A) Pure HA crystals; (B) p-MWCNTs-PVA; (C) f-MWCNTs-PVA; (D) p-MWCNTs-HTAB; (E) f-MWCNTs-HTAB; (F) p-MWCNTs-HTAB (closer observation). In panel (A), the arrows point to the appearance of needle-shaped particles; in panels (B–D) the arrows point to the interaction between the HA and the CNTs. Note, in panel (B) the clusters of needle like HA surrounding the CNTs. In panel (D, E), the growth of HA crystal and the CNTs shows that the crystals have not obtained the needle structure, but have a rod shape (F). |
XRD analysis was performed to determine the crystal structure and phase purity of the HA in the nanocomposite powders during the TEM work. The XRD spectrums of the powders are shown (Figure 2). The data obtained from XRD are consistent with the morphology observed in the TEM images, which indicates that the HA phase was produced in all the samples. Broad diffraction peaks were observed in all the nanocomposite powders, indicating the presence of nanocrystals.
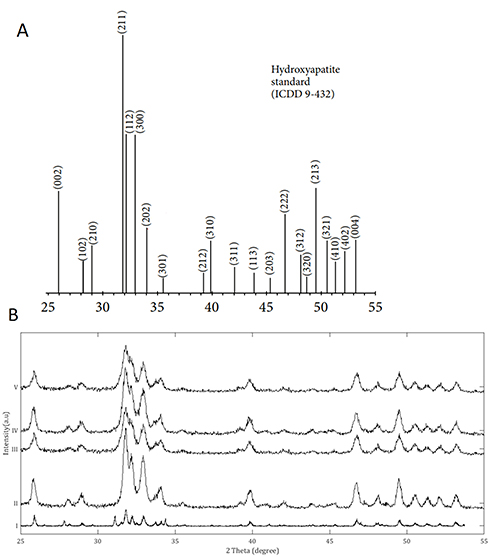 |
Figure 2 X-Ray Diffraction analysis of the nanocomposite powders dried from the stock dispersions prepared for transmission electron microscopy. (A) XRD peaks data for a HA certified reference material from the ICDD no. 9–432;50 (B) XRD analysis of the powders: i) Pure HA control; ii) p-MWCNTs-PVA; iii) f-MWCNTs-PVA; iv) p-MWCNTs-HTAB; v) f-MWCNTs-HTAB. The peaks observed in the powders correspond to the reference HA sample from the ICDD. The spectra lines are from triplicate measurement on two batches. |
FTIR spectroscopy is an effective tool for determining the presence of functional groups. Figure 3 represents the FTIR absorption peaks of the prepared nanocomposite powders and the final composites. All the major characteristic bands of HA are observed in all the nanocomposite powders as shown in Figure 3A. Additionally, a peak at 2917 cm−1 was observed in all the powders, except the pure HA. This is due to C–H bond which is expected from the presence of MWCNTs. The intensity of the peaks suggests a chemisorption nature of the bond. FTIR analysis of the final composites (Figure 3B) shows that all the peaks present in the powders exist in the composites. However, the peak corresponding to the MWCNTs was not visible above the background, and this is not surprising given the µg amounts of MWCNTs in the composites. An important peak was verified at 1141 cm−1 that is attributed to the crystallinity of the PVA. The peaks corresponding to PVA (reference spectra) were observed in all the composites.
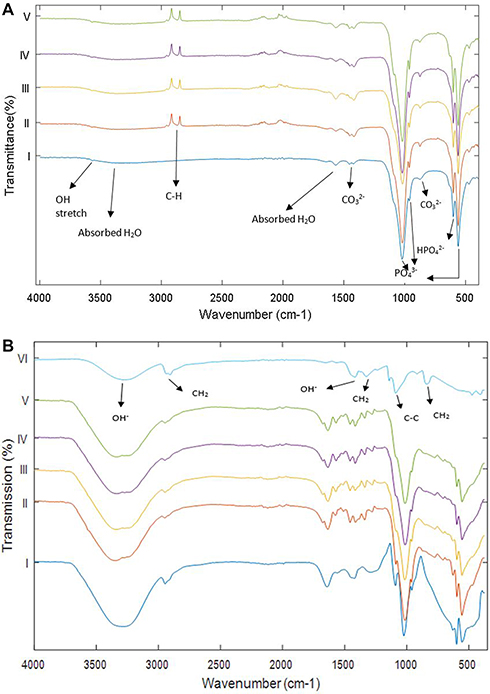 |
Figure 3 (A) Fourier Transform Infrared Spectroscopy analysis of the prepared powders and (B) the final composites. i) Pure HA control; ii) p-MWCNTs-PVA; iii) f-MWCNTs-PVA; iv) p-MWCNTs-HTAB; v) f-MWCNTs-HTAB; vi) PVA (reference). Arrows point to the absorbance peaks for the functional groups present in the powders and the composites. The spectra lines are from triplicate measurement on two batches. |
The tensile and compressive strength test results of the pure HA nanocomposite (control) and all the final composite treatments (p-MWCNTs-PVA, f-MWCNTs-PVA, p-MWCNTs-HTAB and f-MWCNTs-HTAB) after 48 hrs curing are presented in Table 2. The tensile strength of all the final composites was significantly higher than pure HA composites. There was also a significant difference in the tensile strength between all the composites. The maximum tensile strength was observed in the composite made of p-MWCNTS-PVA which was an increase of 215% compared to the pure HA composite. The least tensile strength among the composites containing MWCNTs was observed in the p-MWCNTs-HTAB, which was 49% less than the p-MWCNTS-PVA composite. The maximum compressive strength was also observed from the composite made of p-MWCNTs-PVA, which was significantly higher than the other composites. This was an increase of 100% compared to the pure HA composite. Unlike tensile strength, no significant difference in compressive strength was observed between the composites containing HTAB surfactant, but they were significantly lower compared to the composites containing PVA.
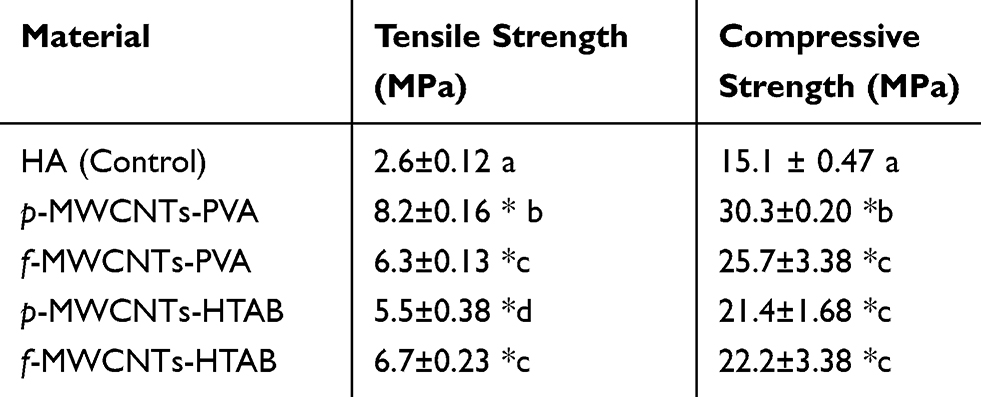 |
Table 2 Comparison Of The Tensile And Compressive Strength Of The Final Composites** |
Morphology And Health Of Cultured Cells
Light microscopy was used to visually monitor cell health during the experiment. The cultured wells after 24 hrs showed no evidence of cell debris, no detachment of cells from the substrate or gross changes in morphology. Routine light microscopy during the rest of the experiment showed cells with normal morphology and with intact cell membranes. The pH of the cell culture media also remained circumneutral, with only a slight acidity (pH 6.8) in the first 24 hrs in the HA and p-MWCNTs-PVA treatments that was quickly corrected at the first media change and remained neutral thereafter. The average pH of the media over the seven days were: media (no cells), 7.7 ± 0.02; blank (cells in DMEM), 7.80 ± 0.03; HA, 7.20 ± 0.05; p-MWCNTs-PVA, 7.12 ± 0.13; f-MWCNTs-PVA, 7.34 ± 0.06; p-MWCNTs-HTAB, 7.41 ± 0.03; f-MWCNTs-HTAB, 7.34 ± 0.03. At the end of the experiments, cells were observed in more detail by SEM (Figure 4). After seven days, the cells displayed a typical osteoblast morphology with regular cell structures in the controls and the treatments. No obvious morphological differences were observed between the cells maintained on the various treatments (Figure 4). The cells had intact homogenous cytoplasm and were attached and flattened on the substrate surface. In all the treatments, the cells spread to cover essentially all available regions of the dish. Since the treatments were on uneven surfaces, the cells were often seen to form bridges between the gaps, as expected for healthy osteoblasts.
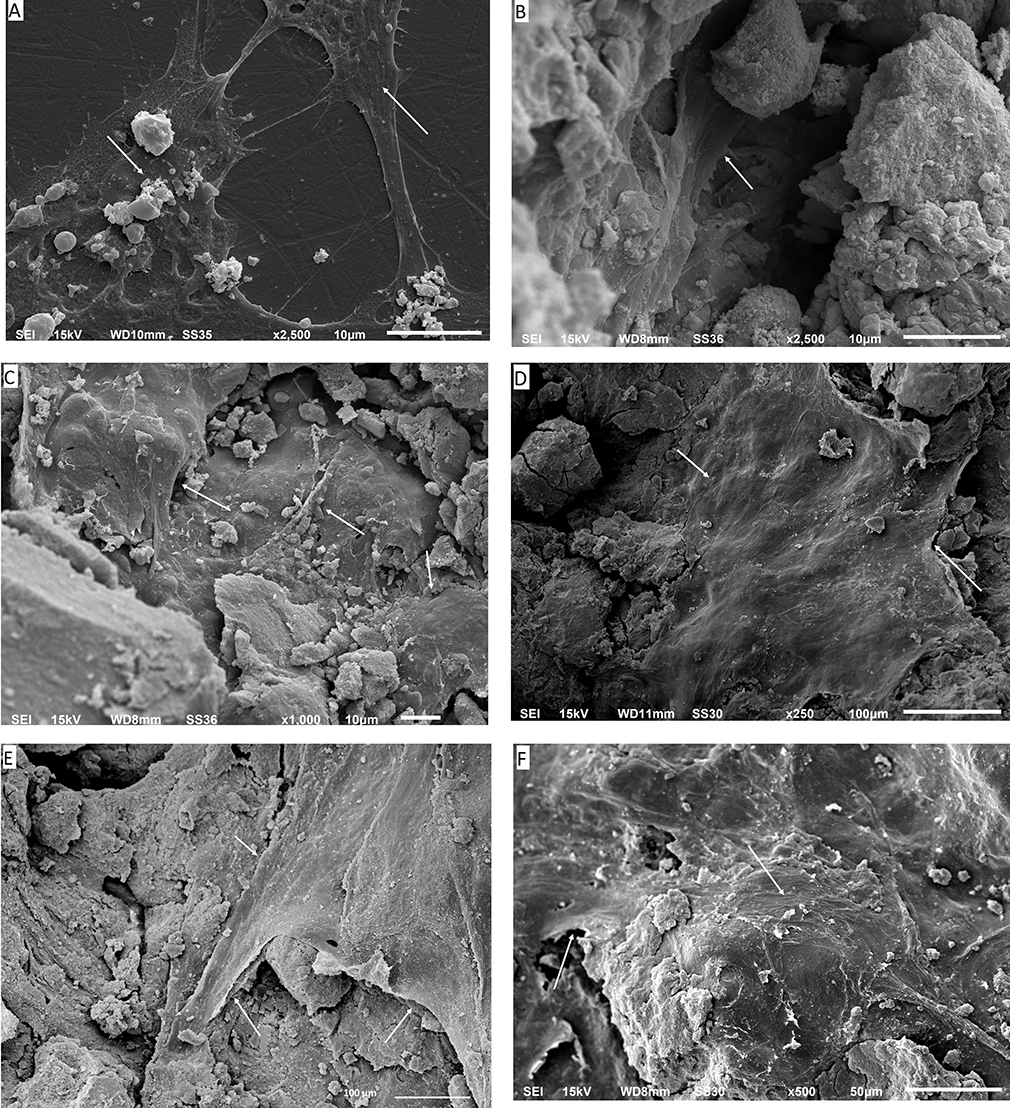 |
Figure 4 Scanning electron microscopy images of the osteoblasts in various magnifications. (A) Control cells grown on the plastic plate; (B) cells grown on pure HA as a control. It can be seen that the cells have infiltrated into the pores; (C) shows cells grown on p-MWCNTs-PVA; (D) shows cells on f-MWCNTs-PVA, note that the cells are flat and extending to cover large surfaces of the substratum; (E) shows cells on p-MWCNTs-HTAB; (F) shows cells on f-MWCNTs-HTAB. The arrows show the presence of the cells on the composites. |
Cell health was also confirmed biochemically by the absence of progressive LDH leak into the external media, with LDH in the media remaining at a background level (<0.1 µmol/min/L, Figure 5). The LDH activity in the cell homogenates was also modest, and although there were some statistical differences between treatments (Figure 5), the LDH activity remained <1 µmol/min/mg protein with no evidence of induction of anaerobic metabolism by the cells. There was also no evidence of osmotic disturbances in the cells with no differences amongst the treatments in the bulk electrolytes (Na+, K+) within the cells (Table 3). The presence of alkaline phosphatase activity in the cell homogenate was used as a measure of metabolically active osteoblast cells (Figure 5). At the end of the experiment, all treatments showed the presence of alkaline phosphatase activity, and although there were some differences between treatments, the activity remained below 0.1 µmol/min/mg protein (Figure 5).
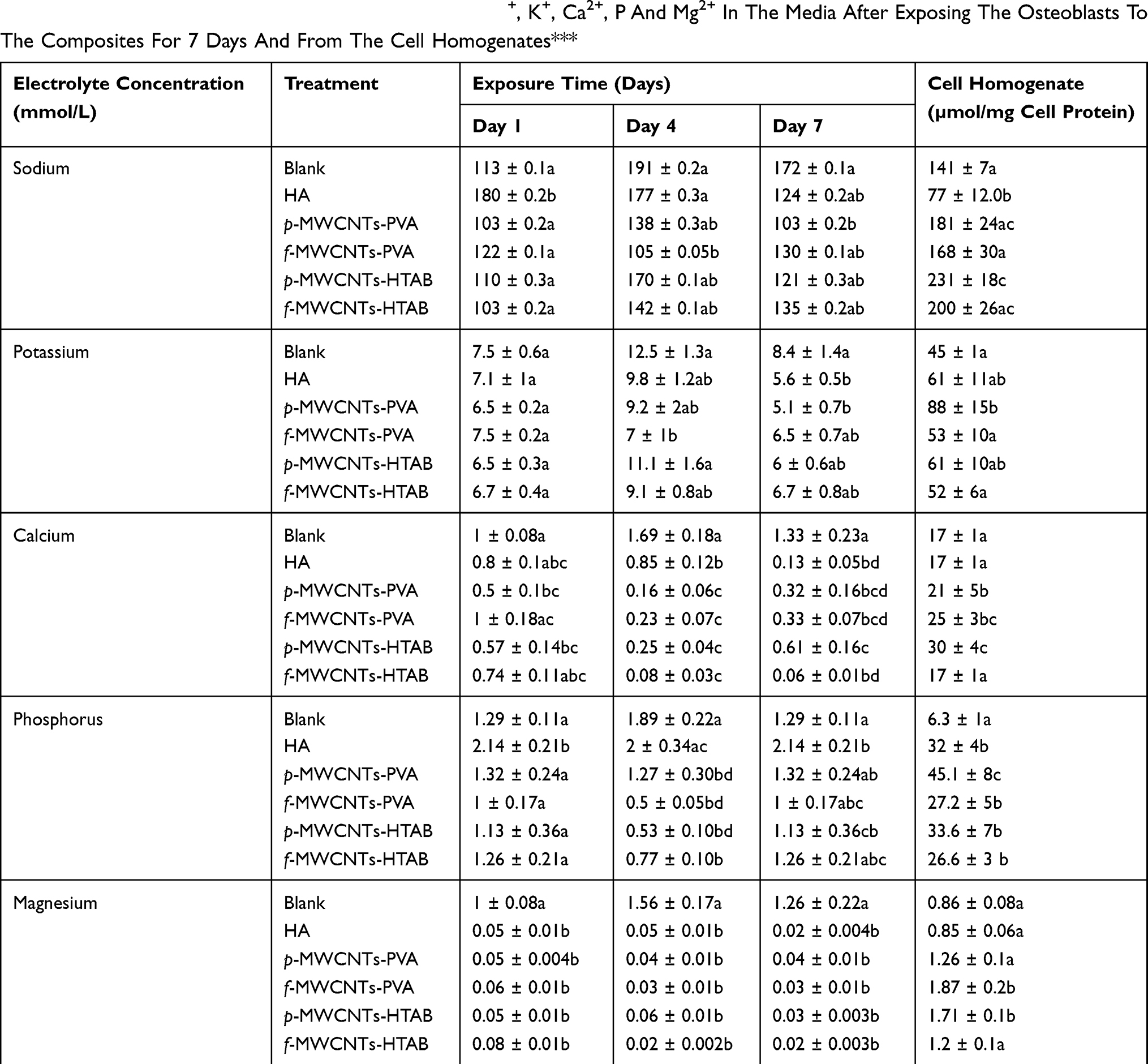 |
Table 3 The Total Concentration Of The Electrolytes, Na+, K+, Ca2+, P And Mg2+ In The Media After Exposing The Osteoblasts To The Composites For 7 Days And From The Cell Homogenates*** |
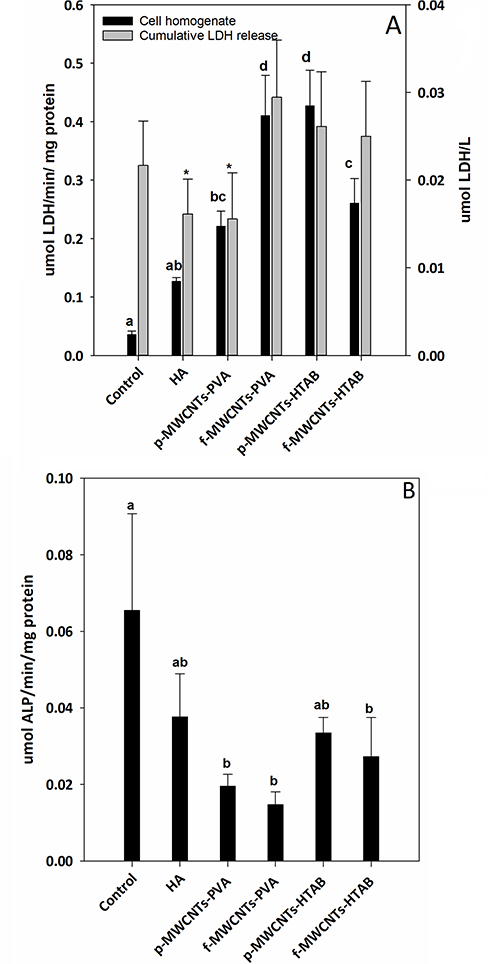 |
Figure 5 LDH and ALP activity release from osteoblast cells after 7 days of growth in DMEM on culture plate. (A) LDH activity released into the media, and the cell homogenate LDH activity for all the treatments and the control. (B) Alkaline phosphatase (ALP) activity from cell homogenates. Data are mean ± S.E.M (n = 6), bars with “*” indicate statistical significance and the different letter a, b, c, d indicates statistical difference from each other (one-way ANOVA, P <0.05). |
Discussion
This study reports the synthesis of MWCNTs reinforced HA composites and the effect of MWCNTs and surfactants on the overall biocompatibility and mechanical properties of the composites. Overall, the data confirm that reinforcing HA with MWCNTs improves the mechanical properties of the composites significantly; the addition of surfactants plays a major role in the formation of HA crystals, and the subsequent strength of the HA–MWCNTs composites has at least doubled, compared to the control. The biocompatibility study demonstrated cells with healthy morphology, with no evidence of LDH leak and the capability to produce ALP activity.
HA Crystal Nucleation And Growth In The Presence Of MWCNTs
The TEM images (Figure 1) demonstrate that HA-MWCNTs nanocomposite was successfully synthesized. However, a difference in the growth of HA on the sidewalls of pristine and functionalized MWCNTs was observed. Relatively higher amount of HA interaction was observed with functionalised MWCNTs compared to pristine MWCNTs. This is due to the fact that functionalising CNTs with nitric acid creates negative charge (–COO−) on the sidewalls of the CNTs which acts as nucleation sites for HA. The Ca2+ ions get attracted to the negative charge allowing the growth of HA on functionalised CNTs. Similarly, the presence of surfactants (PVA and HTAB) also influences the nucleation and growth of HA.
Preliminary trials on the synthesis of HA in the presence of MWCNTs without surfactants were not successful at producing HA at all (not reported here); but the procedure was achieved with the additions of either PVA or HTAB (Figure 1). The synthesis is, therefore, best achieved in the presence of dispersing agent. Of these surfactants, PVA is the preferred choice as only HA synthesised in the presence of PVA produced nano-HA crystals that were the expected needle shaped. Critically, the needle-like morphology of the HA (Figure 1) is consistent with the HA crystals in human bone.37 The needle-like morphology of HA in the presence of PVA could be explained by ligand chemistry. The Ca2+ ions from the additions of calcium acetate likely electrostatically bind to the OH− groups on the PVA polymer forming a –OH-Ca+ moiety (ie, a net positive charge on the surface of the PVA) which is then capable of attracting PO43− ions from the subsequent additions of ammonium phosphate. Thus, the PVA regulates the growth of the HA crystals along the c-axis of the polymer giving the crystals the needle structure.38,39 In the present study, the biocompatibility of the dispersants was also a consideration and a low concentration of PVA was chosen for the study (0.05 wt%). This low concentration of PVA enabled nucleation of the HA to occur with the expected needle-like morphology. Interestingly, Rajkumar et al, have reported that if the concentration of PVA in the solution is increased by 2.5 wt%, it results in the formation of crystals with irregular structures combined with a reduction of the crystal size.38
In contrast to PVA, HTAB being an organic cationic surfactant does aid the dispersion of the MWCNTs in the mixture; but does not promote nucleation. This is because HTAB is positively charged like the Ca2+ involved in the nucleating HA, and will therefore cause a coulombic repulsion.40 Hence, the interaction of the surfactant and HA is weaker compared to that of PVA. However, the absorption density of HTAB on HA is higher compared to anionic surfactants due to the lateral mobility of HTAB. Owing to the higher absorption density of HTAB and the presence of coulombic repulsion, small HA particles are more stable in aqueous solution as observed. However, the presence of PVA or HTAB in the HA-MWCNTs powders cannot be verified using XRD and FTIR due to the minuscule amount being used during the synthesis process.
Factors Contributing To The Mechanical Properties
The main purpose of synthesizing the HA-MWCNTs composites is to improve the mechanical properties of synthetic HA. Significant increase in both tensile and compressive strength of the composites containing MWCNTs was observed compared to control (pure HA) (Table 2). This is due to the interfacial bonding between the HA and the MWCNTs. However, variation in both tensile and compressive strength can be observed between the composites containing MWCNTs. Although literature suggests that functionalisation of MWCNTs improves their wettability which in turn enables the interaction with the HA particles,20 composites containing f-MWCNTs have not exhibited the maximum tensile or compressive strength. This could be explained by the fact that the acidic oxidative treatment causes major alteration to the structural properties of the CNTs. Garg et al, developed a simulation model to predict the effect of acid treatment on the mechanical properties of CNTs. They have showed that acid treatment introduces carbon defects that degrade the mechanical strength of CNTs by an average value of 15%.41 Composites made of p-MWCNTs-PVA have shown the maximum tensile and compressive strength (Table 2). There are two factors contributing toward the higher mechanical properties with pristine MWCNTs. The absence of defective sites in the sidewalls of the MWCNTs contributes to the overall strength of the final composite. The HA particles are significantly longer with a needle shape which contributes to the improved mechanical properties of p-MWCNTs-PVA composites. However, while comparing the interaction of HA particles with MWCNTs (Figure 1) to the mechanical strength of the composites (Table 2), it appears that there is no direct correlation between the two, since, composites containing f–MWCNTs show high degree of interaction with HA particles.
Effects Of Composites On DMEM Media And Cell Health
The pH and electrolyte concentrations were measured in the external media for several reasons. Firstly it is to confirm that the presence of the composite had not compromised the osmotic status of the DMEM in relation to cell health, but also to recognise that synthetic HA contains high concentrations of calcium phosphate that could be released from the material into the external media (e.g., by degradation of the material and/or dissolution). Table 3 shows that the concentration of calcium in the media was significantly lower than the negative control, confirming that there was no dissolution of the composite; but in fact, calcium and phosphorus have been adsorbed from the DMEM, most likely for apatite nucleation. Kokubo et al, have shown apatite nucleation and growth of HA in Simulated Body Fluid (SBF) at physiologic temperature and pH.42 However, the decreasing concentration of calcium in the DMEM observed here was modest and the total Ca concentrations remained in the sub-mmol/L range (Table 3). It was not limiting cell growth or health as confirmed by the normal cell morphology (Figure 3), as well as the presence of expected enzyme activities (ALP and LDH activity, Figure 4). Indeed, leaky cells would be vulnerable to Ca-overload and cell death from a massive inward Ca gradient (the intracellular free Ca2+ concentration is around 0.1 µmol/L in osteoblast43), but this was not observed. However, compared to the HA alone treatment, some of the cell homogenates from the composites showed a higher total Ca concentration (Table 3). This can be interpreted as physiological, where the cells are storing more total intracellular Ca consistent with their role in mineralisation of bone. Similar to calcium, significantly higher quantities of phosphorus were observed in the cell homogenate of the composites suggesting that the osteoblasts were reserving calcium and phosphorus for the mineralization process. The observations on cell homogenates are also corroborated by the reduction of phosphorus in the external media (Table 3).
The concentrations of sodium and potassium in the external media were in the normal range (Table 3). Since sodium makes most of the osmotic content of the media, any changes in the cell homogenates are not likely to be explained by osmotic stress such as loss of volume control (cell swelling not observed), and in any case, the data are normalised to cell protein. Homogenate sodium and potassium concentrations are rarely measured from osteoblasts, but they are broadly similar to homogenates of mammalian cells with nanomaterials (e.g.,34). Interestingly, the HA alone treatment did have a lower total sodium concentration in the homogenate compared to all other treatments (Table 3). This is not likely due to Ca/Na exchange activity on the cell membrane causing Na efflux from the cells,44 because the homogenate Ca was unchanged in this treatment. The homogenate potassium concentration also did not change in this treatment. The effect of the coatings on electrolyte homeostasis needs further investigation.
In addition to normal cell morphology (Figure 4) and the electrolyte concentrations in the cell homogenates, the biocompatibility of the composites was also assessed by measuring the LDH and ALP activity of the cells (Figure 5). Cell viability was confirmed by the absence of LDH leak into the external media. This is a well-established viability marker34 and the absence of leak of this cytosolic enzyme into external media shows that cell membranes are intact (i.e., not leaking). The absence of cumulative increases in LDH activity in the media also would indicate that there are no dead cells releasing their contents (i.e., that cell are alive and healthy). The cumulative LDH leak into the external media was small (<0.1 µmol/L, Figure 5) and a typical background for cell culture (e.g.,34); suggesting the cells were healthy without membrane damage. A modest LDH activity was detected inside the cells (the cell homogenates, Figure 5), as would be expected,45 but the absence of an elevated LDH activity in the homogenates also indicated that the cells were not under metabolic stress (i.e., not using anaerobic metabolism). Alkaline phosphatase activity was also measured (Figure 5B) and the values for the controls (about 60 nmol/mg protein/min) are broadly similar to those reported for osteoblasts (e.g.,46). However, the ALP activity showed some small, but statistically significant decreases in the presence of the composites (Figure 5B) which suggests that, although the cells were proliferated, the expression of ALP was not keeping up; and so some growth dilution of ALP activity may have occurred. The absence of large increases in ALP activity is not surprising in the seven-day duration of the current experiment as the cells were still relatively immature, and ALP induction occurs later on in the cell differentiation process.47 Nevertheless, the presence of ALP activity in the cell homogenate confirms that the cells were functional.
Conclusions And Clinical Perspective
This study successfully synthesised HA-MWCNTs composites and then demonstrated that healthy cells with normal morphology, electrolyte composition and enzyme activities could be grown on the materials over seven days. The study also shows that surfactants play a crucial role in determining the structure of the HA crystals and that the presence of MWCNTs had at least doubled the strength of the composites, with PVA surfactant proving the best coverage of HA over the composite material and the p-MWCNTs PVA composite the strongest material. All of the composites showed biocompatibility in that overt toxicity to osteoblast cells was not observed. However, of the composites examined, the f-MWCNTs HTAB was considered the most biocompatible with osteoblast cells as they had comparatively lower LDH activity release in the media and higher ALP activity in cell homogenate. These in vitro studies are a first step towards meeting the criteria for an acceptable nanomedicine or nanomaterial-containing implant for clinical use.48,49 This study shows the f-MWCNTs HTAB composite to be stronger than HA alone (i.e., better than the implant materials used now) and biocompatible, suggesting that it may at least be safe for bone cells in the short term. In terms of mechanical strengths, it is in line with callous bones with a compressive strength above 20 MPa and tensile strength 6–7 MPa. Therefore, this material can potentially be used as dental or small bone graft. It will need further improvement in mechanical strength to be used as a load-bearing cortical bone replacement. Further research also is needed to demonstrate the biocompatibility of the composites over longer time scales (e.g., several weeks) and then in vivo with an animal model before trials can begin with patients.
Data Sharing Statement
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.
Acknowledgments
This research was supported by Plymouth University Peninsula School of Medicine and Dentistry. We thank our colleague A. Besinis from the School of Engineering, Plymouth University who provided insight and expertise. We thank our colleagues from the Plymouth Electron Microscope Centre for their help with the Scanning and Transmission Electron Microscopy techniques. We would also like to show our gratitude to A. Atfield and L. Cooper from the School of Biological & Marine Sciences, Plymouth University for their assistance with the cell culture experiments.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kini U, Nandeesh BN. Physiology of bone formation, remodeling, and metabolism. In: Fogelman I, Gnanasegaran G, van der Wall H (eds) Radionuclide Hybrid Bone Imaging. Springer, Berlin, Heidelberg, 2012;29–57.
2. Poinern GEJ, Brundavanam RK, Fawcett D. Nanometre scale hydroxyapatite ceramics for bone tissue engineering. Am J Biomed Eng. 2013;3(6):148–168.
3. Woodard JR, Hilldore AJ, Lan SK, et al. The mechanical properties and osteoconductivity of hydroxyapatite bone scaffolds with multi-scale porosity. Biomaterials. 2007;28(1):45–54. doi:10.1016/j.biomaterials.2006.08.021
4. Šupová M. Problem of hydroxyapatite dispersion in polymer matrices: a review. J Mat Sci. 2009;20(6):1201–1213. doi:10.1007/s10856-009-3696-2
5. Murugan R, Ramakrishna S. Development of nanocomposites for bone grafting. Compos Sci Technol. 2005;65(15–16):2385–2406. doi:10.1016/j.compscitech.2005.07.022
6. Natesan K, Shah W, Le H, Tredwin C. A critical comparison on biocompatibility of different phases of sol-gel derived calcium phosphates as bone graft materials. J Biomater Tissue Eng. 2015;5(8):655–664. doi:10.1166/jbt.2015.1364
7. Yang Y, Kim K-H, Agrawal CM, Ong JL. Interaction of hydroxyapatite–titanium at elevated temperature in vacuum environment. Biomaterials. 2004;25(15):2927–2932. doi:10.1016/j.biomaterials.2003.09.072
8. Liu D-M, Yang Q, Troczynski T. Sol–gel hydroxyapatite coatings on stainless steel substrates. Biomaterials. 2002;23(3):691–698. doi:10.1016/s0142-9612(01)00157-0
9. Bellucci D, Sola A, Anesi A, Salvatori R, Chiarini L, Cannillo V. Bioactive glass/hydroxyapatite composites: mechanical properties and biological evaluation. Mat Sci Eng. 2015;51:196–205. doi:10.1016/j.msec.2015.02.041
10. Salehi S, Fathi MH. Fabrication and characterization of sol–gel derived hydroxyapatite/zirconia composite nanopowders with various yttria contents. Ceram Int. 2010;36(5):1659–1667. doi:10.1016/j.ceramint.2010.02.045
11. Mantripragada VP, Lecka‐Czernik B, Ebraheim NA, Jayasuriya AC. An overview of recent advances in designing orthopedic and craniofacial implants. J Biomed Mat Res Part A. 2013;101(11):3349–3364. doi:10.1002/jbm.a.34605
12. Holzwarth JM, Ma PX. Biomimetic nanofibrous scaffolds for bone tissue engineering. Biomaterials. 2011;32(36):9622–9629. doi:10.1016/j.biomaterials.2011.09.009
13. Robinson D, Alk D, Sandbank J, Farber R, Halperin N. Inflammatory reactions associated with a calcium sulfate bone substitute. Ann Transplant. 1999;4(3–4):91–97.
14. Elshereksi NW, Ghazali MJ, Muchtar A, Azhari CH. Perspectives for titanium-derived fillers usage on denture base composite construction: a review article. Adv Mat Sci Eng. 2014;2014:13. doi:10.1155/2014/746252
15. White AA, Best SM, Kinloch IA. Hydroxyapatite–carbon nanotube composites for biomedical applications: a review. Int J Appl Ceram Technol. 2007;4(1):1–13. doi:10.1111/j.1744-7402.2007.02113.x
16. Eatemadi A, Daraee H, Karimkhanloo H, et al. Carbon nanotubes: properties, synthesis, purification, and medical applications. Nanoscale Res Lett. 2014;9(1):1–13. doi:10.1186/1556-276X-9-1
17. Lahiri D, Ghosh S, Agarwal A. Carbon nanotube reinforced hydroxyapatite composite for orthopedic application: a review. Mat Sci Eng C-Mater. 2012;32(7):1727–1758. doi:10.1016/j.msec.2012.05.010
18. Shin US, Yoon IK, Lee GS, Jang WC, Knowles JC, Kim HW. Carbon nanotubes in nanocomposites and hybrids with hydroxyapatite for bone replacements. J Tissue Eng. 2011;2011:674287.
19. Khalid P, Hussain M, Rekha P, Arun A. Carbon nanotube-reinforced hydroxyapatite composite and their interaction with human osteoblast in vitro. Hum Exp Toxicol. 2015;34(5):548–556. doi:10.1177/0960327114550883
20. Khanal SP, Mahfuz H, Rondinone AJ, Leventouri T. Improvement of the fracture toughness of hydroxyapatite by incorporation of carboxyl functionalized single walled carbon nanotubes and nylon. Mat Sci Eng. 2016;60:204–210. doi:10.1016/j.msec.2015.11.030
21. Venkatesan J, Qian Z-J, Ryu B, Ashok Kumar N, Kim S-K. Preparation and characterization of carbon nanotube-grafted-chitosan – natural hydroxyapatite composite for bone tissue engineering. Carbohydr Polym. 2011;83(2):569–577. doi:10.1016/j.carbpol.2010.08.019
22. Maho A, Linden S, Arnould C, Detriche S, Delhalle J, Mekhalif Z. Tantalum oxide/carbon nanotubes composite coatings on titanium, and their functionalization with organophosphonic molecular films: a high quality scaffold for hydroxyapatite growth. J Colloid Interface Sci. 2012;371(1):150–158. doi:10.1016/j.jcis.2011.12.066
23. Abarrategi A, Gutierrez MC, Moreno-Vicente C, et al. Multiwall carbon nanotube scaffolds for tissue engineering purposes. Biomaterials. 2008;29(1):94–102. doi:10.1016/j.biomaterials.2007.09.021
24. Zanello LP, Zhao B, Hu H, Haddon RC. Bone cell proliferation on carbon nanotubes. Nano Lett. 2006;6(3):562–567. doi:10.1021/nl051861e
25. Dawei Z, Changqing Y, Jinchao Z, Yao C, Xinsheng Y, Mengsu Y. The effects of carbon nanotubes on the proliferation and differentiation of primary osteoblasts. Nanotechnology. 2007;18(47):475102. doi:10.1088/0957-4484/18/49/495102
26. Wojtek T, Ki H, Anatoly P, et al. Toxicity induced enhanced extracellular matrix production in osteoblastic cells cultured on single-walled carbon nanotube networks. Nanotechnology. 2009;20(25):255101. doi:10.1088/0957-4484/20/25/255101
27. Constanda S, Stan MS, Ciobanu CS, Motelica-Heino M, et al. Carbon nanotubes-hydroxyapatite nanocomposites for an improved osteoblast cell response. J Nanomater. 2016;2016:10. doi:10.1155/2016/3941501
28. Firme Iii CP, Bandaru PR. Toxicity issues in the application of carbon nanotubes to biological systems. Nanomed. 2010;6(2):245–256. doi:10.1016/j.nano.2009.07.003
29. Datsyuk V, Kalyva M, Papagelis K, et al. Chemical oxidation of multiwalled carbon nanotubes. Carbon. 2008;46(6):833–840. doi:10.1016/j.carbon.2008.02.012
30. Natesan K, Shah W, Le H, Tredwin C. A critical comparison on biocompatibility of different phases of sol–gel derived calcium phosphates as bone graft materials. J Biomater Tissue Eng. 2015;5(8):655–664. doi:10.1166/jbt.2015.1364
31. Klug HP, Alexander LE (eds). X-ray diffraction procedures. Wiley -Inderscience, New York. 1954;2.
32. Berthomieu C, Hienerwadel R. Fourier transform infrared (FTIR) spectroscopy. Photosynth Res. 2009;101(2):157–170. doi:10.1007/s11120-009-9439-x
33. Della Bona Á, Benetti P, Borba M, Cecchetti D. Flexural and diametral tensile strength of composite resins. Braz Oral Res. 2008;22(1):84–89.
34. Gitrowski C, Al-Jubory AR, Handy RD. Uptake of different crystal structures of TiO2 nanoparticles by Caco-2 intestinal cells. Toxicol Lett. 2014;226(3):264–276. doi:10.1016/j.toxlet.2014.02.014
35. Campbell HA, Handy RD, Nimmo M. Copper uptake kinetics across the gills of rainbow trout (Oncorhynchus mykiss) measured using an improved isolated perfused head technique. Aquat Toxicol. 1999;46(3–4):177–190. doi:10.1016/S0166-445X(99)00003-X
36. Sabokbar A, Millett PJ, Myer B, Rushton N. A rapid, quantitative assay for measuring alkaline phosphatase activity in osteoblastic cells in vitro. Bone Miner. 1994;27(1):57–67.
37. Kalia P, Vizcay-Barrena G, Fan JP, Warley A, Di Silvio L, Huang J. Nanohydroxyapatite shape and its potential role in bone formation: an analytical study. J R Soc Interface. 2014;11(93):20140004. doi:10.1098/rsif.2014.0004
38. Rajkumar M, Sundaram NM, Rajendran V. In-situ preparation of hydroxyapatite nanorod embedded poly (vinyl alcohol) composite and its characterization. Int J Eng Sci Technol. 2010;2(6):2437–2444.
39. Mollazadeh S, Javadpour J, Khavandi A. In situ synthesis and characterization of nano-size hydroxyapatite in poly(vinyl alcohol) matrix. Ceram Int. 2007;33(8):1579–1583. doi:10.1016/j.ceramint.2006.06.006
40. Chen C-W, Oakes CS, Byrappa K, et al. Synthesis, characterization, and dispersion properties of hydroxyapatite prepared by mechanochemical-hydrothermal methods. J Mater Chem. 2004;14(15):2425–2432. doi:10.1039/B315095J
41. Garg A, Sinnott SB. Effect of chemical functionalization on the mechanical properties of carbon nanotubes. Chem Phys Lett. 1998;295(4):273–278. doi:10.1016/S0009-2614(98)00969-5
42. Kokubo T, Ito S, Huang ZT, et al. Ca,P-rich layer formed on high-strength bioactive glass-ceramic A-W. J Biomed Mater Res. 1990;24(3):331–343. doi:10.1002/jbm.820240306
43. Schanne FAX, Long GJ, Rosen JF. Lead induced rise in intracellular free calcium is mediated through activation of protein kinase C in osteoblastic bone cells. Biochimica Et Biophysica Acta (BBA) Mol Basis Dis. 1997;1360(3):247–254. doi:10.1016/S0925-4439(97)00006-9
44. Stains JP, Gay CV. Asymmetric distribution of functional sodium-calcium exchanger in primary osteoblasts. J Bone Miner Res. 1998;13(12):1862–1869. doi:10.1359/jbmr.1998.13.12.1862
45. Morrison C, Macnair R, MacDonald C, Wykman A, Goldie I, Grant MH. In vitro biocompatibility testing of polymers for orthopaedic implants using cultured fibroblasts and osteoblasts. Biomaterials. 1995;16(13):987–992. doi:10.1016/0142-9612(95)94906-2
46. Kim GS, Kim CH, Park JY, Lee KU, Park CS. Effects of vitamin B12 on cell proliferation and cellular alkaline phosphatase activity in human bone marrow stromal osteoprogenitor cells and UMR106 osteoblastic cells. Metabolism. 1996;45(12):1443–1446. doi:10.1016/s0026-0495(96)90171-7
47. Collin P, Nefussi JR, Wetterwald A, et al. Expression of collagen, osteocalcin, and bone alkaline phosphatase in a mineralizing rat osteoblastic cell culture. Calcif Tissue Int. 1992;50(2):175–183. doi:10.1007/bf00298797
48. Juillerat-Jeanneret L, Dusinska M, Fjellsbo LM, Collins AR, Handy RD, Riediker M. Biological impact assessment of nanomaterial used in nanomedicine. Introduction to the NanoTEST project. Nanotoxicology. 2015;9(Suppl 1):5–12. doi:10.3109/17435390.2013.826743
49. Besinis A, De Peralta T, Tredwin CJ, Handy RD. Review of nanomaterials in dentistry: interactions with the oral microenvironment, clinical applications, hazards, and benefits. ACS Nano. 2015;9(3):2255–2289. doi:10.1021/nn505015e
50. Shahabi S, Najafi F, Majdabadi A, et al. Effect of gamma irradiation on structural and biological properties of a PLGA-PEG-hydroxyapatite composite. Sci World J. 2014;2014:9. doi:10.1155/2014/420616
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.