Back to Journals » OncoTargets and Therapy » Volume 7
Cancer cell-oriented migration of mesenchymal stem cells engineered with an anticancer gene (PTEN): an imaging demonstration
Authors Yang Z, Tang X, Guo X, Zou D, Sun X, Feng J, Luo J, Dai L ![]() , Warnock G, Wang J
, Warnock G, Wang J
Received 15 December 2013
Accepted for publication 3 January 2014
Published 17 March 2014 Volume 2014:7 Pages 441—446
DOI https://doi.org/10.2147/OTT.S59227
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
The supplementary video shows the real-time dynamic cell migration.
Views: 534
Zhuo-Shun Yang,1,* Xiang-Jun Tang,2,* Xing-Rong Guo,1 Dan-Dan Zou,1 Xu-Yong Sun,3 Jing-Bo Feng,1 Jie Luo,1 Long-Jun Dai,1,4 Garth L Warnock4
1Hubei Key Laboratory of Stem Cell Research, Taihe Hospital, Hubei University of Medicine, Shiyan, People’s Republic of China; 2Department of Neurosurgery, Taihe Hospital, Hubei University of Medicine, Shiyan, People’s Republic of China; 3Guangxi Key Laboratory for Transplant Medicine, 303 Hospital of PLA, Nanning, People’s Republic of China; 4Department of Surgery, University of British Columbia, Vancouver, BC, Canada
*These authors contributed equally to this work
Background: Mesenchymal stem cells (MSCs) have been considered to hold great potential as ideal carriers for the delivery of anticancer agents since the discovery of their tumor tropism. This study was performed to demonstrate the effects of phosphatase and tensin homolog (PTEN) engineering on MSCs’ capacity for cancer cell-oriented migration.
Methods: MSCs were engineered with a PTEN-bearing plasmid and the expression was confirmed with Western blotting. A human glioma cell line (DBTRG) was used as the target cell; DBTRG cell-oriented migration of MSCs was monitored with a micro speed photographic system.
Results: The expression of transfected PTEN in MSCs was identified by immunoblotting analysis and confirmed with cell viability assessment of target cells. The DBTRG cell-oriented migration of PTEN-engineered MSCs was demonstrated by a real-time dynamic monitoring system, and a phagocytosis-like action of MSCs was also observed.
Conclusion: MSCs maintained their capacity for cancer cell-directed migration after they were engineered with anticancer genes. This study provides the first direct evidence of MSCs’ tropism post-anticancer gene engineering.
Keywords: gene therapy, mesenchymal stem cells, phosphatase and tensin homolog, cancer
Introduction
Cancer is one of the most common life-threatening diseases, accounting for an estimated one in four human deaths.1 Despite improved treatment models, many tumors remain unresponsive to conventional cancer therapies. The major obstacle limiting the effectiveness of conventional therapies for cancer is their tumor specificity. An ideal therapeutic strategy would directly target tumors, in both primary and metastatic sites, and possess the ability to act locally over a sustained period of time.
Mesenchymal stem cells (MSCs) were first identified in the stromal compartment of bone marrow by Friedenstein et al in the 1960s.2,3 MSCs have generated considerable biomedical interest since their multilineage potential was first identified in 1999.4 Owing to their easy acquisition, fast ex vivo expansion, and the feasibility of autologous transplantation, MSCs have become the first type of stem cells to be utilized in clinical applications. Recent findings on specific tumor-oriented migration and incorporation of MSCs demonstrate the great potential for MSCs to be used as an ideal carrier for anticancer gene delivery.5–7 Tumor-directed migration and incorporation of MSCs have been demonstrated by a number of preclinical studies using both transwell migration assays (in vitro) and animal tumor models (in vivo). The homing capacity of MSCs has been demonstrated with almost all tested human cancer cell lines, including lung cancer,8 malignant glioma,9 breast cancer,10 colon carcinoma,11 pancreatic cancer,12,13 melanoma,14 and ovarian cancer.10 The high frequency of MSC migration and incorporation was observed during in vitro coculture and in vivo xenograft tumors, respectively; these findings were found to be consistent, and were independent of tumor type, immunocompetence, and delivery route of MSCs.
In order to utilize MSCs as the therapeutic vehicles for cancer treatment, it is critical to maintain the tumor-tropic capacity of these cells. However, whether such a homing property is altered by the engineering process of adding anticancer genes to the MSCs remains largely unknown. In our recent pancreatic cancer study, the cancer cell-oriented migration of MSCs was demonstrated using a real-time cell analyzer system (xCELLigence; Roche Diagnostics, Indianapolis, IN, USA).13 In the present study, imaging techniques were utilized to directly verify the cancer cell-oriented migration of MSCs which were engineered with an anticancer gene, phosphatase and tensin homolog (PTEN), which functions as the central negative regulator of the PI3K-AKT-mTOR (phosphoinositide 3-kinase – protein kinase B – mammalian target of rapamycin) pathway in controlling apoptosis.
Materials and methods
Cells and culture conditions
MSCs were isolated from human pancreas and expanded ex vivo as previously described.13,15 Based on the minimal criteria for defining human MSCs established by the International Society of Cellular Therapy, theses MSCs were verified by both membrane biomarker determination and functional differentiation. They fulfilled the characteristics of human MSCs, exhibiting positive expression of cluster of differentiation (CD) 44+, CD73+, CD95+, and CD105+, and negative expression of CD34–. The results of adipogenic and osteogenic differentiation also met the standards. MSCs were cultured in Minimum Essential Medium (MEM) with 10% fetal calf serum, 2 mM L-glutamine, and 1% penicillin-streptomycin solution (all from Life Technologies, Carlsbad, CA, USA) and incubated at 37°C in a humidified, 5% CO2 atmosphere. A human glioma cell line (DBTRG) was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and used as target cells in the present study. DBTRG cells were maintained as suggested by ATCC and their culture condition was kept consistent with MSCs.
Construction of PTEN-bearing expression vector
Mammalian expression plasmid pDsRed1-N1 was used as the backbone structure. An 18 amino acid leading sequence (MKFPSQLLLLLLFGIPGM) and an eleven amino acid transacting activator of transcription (TAT, YGRKKRRQRRR) were inserted at the multiple cloning site followed by human PTEN (403 amino acids). The fusion protein was designated as TAT-PTEN-red fuorescent protein (RFP) with the predicted molecular weight of 74 kDa.
Plasmid transfection of MSCs
PTEN-bearing expression plasmid was transfected into MSCs with an electroporation method. MSCs were collected and washed with Opti-MEM (Life Technologies, Carlsbad, CA, USA) three times, then resuspended with Opti-MEM at a density of 1×107cells/mL. MSCs suspension (90 μL) was placed into each cuvette and 10 μg plasmid (1 μg/μL) was added. The electroporation was performed according to the manufacturer’s instruction (NEPA21; Nepa Gene Co, Ltd., Chiba, Japan). After a series of trials on this particular cell type, the most optimized transfection conditions were defined as a voltage of 150 V and a 5 millisecond pulse length. The PTEN-engineered MSCs were designated as MSCPTEN. Two days after transfection, the culture media were collected as conditioned media for coculture studies.
Immunoblotting and enzyme-linked immunosorbent assay (ELISA) analysis
Immunoblotting analysis was used to detect the cellular expression of PTEN in the MSCs. The MSCs transfected with PTEN or mock infected were harvested in lysis solution. Whole-cell lysates (50 μg) were separated through 12% denaturing sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. The membrane was incubated overnight with mouse anti-PTEN antibody (1:1000; R&D Systems Inc., Minneapolis, MN, USA). A separate membrane was prepared and incubated overnight with mouse anti-red fluorescent protein (RFP) antibody (1:1000; Abcam PLC, Cambridge, England). This was followed by a 1 hour incubation with goat-anti-mouse immunoglobulin (Ig)G conjugated to horseradish peroxidase (HRP) (1:2500). The blots were developed using enhanced chemiluminescence detection (Amersham Bioscience, Baie D’Urfe, QC, Canada).
The soluble PTEN in the culture supernatants was measured using ELISA as per the manufacturer’s instruction (Santa Cruz Biotechnology Inc., Dallas, TX, USA). Conditioned media collected from the corresponding cultures were equally concentrated using 10,000 molecular weight cut off (MWCO) (cat # 42406; EMD Millipore, Billerica, MA, USA) and protein concentrations were determined using a Lowry based method (DC assay; Bio-Rad Laboratories Inc., Hercules, CA, USA). All samples were studied together in duplicate. The protein samples (4.8 μg each in distilled H2O) were added into 384-well ELISA plates; the covered plates were incubated for 5 hours at 37°C. The wells were then blocked with 5% milk in Tris-buffered saline (TBS: 10 mM Tris-HCl, 140 mM NaCl, pH 7.4) for 1 hour at room temperature. After washing with wash buffer (0.05% Tween 20 in TBS), 20 μL mouse anti-PTEN antibody (1:100, R&D Systems Inc.) was added to each well. After overnight incubation at 4°C, the wells were washed five times with wash buffer. Secondary antibody (20 μL goat-anti-mouse IgG-HRP, 1:1000; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) was added and incubated for 1 hour at room temperature. After washing five times, 20 μL ABTS (2,2′-azino-bis[3-ethylbenzothiazoline-6-sulphonic acid]) was added into each well and incubated for 30 minutes at room temperature. Absorbance was measured at 405 nm using an ELISA reader. A qualitative comparison was made with corresponding controls.
Fluorescence microscopy
The cell viability was detected using a LIVE/DEAD Viability/Cytotoxicity Assay Kit (Life Technologies) as per the manufacturer’s instruction with a slight modification. Briefly, a total of 1×105 DBTRG cells were plated onto 24-well plates in 500 μL of MEM medium on day 0. The media were replaced with 50% or 100% conditioned media on day 1. On day 4, the cultures were washed twice with phosphate-buffered saline. Freshly prepared working solution (250 μL per well on 24-well plates, containing 1 μM acetomethoxy derivate of calcein and 2 μM ethidium homodimer-1) was then added directly to the cultures and incubated at room temperature for 10 minutes in the dark. The images were taken using a fluorescence microscope (IX71; Olympus Corporation, Tokyo, Japan) and the related analysis was performed through ImageJ (provided online by the National Institute of Health).
Direct monitoring of MSC migration
A micro speed photographic system (LEICA DMIRE2; Leica Microsystems, Wetzlar, Germany) was used to monitor MSC migration.
Statistical analysis
Numerical data were expressed as mean ± standard error. Statistical differences between the means for the different groups were evaluated with Prism 4.0 (GraphPad Software, Inc., La Jolla, CA, USA) using the Student’s t-test with the level of significance at P<0.05.
Results
PTEN expression in engineered MSCs
Figure 1 provides images of cells at 24 hours after electrotransfection. More than 60% of MSCs transfected with the plasmid control (pDsRed1-N1) were detected with red fluorescence (Figure 1A1 and A2). Under the same imaging condition, there was no visible positive cell on PTEN-RFP-transfected MSCs (Figure 1A3 and A4). The unexpected lack of red fluorescence in PTEN-RFP-engineered MSCs might be due to the interference of PTEN insert on the fusion protein. However, the immunoblotting results with anti-PTEN antibody showed an additional strong signal (~74 kDa) in the cell lysate from PTEN-RFP-engineered MSCs (Figure 1B1), in addition to the endogenous PTEN expression (~52 kDa). The immunoblotting results with anti-RFP antibody were consistent with the results of anti-PTEN antibody (Figure 1B2). ELISA analysis confirmed the presence of soluble PTEN in the culture supernatants (Figure 2). The PTEN content in the supernatants from MSCPTEN was significantly higher than that from the MSC control (P<0.05).
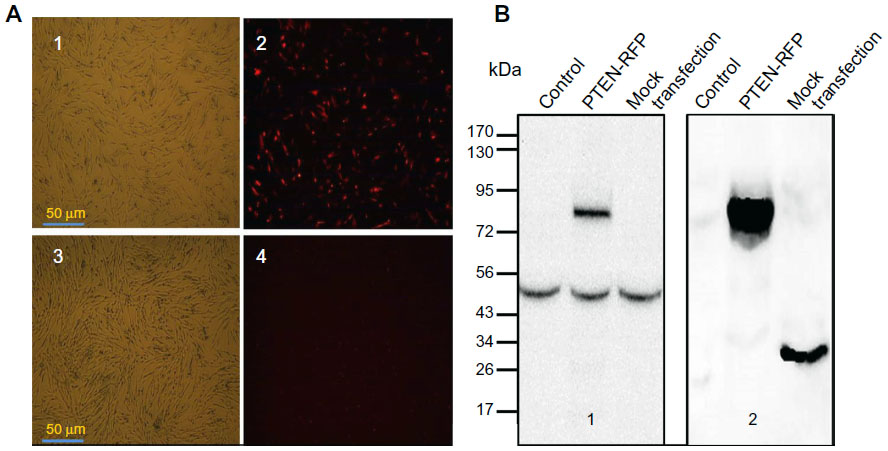 | Figure 1 PTEN expression and identification on PTEN-engineered MSCs. |
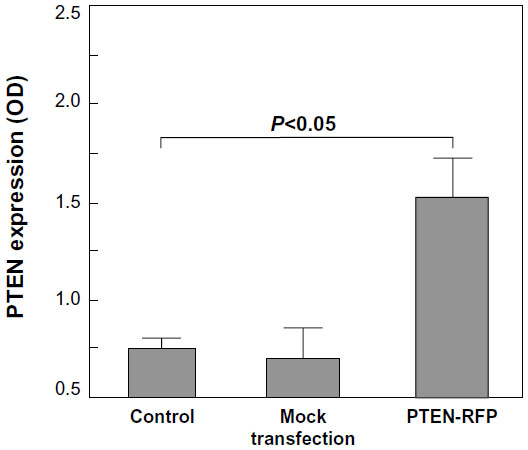 | Figure 2 ELISA analysis of PTEN in MSC culture media. |
MSCPTEN-mediated DBTRG cell death in indirect cocultures
As shown in Figure 3, DBTRG cell death was proportionally related to the conditioned media from MSCPTEN. The dose-dependent cell death indicates that MSCPTEN-derived PTEN is an important mediator responsible for DBTRG cell death during this indirect coculture. Marked cell death was not detected with conditioned media from native MSCs under the same experiment condition. It is worth noting that LIVE/DEAD assay only applies to the cells which remain on the culture surface during the staining. The detached cells, most of which are dead cells, are not included in the assessment.
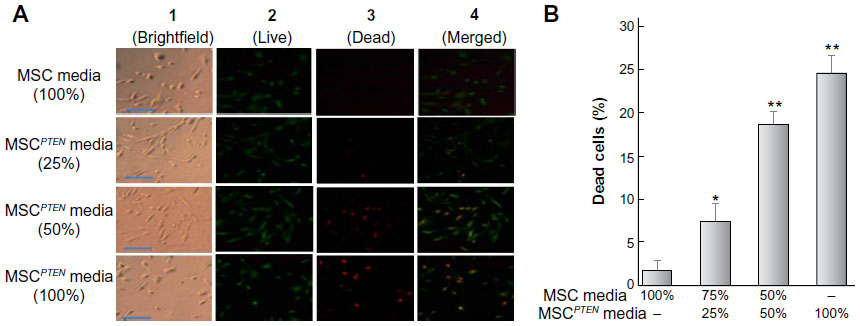 | Figure 3 DBTRG cell viability of indirect cocultures. |
MSCPTEN migration toward DBTRG cells
Figure 4 demonstrates the process of MSCPTEN migration toward DBTRG cells. A typical cell migration is highlighted in the red boxes. An MSC formed pseudopodium near a DBTRG cell. It took about 10 hours for MSCs to reach their targets (Figure 4A and B). Interestingly, a phagocytic phenomenon was observed in the real-time video. As indicated in the blue boxes, a phagocytosis-like action was clearly displayed. The real-time dynamic process can be viewed at Supplementary video.
 | Figure 4 Imaging demonstration of MSC’s migration toward DBTRG cells. |
Discussion
An MSC-mediated therapeutic strategy holds great potential to become a practically meaningful personalized treatment for cancer.5,6 There are several benefits to using an MSC-mediated therapy: 1) cancer targets can be specifically identified through multiple mechanisms; 2) the sensitivity of anticancer agents can be predetermined for a given cancer patient; 3) autologous MSCs eliminate ethical concerns surrounding heterologous stem cells; and 4) cell delivery route and administration frequency are flexible and can be customized according to the individual situation. The presence of MSCs in the tumor sites is critical to the success of the proposed strategy. As previously described, the capacity of MSCs for tumor-directed migration and incorporation has been widely demonstrated by both in vitro and in vivo studies. The present study was designed to prove the same properties of MSCs at the cellular level after they are engineered with anticancer genes.
PTEN functions as the central negative regulator of the PI3K-AKT-mTOR pathway in controlling apoptosis. The PI3K-AKT signaling pathway is the most frequently activated pathway in human cancers. It promotes cell growth, survival, and proliferation. It contributes to the evasion of apoptosis, loss of cell cycle control, and genomic instability during tumorigenesis through numerous mechanisms.16 PTEN dephosphorylates phosphatidylinositol (3,4,5)-trisphosphate (PIP3) to phosphatidylinositol 4,5-bisphosphate (PIP2), thereby directly opposing the activity of PI3K. In addition, PTEN plays a critical role in regulating the apoptotic threshold to multiple stimuli, including death ligands and chemotherapeutic agents.17 The loss of PTEN expression in a wide range of cancer cells reflects its importance in the maintenance of cancer cell survival.18 The frequency of its loss of expression in human glioblastoma is extremely high (over 50%).18 PTEN function restoration would inhibit cancer cell growth and might induce cell death under certain circumstances. In the present study, DBTRG cells were used as the target cells. This cell line originated from a female patient with glioblastoma. The majority of DBTRG cells lack chromosome 10 where the PTEN gene is located. Presumably, PTEN-engineered MSCs and DBTRG cells are an ideal model to reveal the practicability of MSC-mediated therapeutic strategy. The intracellular targets of PTEN include PIP3, mTOR, extracellular signal-regulated Kinase (ERK), and AKT. The intracellular sites of PTEN action involve all parts of the cell, including the sub-membrane, cytoplasm, and nucleus. The detailed mechanisms by which PTEN functions have been described by Chalhoub and Baker.19
In consideration of potential clinical applications, a secreting form of PTEN was integrated with a regular plasmid. Encouraging clinical trials were recently reported by Breitbach et al20 and Porter et al.21 In their studies, oncolytic poxvirus and lentiviral vector were successfully used to treat cancer patients. Our proposed strategy uses regular plasmids as vectors, thereby avoiding virus-related concerns. The upregulated PTEN expression was confirmed by immunoblotting and ELISA analysis (Figures 1 and 2) and functional experiments (Figure 3). Using the same expression vector, the shuffling of PTEN between MSCs and cancer cells was verified and reported at the 2012 World Congress on Engineering and Technology (Beijing, People’s Republic of China). According to the type of cancer, a series of anticancer genes can be transfected for specific sensitivity tests, including TNF-related apoptosis-inducing ligand, interferon (IFN)-α, IFN-β, IFN-γ, interleukin (IL)-2, and IL-12. Interested parties should refer to our recent review article.6 In order to make this MSC-mediated strategy therapeutically meaningful, it is critical to prove the homing capability of MSCs after they are engineered with anticancer genes, since the presence of engineered MSCs in the tumor microenvironment is the foundation for them to exert anticancer actions directly and/or indirectly.7 Figure 4 demonstrates the dynamic process of MSC migration toward DBTRG cells. The results provide direct evidence showing MSCPTEN’s cancer cell-directed migration under the culture conditions.
Although tumor-directed migration and integration of MSCs have been well recognized, their anticancer effects have not been precisely documented. The relationship between MSCs and cancer is complicated and sometimes contradictory; it is not an overstatement to describe MSCs as a “double-edged sword” in this regard.6 Nevertheless, we have demonstrated here that the combination of anticancer engineering and the homing capability of MSCs is able to improve cancer-killing effectiveness by releasing soluble anticancer agents directly to the target. The direct effects of MSCs on cancer cells through cell-to-cell interaction have not been investigated. The phagocytosis-like phenomenon which was observed in the present study needs to be further studied; MSCs are able to enhance the phagocytic activity of some types of cells, such as monocytes,22 microglia cells,23 and macrophages.24 A further investigation of MSCs’ phagocytosis activity may be beneficial to our understanding of these particular type of cells.
Conclusion
The expression of transfected PTEN in MSCs was identified by ELISA and immunoblotting analysis and confirmed with cell viability assessment of target cells. The cancer cell-oriented migration of PTEN-engineered MSCs was demonstrated by a real-time dynamic monitoring system. This study provides the first direct evidence of MSCs’ tropism post-anticancer gene engineering.
Acknowledgments
This work was supported by the Taihe Hospital Foundation, the University of British Columbia, the Vancouver General Hospital Foundation, and the Guangxi Natural Science Foundation (2013GXNSFAA019253). The authors are grateful to Crystal Robertson for her assistance in preparing the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | |
Friedenstein AJ, Piatetzky-Shapiro II, Petrakova KV. Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol. 1966;16(3):381–390. | |
Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968;6(2):230–247. | |
Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. | |
Loebinger MR, Janes SM. Stem cells as vectors for antitumour therapy. Thorax. 2010;65(4):362–369. | |
Dai LJ, Moniri MR, Zeng ZR, Zhou JX, Rayat J, Warnock GL. Potential implications of mesenchymal stem cells in cancer therapy. Cancer Lett. 2011;305(1):8–20. | |
Sun XY, Nong J, Qin K, Warnock GL, Dai LJ. Mesenchymal stem cell-mediated cancer therapy: A dual-targeted strategy of personalized medicine. World J Stem Cells. 2011;3(11):96–103. | |
Loebinger MR, Kyrtatos PG, Turmaine M, et al. Magnetic resonance imaging of mesenchymal stem cells homing to pulmonary metastases using biocompatible magnetic nanoparticles. Cancer Res. 2009;69(23):8862–8867. | |
Sasportas LS, Kasmieh R, Wakimoto H, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci U S A. 2009;106(12):4822–4827. | |
Kidd S, Spaeth E, Dembinski JL, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27(10):2614–2623. | |
Menon LG, Picinich S, Koneru R, et al. Differential gene expression associated with migration of mesenchymal stem cells to conditioned medium from tumor cells or bone marrow cells. Stem Cells. 2007;25(2):520–528. | |
Zischek C, Niess H, Ischenko I, et al. Targeting tumor stroma using engineered mesenchymal stem cells reduces the growth of pancreatic carcinoma. Ann Surg. 2009;250(5):747–753. | |
Moniri MR, Sun XY, Rayat J, et al. TRAIL-engineered pancreas-derived mesenchymal stem cells: characterization and cytotoxic effects on pancreatic cancer cells. Cancer Gene Ther. 2012;19(9):652–658. | |
Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62(13):3603–3608. | |
Sun XY, Nong J, Qin K, et al. MSC(TRAIL)-mediated HepG2 cell death in direct and indirect co-cultures. Anticancer Res. 2011;31(11):3705–3712. | |
Liu W, Zhou Y, Reske SN, Shen C. PTEN mutation: many birds with one stone in tumorigenesis. Anticancer Res. 2008;28(6A):3613–3619. | |
Yuan XJ, Whang YE. PTEN sensitizes prostate cancer cells to death receptor-mediated and drug-induced apoptosis through a FADD-dependent pathway. Oncogene. 2002;21(2):319–327. | |
Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J. 2004;382(Pt 1):1–11. | |
Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. | |
Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477(7362):99–102. | |
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. | |
Krasnodembskaya A, Samarani G, Song Y, et al. Human mesenchymal stem cells reduce mortality and bacteremia in gram-negative sepsis in mice in part by enhancing the phagocytic activity of blood monocytes. Am J Physiol Lung Cell Mol Physiol. 2012;302(10):L1003–L1013. | |
Giunti D, Parodi B, Usai C, et al. Mesenchymal stem cells shape microglia effector functions through the release of CX3CL1. Stem Cells. 2012;30(9):2044–2053. | |
Kim J, Hematti P. Mesenchymal stem cell-educated macrophages: a novel type of alternatively activated macrophages. Exp Hematol. 2009;37(12):1445–1453. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.